
Abstract
Hyaluronic acid (HA) is a biopolymer used in several industries. There is increasing global demand. HA is normally produced on a large scale using attenuated strains of group C streptococci that are pathogenic and fastidious. Accordingly, it is of interest to use a “generally recognized as safe” (GRAS) organism such as Bacillus subtilis for HA production. Here, we report an engineered B. subtilis strain named WmB that produces different molecular weights (MW) and titers of HA at different temperatures. The faster the bacteria grew, the lower the MW of HA produced and the higher the titer. The MW of HA obtained ranged from 6.937 MDa at 47 °C to 0.392 MDa at 32 °C. At 32 °C, the HA titer reached 3.65 ± 0.13 g/L. We have engineered a strain that can produce high-molecular-weight and medium-molecular-weight HA at different growth temperatures. This GRAS B. subtilis strain can be applied in industry and provides a new strategy for production of HA with different molecular weights.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hyaluronic acid (HA) is a high-value glycosaminoglycan composed of alternating N-acetyl-d-glucosamine and d-glucuronic acid monomers (Chong et al. 2005). The market for HA is expected to increase quickly with the escalating demand for viscosupplements and dermal fillers (Liu et al. 2011). Different molecular weight has had different functions. High-molecular-weight HA (HMW, Mr > 2×106 Da) has good viscoelasticity, inhibits inflammatory reactions, and has other functions, and can be used in ophthalmic surgery, and bone and joint injection preparations. Medium-molecular-weight HAs (MMW, Mr 105–106 Da) have good moisture retention, lubricity, and so on. MMW HAs are widely used in cosmetics, eye drops, and skin burn healing products, and for postoperative anti-adhesion. Low-molecular-weight HAs (LMW, Mr < 104 Da) can be absorbed by the intestine, supplement deficiency of HA in the body, and are used in health and beauty products (Yang et al. 2005).
Traditionally, HA has been commercially obtained from cockscomb and certain attenuated strains of group A and group C streptococci, which can produce HA natively. In the past few years, the biosynthesis of HA in recombinant microorganisms has become attractive. The typical hosts include Bacillus sp. (Chien and Lee 2007a; Widner et al. 2005), Lactococcus lactis (Chien and Lee 2007b), Agrobacterium sp. (Mao and Chen 2007), Escherichia coli (Yu and Stephanopoulos 2008), and Corynebacterium glutamicum (Cheng et al. 2017). B. subtilis is a model Gram-positive bacterium, which is identified as “generally recognized as safe” (GRAS) by the US Food and Drug Administration (Schallmey et al. 2004). Therefore, it can be an optimal host for HA production. However, B. subtilis, like all other heterologous producers of HA investigated to date, produces lower MW HA than group C streptococci. The highest MW of HA produced to date by B. subtilis is 4.5 MDa (Jia et al. 2013). It is a major challenge to increase the MW of heterologously produced HA.
Bacillus subtilis usually has properties, such as production of various proteinases, sporulation in nutrient deficiency conditions, and high maintenance metabolism, which make its use challenging in industrial operations. Consequently, as the starting strain in the present study, we chose B. subtilis WB600 (Fig. 1) which has only 0.32% of the extracellular protease activity of B. subtilis 168 because of the inactivation of genes encoding neutral protease A, subtilisin, extracellular protease, metalloprotease, bacillopeptidase F, and neutral protease B (Wu et al. 1991). In B. subtilis, sigma factor F (sigF) is the first prespore-specific transcription factor and controls genes required for the early stages of spore germination (Errington 2003). A relatively small number of genes were found to be differentially expressed in non-sporulation conditions in comparison with sporulation conditions (Overkamp and Kuipers 2015). Therefore, deleting sigF is expected to be an adequate solution to the problem of sporulation in industrial fermentation.

