
Abstract
Mutations in the gene encoding Cu/Zn Superoxide Dismutase 1 (SOD1) protein are contemplated to be a protruding reason for Amyotrophic lateral sclerosis (ALS), which leads towards protein aggregation, misfolding and destabilization. Thus, we investigated the systematic action of entire mutations reported on electrostatic loop of SOD1 protein through thermodynamical and discrete molecular dynamics (DMD) studies. Accordingly, we analyzed the outcomes distinctly for screening the mutant structures having both, deleterious and destabilizing effect. Progressively, the impacts of those mutations on SOD1 were studied using DMD program. Surprisingly, our results predicted that the mutants viz., L126S, N139H and G141A to be the most destabilizing, misfolded and disease-causing compared to other mutants. Besides, the outcomes from secondary structural propensities and free energy landscapes, together assertively suggested that L126S, N139H and G141A tend to increase the formation of aggregates in SOD1 relative to other mutants. Hence, this study could provide an insight into the sprouting neurodegenerative disorder distressing the humans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Electrostatic interactions are considered to be key components in proteins that are highly specific in nature, which governs protein folding and function. Despite the impact on protein stability, their total actions also aid in preserving the 3D structure of protein [1,2,3].
Denervation of the upper and lower motor neurons results in the fatal neurodegenerative disorder, Amyotrophic lateral sclerosis (ALS). ALS is found to be unveiled in two different forms namely, Familial and Sporadic. The cause for the familial ALS is found to be the mutations in the gene encoding SOD1 protein while the causative factor the later remains elusive. Thus, we focused our study in elucidating the impact of the mutations in the SOD1 protein causing misfolding, aggregation and destabilization [4,5,6].
From the structural point of view, SOD1 is a homo dimer with 153 amino acids in with the Cu and Zn metal ions each monomer. The metal ions are found to stabilize the protein structure and aid in the dismutase activity [7,8,9,10,11]. Most importantly, the presence of the functionally essential loops i.e., Zn binding loop (49–82) and electrostatic loop (121–142) aid in retaining the structural architecture of the protein [12]. Further, reports from the earlier studies have stipulated that the presence of the Zn binding loop aids in upholding the coordination of the metal ions in the SOD1 protein. However, various disease-causing missense mutations were being reported on the Zn binding loop. We have previously analyzed the effect of those mutations on the Zn binding loop using the computational approaches that were agreeing with the various experimental studies [13,14,15,16]. Therefore, in this study we focused on the mutations reported in the electrostatic loop. From in-depth literature survey, numerous studies have suggested that enzyme-substrate encounter is improved by electrostatic support governed mainly by charged residues present in the electrostatic loop around Cu binding site. Moreover, an exceedingly conserved cluster of charged residues that are located on the surface of electrostatic loop forms an active site channel for the effectuate long-range assistance of superoxide [17, 18]. Besides, the location of charged residues and the structure of the electrostatic loop are also essential for upholding catalytic activity and enzymatic behavior of SOD1. The mutations in electrostatic loop residues are found to change in SOD1 activity that in turn aids towards the cause of fatal neurodegenerative disorder familial amyotrophic lateral sclerosis (FALS) [19, 20]. Varying studies from experimental and theoretical background stipulated that the mutations on the electrostatic loop often leads to structural and dynamic changes of SOD1, thereby improvising the disease onset [21,22,23,24]. Moreover, the mutations in electrostatic loop residues cause protein misfolding and increased intermolecular interaction, thereby leading to the formation of protein aggregates [25].
In this study, we focused on an entire set of mutations reported on the electrostatic loop residues from ALSoD1 database [26]. ALSoD1 (http://alsod.iop.kcl.ac.uk), is a database that is freely available with the records of various information regarding mutations, genotype, phenotype and geographical information of various genes involved in both, the sporadic and familial ALS from the data reported all over the world. We computationally analyzed the effect of each mutation on SOD1 using various parameters depending on protein function, stability, thermodynamics and molecular dynamics. Thus, we determined the most lethal mutant regarding aggregation and misfolding in SOD1 protein using computational approach. Hence, the study aid in profound enlightening knowledge on the effect of mutations on the electrostatic loop residues, which could provide an initiative for understanding the biological relevance of the electrostatic loop in SOD1.
2 Materials and Methods
2.1 Data Collection
The sequence of Human SOD1 protein with UNIPORT id: P00441 was obtained from UniProt database [27]. Besides, the structure of wild type SOD1 protein [PDB ID: 2V0A(A)] were acquired from the PDB database [28]. Mutant structures of electrostatic loop residues were modeled, using SWISS-PDB viewer [29]. Further, the obtained structures were energy-minimized, using YASARA program. The wild type and mutant structures were energy minimized, using AMBER14 force field in YASARA program. The entire system was solvated within the cubic box with a dimension of 1.0 nm. Addition of a Na+ ion neutralized the charge of the system. The temperature was maintained at 298 K. Periodic boundary conditions were included during the energy minimization of all the structures. Particle Mesh Ewald [30] (PME) and van der Waal’s interaction were included for the long–range interactions.
2.2 Effect of Mutation on Protein Function
Consequently, the electrostatic loop mutations reported on SOD1 was evaluated with PREDICT SNP [31]. PREDICT SNP is a consensus classifier, which combines the prediction of best six prediction tools (MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT and SNAP), for accurate predictions along with prediction score.
2.3 Predicting the Structural Stability Upon Mutation
Stability of SOD1 upon mutations in the electrostatic loop residues was determined, using DynaMut web server [32]. The web server examines and visualizes the protein dynamics by sampling conformations and assesses the impact of mutations on protein dynamics and stability resulting from vibrational entropy changes, using normal mode approaches. Moreover, it also integrates graph-based signatures to create a consensus prediction of the influence of a mutation on protein stability.
2.4 Discrete Molecular Dynamics (DMD) Simulation
DMD is a distinctive molecular dynamic simulation, which uses the swift processing of event-driven molecular dynamics with the potential function for calculating interactions. Medusa force field was used for performing the dynamic calculations [33, 34]. Consequently, united-atom model was used for the representations. Besides, the system were solvated using the Lazaridis–Karplus implicit solvation [35]. Hydrogen bonds interactions were modeled using the Reaction-like algorithm [36]. Furthermore, the charge–charge interactions were modeled using the Debye–Hu ̈ckel approximation, with Debye length setting to 10 Å. The simulations were carried out with periodic boundary conditions. The temperature was maintained using Andersen’s thermostat [37]. Further, the binding of metal ions was modeled by assigning the distance constraints between each metal atom and the corresponding metal-coordinating atoms. The distance and coordination dependence of disulfide bond establishment was modeled, using a reaction algorithm [38]. Moreover, the previous reports have profoundly suggested that the medusa force field is more appropriate than other force filed in performing the simulation with the divalent metal ions, Cu and Zn in SOD1. The time units in DMD simulations refer to the unit of time [T] that is determined by units of mass [M], length [L] and energy [E], which are Dalton (1.66 × 10−24 g), angstrom (10−10 m), and kcal/mol (6.9 × 10−22 J), respectively. Therefore, each time unit corresponds to approximately 50 fs as relative with classical MD [36]. Thus, the time period of the entire simulations was of 5 ns. Consequently, the snapshots were secured for every 100-timeunits (tu) for wild type and mutant SOD1, over the simulation time.
2.5 Geometrical Assessment
The trajectories obtained from DMD simulation for wild type and mutant SOD1 were subjected to geometrical evaluation, using g_rms (conformational deviation), g_rmsf (conformational flexibility) and g_gyrate (protein gyration/Rg) from GROMACS tools.
2.6 Cross-Correlation Matrix
The dynamic cross-correlation matrix (DCCM), DCCij that reflects the fluctuations of Cα atoms of protein over the dynamic period was computed using g_covara, to examine the collective motions of wild type and mutant protein systems. The covariance matrix was made between atoms, j and i that measures the correlative nature of atomistic fluctuations. The equation to calculate the cross-correlation is as follows.
where Δri and Δrj correspond to atomic displacement vectors for atoms, i and j, respectively, from their mean position concerning time interval [39].
2.7 Free Energy Landscape
Free energy landscape of protein was acquired using conformational sampling method, which provides the near-native structural conformation. Herein, we used DMD to sample the conformations of wild type and mutant SOD1, correspondingly. To obtain the free energy landscape, we utilized two essential components such as conformational deviation and protein gyration as the reaction coordinates in our study. The energy landscape was computed with these two components using the equation,
where ∆G is the Gibbs free energy of state, kB is the Boltzmann constant, T is the temperature of simulation. Considering two different reaction coordinates, p1 and p2, the two-dimensional free-energy landscapes were obtained from the joint probability distributions, P(p1, p2) of the system [40].
2.8 Statistical Validation
Statistical method on dynamic evaluation stipulates an significant assessment with the experimental studies [41]. Therefore, the outcomes obtained from the simulation studies were exposed to statistical substantiation using STATPLUS program.
3 Results and Discussions
Modification of particular amino acid in proteins may alter the biochemical functions, folding and stability leading to various disease state conditions in humans [42, 43]. Accordingly, the present study provides insight towards the effect of point mutations reported on the electrostatic loop residues of SOD1, which is found to be a causative factor for FALS, due to aggregation and misfolding. Further, we computationally analyzed the mutational effect on SOD1 regarding functionality and stability to elucidate the disease-causing fatal mutants, which could alter the structure and function of the SOD1 protein.