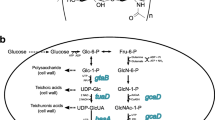
Biosynthetic pathway of hyaluronic acid in Bacillus subtilis. The six extracellular proteases in the dotted box exist in B. subtilis 168 but not in WB600. Important genes are: hasA (coding for hyaluronan synthase), hasB (tuaD, coding for UDP-glucose 6-dehygrogenase), and hasC (gtaB, coding for glucose-1-P uridyltransferase)
Target metabolite production can be heightened remarkably by strain engineering, metabolic engineering, synthetic biology, and molecular biology (Keasling 2010). Enhanced expression of key genes aims to direct carbon flux toward target metabolite formation (Lee et al. 2012). For example, coexpression of HA pathway genes, i.e., UDP-glucose-1-dehydrogenase (TuaD; encoded by tuaD/hasB) and UDP-glucose pyrophosphorylase (GtaB; encoded by gtaB/hasC), moderately improved HA production (Widner et al. 2005).
In general, researchers are accustomed to culturing B. subtilis at 37 °C. However, changing the culture temperature may bring about a change in the distribution and content of cardiolipin (CL), a dimer of the phospholipid phosphatidylglycerol (Kawai et al. 2006; Salzberg and Helmann 2008). It was first observed by Tlapak-Simmons et al. that the activity of Class I hyaluronan synthase (HAS) depended on CL when the solubilized HAS of Streptococcus pyogenes (SpHAS) was recovered (Triscott and Rijn 1986). It was proposed that a single SpHAS or SeHAS (HAS of S. equisimilis) protein needs to interact with ~ 16 CL molecules, and that loss of even a single CL molecule can cause enzyme inactivation. CL may play a role in assisting HAS to create an internal pore-like passage for HA translocation (Tlapak-Simmons et al. 1998), which is similar to the function of CL proposed for several mitochondrial membrane proteins that lack transport capability in its absence (Dowhan 1997). Furthermore, there is an HA·HAS retain–release model for the control of HA product size by Class I HAS (Weigel and Baggenstoss 2012). The interactions between CL and HA may increase the net retention force, allowing the HA chain to grow until the release force exceeds the net retention force, which then results in release of the growing HA chain (Weigel and Baggenstoss 2012; Tlapak-Simmons et al. 1999). Westbrook et al. modified the content and distribution of CL in the cell membrane, and improved both the titer and MW of HA (Westbrook et al. 2018). Therefore, the MW and titer of HA may change just by cultivating the producing cells at different temperatures. Some other factors may also affect the MW of HA; for example, the carbon source used in B. subtilis (Westbrook et al. 2016). Leech hyaluronidase was expressed in B. subtilis for producing HA with an MW range of 2.2 kDa to 1.42 MDa (Jin et al. 2016).
Here, we aimed to increase the MW of HA as conveniently as possible. We used B. subtilis WB600 as our starting strain and deleted the spore formation-related gene sigF to make the strain better for industrial use. We obtained a strain that produced higher MW HA, and different MWs of HA at different culture temperatures.
Materials and methods
Bacterial strains, plasmids, and primers
Escherichia coli DH5α chemically competent cells were prepared as described previously (Sambrook and Russell 2001) and used as hosts for plasmid construction. The B. subtilis strains and the plasmids used in this study are listed in Table 1. The primers used in this study are listed in Table 2.
Plasmid construction
Fragments containing upp, sigF, hasA, and hasABC were obtained using standard DNA manipulation techniques (Sambrook and Russell 2001). The sigF fragment was amplified from B. subtilis WB600 DNA using primers sigF-F and sigF-R. The obtained sigF fragment was cloned into the deletion plasmid pCU, a kind gift from the Chen Lab, Tianjin University, to produce pCU-sigF, which contains the genes for a counter-selectable marker (upp), and ampicillin and chloromycetin resistance. DNA for hasA, encoding S. ubris hyaluronan synthase (HAS), was synthesized (Genewiz, Suzhou, China). The hasA fragment (1603 bp) (Fig. S1) was amplified from synthetic strain LA1515-1 using primers hasA-F and hasA-R. Fragments containing hasA, hasB, and hasC were amplified from genomic DNA of LA1515-1 and B. subtilis WB600 using primers hasABC-F1/hasABC-R1, hasABC-F2/hasABC-R2, and hasABC-F3/hasABC-R3, respectively. Then, these three DNA fragments were linked together using overlapping PCR to form the complete hasABC fragment (5142 bp) (Fig. S2). The hasA and hasABC fragments were, respectively ligated into the E. coli–B. subtilis shuttle vector pHY300plk which contains genes for ampicillin and tetracycline resistance to produce pHY300plk-hasA and pHY300plk-hasABC, respectively. These plasmids were transformed into E. coli DH5α and verified by colony PCR, and the PCR products were sequenced by Genewiz for further confirmation. The constructed strains were maintained as glycerol stocks stored at − 80 °C.