3.1 Mutation Functionality and Stability Prediction
Initially, the influence of mutation on the SOD1 structural functionality and stability were predicted using the PREDICT SNP and DynaMut web-servers. Further, the predicted results from PREDICT SNP implied the deleterious effect on SOD1 function upon mutation except for E121G. Therefore, the outcomes from the predictions unveiled the influence of mutation on SOD1 function.
Mutations that were screened from PREDICT SNP were subjected to the stability analysis. Previous findings have stated that the single point mutation in a protein might lead to variations in protein stability [44,45,46]. Therefore, we monitored the structural changes in the stability of the mutant SOD1, using DynaMut program (Table 1). Remarkably, several mutations viz., L126S, G138E, N139D, N139H, G141A and G141E were found to destabilize the SOD1 protein, while the other mutants exhibited the stabilizing effect on SOD1 protein. The use of PREDICT SNP and DynaMut programs were reported to have more predictive accuracy and more reliability as compared to other prediction programs reported for the protein mutational studies concerning the function and stability features. Therefore, the screened mutants having both deleterious and destabilizing effect on SOD1 function and stability were used for further molecular dynamics studies.
3.2 Mutational Effect on the Protein Structure Over the Dynamic Period
The influence of mutations on SOD1 protein was exclusively studied using discrete molecular dynamics simulation in order to elucidate the change in the structural stability, flexibility compactness, FEL and overall secondary structural features.
3.2.1 Conformational Structural Studies on SOD1
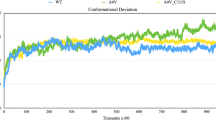
Conformational ensembles of the wild type and mutant SOD1 proteins (L126S, G138E, N139D, N139H, G141A and G141E) were inspected via different factors to decrypt the effect of the mutation on the structural and residual level. Initially, the conformational stability was computed using RMSD parameter, which suggested the overall conformational deviation of the wild type and mutant SOD1 protein (Table 2). The c-alpha carbon atom of protein structures was utilized for computing the conformational deviation (Fig. 1a). From the overall examination, we deciphered that L126S and N139H attained greater RMSD values with 0.50 and 0.54 nm as considered to that of wild type with 0.45 nm. Moreover, the RMSD values of other mutant proteins exhibited a relative deviation as shown in the descending order, N139D (0.48) > G138E (0.47) > G141E (0.44) > G141A (0.43 nm). Thus, suggesting that the mutation in L126S and N139H has rigorously affected the proteins conformation deviation compared to all the other mutants. Further, the disparity in the movement of the electrostatic loop was monitored by computing the RMSD of the residues ranging from (121–142) in all the protein structures. Unexpectedly, an increased RMSD was found in mutants, L126S and N139H with 0.76 and 0.68 nm, respectively, than the wild type (0.53 nm) (Fig. 1b). Exceptionally, the graphical representation showed a larger deviation for L126S and N139H as compared to the wild type and other mutants over the period of simulation. Besides, G138E, N139D, G141A and G141E showed a marginally similar RMSD to that of wild type with 0.51, 0.51, 0.53 and 0.52 nm respectively. Therefore, we could endorse that L126S and N139H had radically altered the motion of electrostatic loop that supported for the increased RMSD in mutant SOD1, when contrasted with wild type SOD1. Furthermore, the results from both the studies were found to be statistically significant with p value < 0.001.

Geometrical parameters used for analyzing the mutational effect on SOD1 protein. a Overall conformational change in the backbone of SOD1 upon various mutations computed over the trajectory. b Influence of mutations altering the deviation on the electrostatic loop of SOD1. c Protein compactness computed for SOD1 and its mutant types thought the simulation time. d Residual fluctuation of the wild type and mutant SOD1 protein
Similarly, the total compactness of wild type and mutant SOD1 proteins were examined, using the g_gyrate tool. The results obtained were mapped using Xmgrace tool as portrayed in Fig. 1c. From the graph 1c, we could decipher that L126S, N139H and G141A showed greater deviation in protein compactness as compared to wild type and other mutant proteins. Indeed, the average compactness was also found to be higher in those mutant proteins with 1.49 nm, 1.48 nm and 1.49 nm, respectively, relative to wild type (1.47 nm). Other mutant proteins, G138E, N139D and G141E showed an average Rg value with 1.47 nm, 1.46 nm and 1.46 nm, respectively. In general, we found that the mutation at residue position 126 and 139 (H) had directed the distorted conformational movement and hammering the compactness of SOD1 protein as compared to wild type.
3.2.1.1 Residual Fluctuation
The mobility of wild type and mutant SOD1 proteins was scanned through root mean square fluctuation (RMSF) that measures the flexibility of individual residue of a protein. The RMSF profiles of all proteins were combinedly plotted and represented in Fig. 1d. From Fig. 1d, we found that the mutants G138E and N139D showed less fluctuation relative to wild type, throughout the simulation studies. Whereas, the mutants, L126S and N139H exhibited increased residual flexibility comparatively of other mutant forms.
On the other hand, the residual flexibility of G141A and G141E were marginally similar to wild type. With the outcomes from RMSF studies, we could infer that the flexibility of 153 residues in SOD1 was drastically altered upon the substitution of Ser and His at 126th and 139th residual position. From this result, we noticed that the substitution of residue Ser and His at L126 and N139 in SOD1 not only affected its residue flexibility but distorted the flexibility of nearby residues as well, perchance by the shift in their secondary structural states.
Further, we computed the average RMSF values for the wild type and mutant forms of SOD1. Collectively, the results elucidated that the average residual flexibility of L126S (0.20 nm) and N139H (0.20 nm) were relatively greater than all the other forms of mutants viz., G138E (0.16 nm), N139D (0.16 nm), G141E (0.19 nm), G141A (0.19 nm) and wild type (0.19 nm). This result confirmed that the mutation in the electrostatic loop of SOD1 (L126S and N139H) could augment the overall flexibility thus urging towards the loss in protein conformational stability, which could be a root cause for protein misfolding. However, in comparison of all the RMSF fluctuation peaks, N139H and L126S showed quit higher fluctuation, than all other proteins, which corroborates with our results from structural conformation studies. The structural representation of wild type SOD1 with the position of mutations (ball and stick) in the electrostatic loop regions (orange) were shown in Fig. 2.

3-D structural representation of wild type SOD1 (grey) with the electrostatic loop highlighted in orange color. The residual position of mutations in the electrostatic loop represented within the box. (Color figure online)
3.2.1.2 Quantifying the Effect of Mutation Around the Electrostatic Loop Region
To enumerate the effect of mutation in residues surrounding the electrostatic loop, we selected the average structure with least energy from the entire trajectories of wild type and mutant forms of SOD1. Further, we graphed the residues around 6 Å of every mutation and mapped the structural changes in comparison with wild type (Fig. 3). From the results, we could conclude that the substitution of Ser, His and Ala at the residual position 126, 139 and 141 had utterly destroyed the formation of helix in the electrostatic loop, which governs for the structural stability of SOD1. Moreover, the number of adjacent residues within 6 Å of the mutated region in L126S (16), N139H (16), N139D (18) and G141A (23) were found to be reduced relative to wild type (33), G138E (25) and G141E (25). We also computed the difference in the RMSD of the proximal residues around the electrostatic loop of the mutant forms with the wild type. Remarkably, the outcomes exposed that L126S (5.16 nm), N139H (5.74 nm) and G141A (5.05 nm) showed greater structural deviation than the other mutants with wild type SOD1. Hence, our results agreed with our previous findings of conformational stability and the flexibility suggesting that the SOD1 bearing the mutations L126S, N139H and G141A could radically alter the stability and flexibility thereby admonishing towards the misfolded conformations of SOD1.

Local structural (cartoon) and residual (wireframe) change represented within 6 Å of the electrostatic loop for mutant SOD1 proteins in comparison with wild type (grey)
3.2.1.3 Concerted Motion of Wild Type and Mutant Proteins
Concerted dynamics of the protein over the trajectory was accomplished to investigate the conformational changes over the backbone atoms of wild type and mutant SOD1. The diagonalized covariance matrix using atomic fluctuation in coordinate space was computed by employing the g_covar module. This module endows a set of eigenvectors and resultant eigenvalues, which describes the collective motions every atom along with its directions. The tenacity of covariance matrix analysis is to unearth the correlated and anti-correlated motions of atoms. Thus, the positive correlation between two atoms reveals a sterenous motion by the same direction, while the negative correlation signifies an opposite direction of motion. The outcomes from the matrix are visualized as red and grey color. These colors define whether the residues moves together (red) or vice-versa (grey) (Fig. 4). Here, the covariance matrices were structured and analyzed to bestow a perception about the correlated and anti-correlated conformational motion of wild type and mutant SOD1. From our study, we observed that the anti-correlation regions were conspicuously seen in mutant as compared to wild type SOD1. Moreover, we could visualize a dissimilar pattern in the correlation of mutant SOD1, thus evoking the cause for the discrepancies in the conformational stability and flexibility of SOD1 relative to wild type. Therefore, the concerted motion of wildtype SOD1 exhibiting the strong correlation in the region of the electrostatic and Zn binding loops was found altered, thus leading to the loss in the atomistic motion of SOD1 upon the notable mutations in the electrostatic loop region.