Culture conditions and media
Luria–Bertani broth (LB, tryptone 10 g/L, yeast extract 5 g/L, and NaCl 10 g/L) was used for seed cultivation, with ampicillin (100 mg/L), chloromycetin (50 mg/L), and tetracycline (50 mg/L) added as necessary. E. coli strains were cultured overnight at 37 °C. B. subtilis strains were cultured at 27, 32, 37, 42, and 47 °C. They were grown to mid-exponential growth phase and transferred into fermentation medium (sucrose 20 g/L, (NH4)2SO4 3 g/L, KH2PO4 6.5 g/L, Na2HPO4 4.5 g/L, sodium citrate 2 g/L, MgSO4·7H2O 3 g/L, and CaCl2·2H2O 0.5 g/L); 6 mL/L of a trace metallic elements solution was added (citric acid 100 g/L, FeSO4·7H2O 20 g/L, MnSO4·H2O 5 g/L, CuSO4·5H2O 2 g/L, and ZnCl2 2 g/L). B. subtilis strains were incubated for 54 h in 5 L fermenters or shake flasks.
Competent cell preparation and electroporation of B. subtilis
Constructed plasmids were electrotransformed into B. subtilis using electric transfer buffer (ETM, 0.5 M sorbitol, 0.5 M mannitol, and 10% glycerol). First, B. subtilis was cultured overnight at 37 °C in 5 mL LB with corresponding antibiotics and then transferred into 50 mL LB and grown to an OD600 between 0.45 and 0.65. Cells were centrifuged at 5000 × g for 10 min and the supernatant was discarded. The pellet was resuspended in ETM buffer and centrifuged at 5000 × g for 10 min. This wash step was repeated three times, and then, the cells were resuspended in 500 µL ETM. Plasmid DNA (2–5 µg) was added to the cells in a 1-mm cuvette and they were incubated in an ice bath for 30 min. Electroporation was performed at 1.8 kV, 25 µF, and 600 Ω. Cells were cultured in LB with tetracycline after electroporation. Successful transformants were verified by colony PCR.
Construction of the markerless deletion system
Uracil phosphoribosyl-transferase (UPRT), encoded by the upp gene, was used as a counter-selectable marker to construct a markerless deletion system. UPRT can catalyze the conversion of pyrimidine analog 5-fluorouracil (5-FU) into 5-fluoro-UMP, a toxic substrate for the cell (Fabret et al. 2002). Therefore, the strain-containing upp could not survive on the LB-agar with 5-FU (20 μM). When the upp is knocked out, it can grow well. Using this mechanism, it is possible to screen for double-crossover mutants. The upper and lower homologous arms of upp were amplified from strain WB600 genomic DNA using primers upp-upF/upp-upR and upp-downF/upp-downR. Then, the upp fragment comprising upper and lower homologous arms was obtained by overlapping PCR using primers upp-upF/upp-downR. Next, the upp fragment was electrotransformed into WB600. The mix was spread on LB-agar containing 5-FU and single colonies were verified by colony PCR. The strain with the upp gene deleted was named WB600Δupp. The deletion plasmid pCU that contains the gene upp, and genes for ampicillin and chloromycetin resistance can be electrotransformed into WB600Δupp. It was found that WB600Δupp could live on LB containing 5-fluoracil, and thus, this system was verified as being useful for deleting genes with no marker (Fig. S3).
HA quantification
Sodium dodecyl sulfonate (SDS, 0.1% w/v) was added to fermentation broth, which was then centrifuged at 10,000 × g for 20 min. The supernatant was collected and mixed with two volumes of absolute ethanol and incubated at 4 °C for 1 h. The precipitant was collected by centrifugation at 5000 × g for 20 min and redissolved in one volume of distilled water. This purification process was performed three times. The HA titer was routinely determined with the modified uronic acid carbazole reaction (Bitter and Muir 1962). As a control, B. subtilis strain WB600 was treated in the same conditions, and used to remove background signals.