Diagonalized covariance matrix computed at the atomistic level for wild type and mutants SOD1 proteins from DMD trajectory
We further analyzed the significance of concerted motions by the projection of eigenvector 1 and 2, which corresponds to the eigen values defining the active involvement of each component to the motion. The projection of eigen vector 1 (PCA1) and eigen vector 2 (PCA2) was computed and plotted on 2D plane (Fig. 5) for the screened mutant SOD1 proteins in comparison with wild type. The results mapped on 2D graph suggest that the wild type has a confined conformational basin with stronger density, thus exhibiting folded and defined motion of SOD1. In contrast to wild type, a clear estrangement in the conformational behavior with the distinct pattern of wider conformational basin was observed in L126S, N139H and G141A. Moreover, the change in concerted motion and the discrepancies in conformational dynamics of these mutants could be due to the loss in their residual flexibility and stability of SOD1. Notably, the transition in the secondary structural features could be seen in these mutants, owing to the loss in the frustration along PCA1 and PCA2. Whereas, the other mutant forms G138E, N139D and G141E showed only a marginal displacement in their concerted motions with relatively spread basins in comparison with wild type. With the aforementioned results, we could conjecture that the substitution of amino acids in L126S, N139H and G141A are found to be most significant that could alter the folding pattern and structural geometries of SOD1.

Concerted motions of mutant SOD1 computed over the simulation time were mapped using the principal component of eigenvectors 1 and 2 in comparison with wild type (black)
3.2.2 Secondary Structure Composition
To broaden our study, we determined the propensity of secondary structure by utilizing DSSP program for the ensemble of wild type and mutant SOD1 proteins (Table 3). Analysis from the outcomes signified that the arrangement of alpha helix in wild type SOD1 (5%) was marginally similar with G138E (5%), N139D (4%) and G141E (5%), discretely. Besides, L126S, N139H and G141A do not exhibit the formation of helix over the simulation time. Moreover, the formation of helix structure profoundly seen in the electrostatic loop residues of wild type SOD1 was lost due to the substitution of Ser, His and Ala at residual position 126, 139 and 141 of SOD1. To support the aforementioned results, we mapped the structural snapshots of wild type and mutant SOD1 proteins throughout simulation time. Remarkably, the pictorial representations exposed the loss in the helical structure of SOD1 upon the mutation L126S, N139H and G141A in comparison with wildtype (Fig. 6). Moreover, the aforesaid statement could be the causative factor for the augmented deviation in the electrostatic loop of L126S and N139H as compared to other mutants and wild type SOD1. On the other hand, the propensity of beta-sheets was discovered to be higher in L126S (43%) and N139H (44%) than wild type SOD1 (41%). Consequently, we could suggest that the change of alpha helix has provoked the increase of beta sheets in these mutants. Further, the alteration of helix into beta-sheets are considered to be the major concerns in the formation of amyloid fibrils that unswervingly associates to the escalation in aggregation propensity [47, 48]. Furthermore, the content of beta-sheets in others mutants, N139D > G138E > G141A > G141E was found reduced with 41%, 38%, 37% and 33%, respectively, as compared to wild type. Reports have shown that the beta-sheet endure a significant element in protein folding [49]. Thus, the change in beta sheets could bring about protein misfolding, ultimately provoking to the diseased states [50]. Besides, the propensity of coils exhibited an augmented content in L126S, G138E, N139D, N139H, G141A and G141E with 53%, 55%, 50%, 52%, 59% and 59%, respectively, compared to wild type (51%). In addition, the content of beta-bridge also followed a similar trend within the range of 4–6% in mutants relative to wild type (3%). Overall, our investigations signified that these mutants could be destructive not only in the alteration of protein stability and functionality, however, prompt towards misfolding in SOD1 that eventually, ends in neurodegenerative disorder [51, 52].

Time-lapse shots of mutant SOD1 proteins superimposed to wild type (grey) along with the computed average structure over the simulation period
3.2.3 2-D Free Energy Landscape
To provide a better understanding of the profound effect of mutations urging towards the proteins misfolding and aggregation, we mapped the folding free energy funnel for wild type and mutant SOD1. Further, we computed the free energy landscape funnel by utilizing RMSD and Rg coordinates of wild type and mutant SOD1, individually (Fig. 7). From Fig. 7, we could decipher that the folding free energy funnel of wild type protein was confined within one particular region exhibiting a folded state of the protein. Further, the coordinates of RMSD and Rg values was found confined within 0.20 nm and 1.50 nm, respectively. Unfortunately, L126S, N139H and G141A showed an increased number of misfolded conformers by forming multiple metastable states with lower Gibbs free energy. Moreover, RMSD and Rg coordinate values in these mutations were also seen to be greater than wild type falling within a range of 0.20–0.30 nm and 1.50–1.55 nm, respectively. Reports from our previous studies have stipulated that the formation of multiple metastable states could be a causative factor in forming the toxic aggregates in protein [53,54,55]. Our results could suggest that L126S, N139H and G141A could lead to the formation of toxic aggregates in SOD1 and improvise the death rate of patients as compared to wild type. On the other hand, G138E, N139D and G141E displayed altered folding free energy funnel, which could be due to single point substitution mutations on SOD1. The folding free energy funnels of these mutants were different as compared to wild type. Further, there were differences in the folding patterns and formation of metastable states in these mutations. RMSD and Rg values of these mutants were also found to have some discrepancies relative to wild type. Therefore, we could categorize these mutants as aggregation (L126S, N139H, and G141A) and misfolding (G138E, N139D and G141E) prone mutants in comparison with wild type. Overall, our study aided in providing a better outlook for studying the effect of mutations on the functionally important electrostatic loop residues.

Change in 2-D free energy landscape compared between wild type and mutant SOD1 with varying Gibbs free energy
Therefore, the present study outlined the effect of distinct mutations reported on functionally important electrostatic loop, using various computational tools. Initially, we screened the mutations in electrostatic loop of SOD1 that affect the protein functionality and stability, using PREDICTSNP and DynaMut programs discretely. Further, the mutations with both, the deleterious and destabilizing effect were screened to broaden our study. The screened mutations were subjected towards the DMD studies in comparison with the wild type SOD1. Subsequently, we analyzed the impact of mutations on SOD1 from the obtained trajectory using different geometrical parameters, thereby postulating an overall understanding on protein stability, compactness, interatomic contacts, structural disparities, secondary structural propensity and free energy landscapes. Accordingly, the factors above pointed out that the mutations (L126S and N139H) had directed towards the structural destabilization with augmented beta-sheet propensity relative to wild type SOD1.
Moreover, the results from the free energy landscape also suggested that L126S, N139H and G141A led to the formation of more toxic aggregates in SOD1 as compared to other mutants. In general, our results provided better reasoning and noticeable changes in the structural features of SOD1 upon mutations in electrostatic residues causing fALS in human beings. Overall, the study stipulates a perception over the deleterious and destabilizing mutants on electrostatic loop of SOD1 by computationally clarifying their effect on protein structural features along with free energy landscapes. Hence, our study could be helpful in providing an insight into the mutational effect on the electrostatic loop of SOD1 causing the sprouting neurodegenerative disorder ALS that distresses the life of human.
References
Kumar S, Nussinov R (2002) Close-range electrostatic interactions in proteins. ChemBioChem 3:604–617. https://doi.org/10.1002/1439-7633(20020703)3:7%3C604::AID-CBIC604%3E3.0.CO;2-X.
Zhou H-X, Pang X (2018) Electrostatic interactions in protein structure, folding, binding, and condensation. Chem Rev 118:1691–1741. https://doi.org/10.1021/acs.chemrev.7b00305
Nakamura H (1996) Roles of electrostatic interaction in proteins. Q Rev Biophys 29:1–90. https://doi.org/10.1017/S0033583500005746
Tan W, Pasinelli P, Trotti D (2014) Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim Biophys Acta 1842:1295–1301. https://doi.org/10.1016/j.bbadis.2014.02.009
Karch CM, Borchelt DR (2008) A limited role for disulfide cross-linking in the aggregation of mutant SOD1 linked to familial amyotrophic lateral sclerosis. J Biol Chem 283:13528–13537. https://doi.org/10.1074/jbc.M800564200
Kunishige M, Hill KA, Riemer AM, Farwell KD, Halangoda A, Heinmöller E, Moore SR, Turner DM, Sommer SS (2001) Mutation frequency is reduced in the cerebellum of Big Blue® mice overexpressing a human wild type SOD1 gene. Mutat Res 473:139–149. https://doi.org/10.1016/S0027-5107(00)00120-2
Osredkar J (2011) Copper and zinc, biological role and significance of copper/zinc imbalance. J Clin Toxicol. https://doi.org/10.4172/2161-0495.S3-001
Radunović A, Leigh PN (1996) Cu/Zn superoxide dismutase gene mutations in amyotrophic lateral sclerosis: correlation between genotype and clinical features. J Neurol Neurosurg Psychiatry 61:565–572. https://doi.org/10.1136/jnnp.61.6.565
Valentine JS, Doucette PA, Zittin Potter S (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 74:563–593. https://doi.org/10.1146/annurev.biochem.72.121801.161647
Furukawa Y, O’halloran TV (2006) Posttranslational modifications in Cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal 8:847–867. https://doi.org/10.1089/ars.2006.8.847
Galaleldeen A, Strange RW, Whitson LJ, Antonyuk SV, Narayana N, Taylor AB, Schuermann JP, Holloway SP, Hasnain SS, Hart PJ (2009) Structural and biophysical properties of metal-free pathogenic SOD1 mutants A4V and G93A. Arch Biochem Biophys 492:40–47. https://doi.org/10.1016/j.abb.2009.09.020
Rakhit R, Chakrabartty A (2006) Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:1025–1037. https://doi.org/10.1016/j.bbadis.2006.05.004
Srinivasan E, Sethumadhavan R, Rajasekaran R (2017) A theoretical study on Zn binding loop mutants instigating destabilization and metal binding loss in human SOD1 protein. J Mol Model 23:103. https://doi.org/10.1007/s00894-017-3286-z
Srinivasan E, Rajasekaran R (2017) Computational simulation analysis on human SOD1 mutant (H80R) exposes the structural destabilization and the deviation of Zn binding that directs familial amyotrophic lateral sclerosis. J Biomol Struct Dyn 35:2645–2653. https://doi.org/10.1080/07391102.2016.1227723
Srinivasan E, Rajasekaran R (2018) Deciphering the loss of metal binding due to mutation D83G of human SOD1 protein causing FALS disease. Int J Biol Macromol 107:521–529. https://doi.org/10.1016/j.ijbiomac.2017.09.019
Srinivasan E, Rajasekaran R (2017) Exploring the cause of aggregation and reduced Zn binding affinity by G85R mutation in SOD1 rendering amyotrophic lateral sclerosis. Proteins 85:1276–1286. https://doi.org/10.1002/prot.25288
Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA (1992) Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature 358:347–351. https://doi.org/10.1038/358347a0
Polticelli F, Bottaro G, Battistoni A, Carrì MT, Djinovic-Carugo K, Bolognesi M, O’Neill P, Rotilio G, Desideri A (1995) Modulation of the catalytic rate of Cu, Zn superoxide dismutase in single and double mutants of conserved positively and negatively charged residues. Biochemistry 34:6043–6049
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. https://doi.org/10.1038/362059a0
Culotta VC, Yang M, O’Halloran TV (2006) Activation of superoxide dismutases: putting the metal to the pedal. Biochim Biophys Acta 1763:747–758. https://doi.org/10.1016/j.bbamcr.2006.05.003
Rotunno MS, Bosco DA (2013) An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. https://doi.org/10.3389/fncel.2013.00253
Healy EF, Cervantes L (2016) An in silico study of the effect of SOD1 electrostatic loop dynamics on amyloid-like filament formation. Eur Biophys J 45:853–859. https://doi.org/10.1007/s00249-016-1163-9
Keerthana SP, Kolandaivel P (2015) Structural investigation on the electrostatic loop of native and mutated SOD1 and their interaction with therapeutic compounds. RSC Adv 5:34630–34644. https://doi.org/10.1039/c5ra00286a
Ciriolo MR, Battistoni A, Falconi M, Filomeni G, Rotilio G (2001) Role of the electrostatic loop of Cu, Zn superoxide dismutase in the copper uptake process. Eur J Biochem 268:737–742. https://doi.org/10.1046/j.1432-1327.2001.01928.x
Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ (2003) Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol 10:461–467. https://doi.org/10.1038/nsb935
Wroe R, Wai-Ling Butler A, Andersen PM, Powell JF, Al-Chalabi A (2008) ALSOD: the amyotrophic lateral sclerosis online database. Amyotroph Lateral Scler 9:249–250. https://doi.org/10.1080/17482960802146106
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:D115–D119. https://doi.org/10.1093/nar/gkh131
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Kaplan W, Littlejohn TG (2001) Swiss-PDB viewer (deep view). Brief Bioinform 2:195–197. https://doi.org/10.1093/bib/2.2.195
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N⋅log (N) method for Ewald sums in large systems. J Chem Phys 98:10089. https://doi.org/10.1063/1.464397
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10:e1003440. https://doi.org/10.1371/journal.pcbi.1003440
Rodrigues CHM, Pires DEV, Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46:W350–W355. https://doi.org/10.1093/nar/gky300
Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI (1998) Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des 3:577–587. https://doi.org/10.1016/S1359-0278(98)00072-8
Shirvanyants D, Ding F, Tsao D, Ramachandran S, Dokholyan NV (2012) Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys Chem B 116:8375–8382. https://doi.org/10.1021/jp2114576
Lazaridis T, Karplus M (1999) Effective energy function for proteins in solution. Proteins 35:133–152
Ding F, Tsao D, Nie H, Dokholyan NV (2008) Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 16:1010–1018. https://doi.org/10.1016/j.str.2008.03.013
Andersen HC (1980) Molecular dynamics simulations at constant pressure and/or temperature. J Chem Phys 72:2384–2393. https://doi.org/10.1063/1.439486
Ding F, Dokholyan NV (2008) Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci USA 105:19696–19701. https://doi.org/10.1073/pnas.0803266105
McCammon JA (1984) Protein dynamics. Rep Prog Phys 47:1. https://doi.org/10.1088/0034-4885/47/1/001
Papaleo E, Mereghetti P, Fantucci P, Grandori R, De Gioia L (2009) Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: the myoglobin case. J Mol Graph Model 27:889–899. https://doi.org/10.1016/j.jmgm.2009.01.006
Likic V, Gooley PR, Speed TP, Strehler EE (2005) UBER VIP P value A statistical approach to the interpretation of molecular dynamics simulations of calmodulin equilibrium dynamics. Protein Sci 14:2955–2963. https://doi.org/10.1110/ps.051681605
Teng S, Wang L, Srivastava AK, Schwartz CE, Alexov E (2010) Structural assessment of the effects of amino acid substitutions on protein stability and protein-protein interaction. Int J Comput Biol Drug Des 3:334–349. https://doi.org/10.1504/IJCBDD.2010.038396
Reva B, Antipin Y, Sander C (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39:e118. https://doi.org/10.1093/nar/gkr407
Tokuriki N, Tawfik DS (2009) Stability effects of mutations and protein evolvability. Curr Opin Struct Biol 19:596–604. https://doi.org/10.1016/j.sbi.2009.08.003
Beckerman M (2015) Fundamentals of neurodegeneration and protein misfolding disorders. Springer, New York
Ye L, Wu Z, Eleftheriou M, Zhou R (2007) Single-mutation-induced stability loss in protein lysozyme. Biochem Soc Trans 35:1551–1557. https://doi.org/10.1042/BST0351551
Cerdà-Costa N, Esteras-Chopo A, Avilés FX, Serrano L, Villegas V (2007) Early kinetics of amyloid fibril formation reveals conformational reorganisation of initial aggregates. J Mol Biol 366:1351–1363. https://doi.org/10.1016/j.jmb.2006.12.007
Dong M, Li H, Hu D, Zhao W, Zhu X, Ai H (2016) Molecular dynamics study on the inhibition mechanisms of drugs CQ1–3 for Alzheimer amyloid-β40 aggregation induced by Cu2+. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.5b00343
Khandogin J, Brooks CL (2007) Linking folding with aggregation in Alzheimer’s β-amyloid peptides. Proc Natl Acad Sci USA 104:16880–16885. https://doi.org/10.1073/pnas.0703832104
Gross M (2000) Proteins that convert from alpha helix to beta sheet: implications for folding and disease. Curr Protein Pept Sci 1:339–347. https://doi.org/10.2174/1389203003381289
Meiering EM (2008) The threat of instability: neurodegeneration predicted by protein destabilization and aggregation propensity. PLoS Biol 6:1383–1385. https://doi.org/10.1371/journal.pbio.0060193
Stevens JC, Chia R, Hendriks WT, Bros-Facer V, Van Minnen J, Martin JE, Jackson GS, Greensmith L, Schiavo G, Fisher EMC (2010) Modification of superoxide dismutase 1 (SOD1) properties by a GFP tag—implications for research into amyotrophic lateral sclerosis (ALS). PLoS ONE 5:e9541. https://doi.org/10.1371/journal.pone.0009541
Srinivasan E, Rajasekaran R (2018) Cysteine to serine conversion at 111th position renders the disaggregation and retains the stabilization of detrimental SOD1 A4V mutant against amyotrophic lateral sclerosis in human—a discrete molecular dynamics study. Cell Biochem Biophys 76:231–241. https://doi.org/10.1007/s12013-017-0830-5
Srinivasan E, Rajasekaran R (2017) Probing the inhibitory activity of epigallocatechin-gallate on toxic aggregates of mutant (L84F) SOD1 protein through geometry based sampling and steered molecular dynamics. J Mol Graph Model 74:288–295. https://doi.org/10.1016/j.jmgm.2017.04.019
Srinivasan E, Rajasekaran R (2016) Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Adv 6:102744–102753. https://doi.org/10.1039/c6ra21927f
Acknowledgements
The authors thank the management of VIT (deemed to be University) for providing the VIT-SEED GRANT, facilities and encouragement to carry out this research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Srinivasan, E., Rajasekaran, R. Computational Investigation on Electrostatic Loop Mutants Instigating Destabilization and Aggregation on Human SOD1 Protein Causing Amyotrophic Lateral Sclerosis. Protein J 38, 37–49 (2019). https://doi.org/10.1007/s10930-018-09809-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-018-09809-0