HA molecular weight measurement
The MW of HA was measured with an MALLS-SEC system consisting of a pump (515 HPLC, Waters, Milford, MA, USA), a Rheodyne 7725i injector (Waters) with a 200 μl injection loop (Rheodyne®, IDEX Corporation, USA), a size-exclusion chromatography (SEC) column (Waters Ultrahydrogel™ linear column, 300 mm × 7.8 mm i.d.; Waters), a multiple-angle laser light scattering detector (MALLS, Wyatt Technology DAWN HELEOS, Santa Barbara, CA, USA), and a differential refractometer detector (RI, Optilab Rex, Wyatt Technology Co., USA). The mobile phase was an aqueous solution of 0.1 M phosphate-buffered saline (PBS) used at a constant flow rate of 0.6 mL/min. The temperature of the column was maintained at 30 °C and the injection volume was 200 μL. The signals measured with the detector were analyzed to calculate the molecular weights. Bovine serum albumin was determined every time and used as a reference. Each determination was made at least three times and the average value was used as the result.
qRT-PCR
Cells were harvested in the exponential growth phase for RNA extraction. Total RNA was isolated using the RNAprep Pure Bacteria Kit (Tiangen, Beijing, China) as per the manufacturer’s instructions. cDNAs were synthesized using the FastKing RT Kit (Tiangen). Sequence-specific primers were used for reverse transcription of clsA mRNA and internal control 16S mRNA at a final concentration of 1 μM. Real-time quantitative reverse transcription PCR (qRT-PCR) was carried out using Bestar™ qPCR MasterMix (DBI® Bioscience, Ludwigshafen, Germany) in a Two Color Real-time PCR Detection System as per the manufacturer’s instructions. Sequence-specific primers were used for amplification of clsA. Data analysis to quantify relative expression in cultures at different temperatures was performed as previously described (Ruijter et al. 2009). All experiments were performed in triplicate.
Results and discussion
Construction of markerless sigF deletion mutants
To obtain a strain that can express foreign proteins stably and be applied safely and effectively in industry (Overkamp and Kuipers 2015), we deleted the spore formation-related gene sigF which encodes a specific transcription factor that controls genes required for the early stages of prespore development (Hilbert and Piggot 2004), from B. subtilis WB600 using a markerless system (Fig. 2a). The sigF-deficient mutant was named Wm. Deletion of sigF was verified by PCR (Fig. 2b). No sporulation was observed in Wm (Fig. S4).

Construction of strain Wm a Illustration of sigF markerless knockout construction in B. subtilis WB600; b Gel electrophoresis of colony PCR products from amplified target gene sigF using primers sigF-upF/sigF-downR The wild-type genotype results in a product of 2139 bp, whereas the deletion of the sigF is confirmed by a shift of the product to 1465 bp
Constructing strains to produce HA
We constructed vector pHY300plk-hasA encoding HAS from S. ubris, and electrotransformed it into B. subtilis strains Wm, and WB600Δupp, from which UPRT encoded by the upp gene was deleted. The strains obtained were named WmA and WA, respectively. Both of them have a mucoid phenotype (Fig. S5) that is characteristic of HA production in B. subtilis (Widner et al. 2005), and they were genetically stable based on the persistence of the mucoid phenotype upon repetitive revival. In general, B. subtilis strains are incubated at 37 °C. Thus, initially, we cultured strains WA and WmA in liquid LB with tetracycline at 37 °C. The HA titer of WmA was increased by 30.03% in comparison with that of WA (Fig. 3a).

a HA titers and b growth curves of WA, WmA, and WmB when they were incubated in LB at 37 °C. WA was used as a control strain data are average of three independent experiments and error bars represent ± SD (*p < 0.05; **p < 0.01; ***p < 0.001)
To further improve the titer of HA, we constructed strain WmB that coexpressed the HA synthetic pathway genes hasA, encoding HAS from S. ubris, hasB, encoding UDP-glucose-1-dehydrogenase from B. subtilis WB600, and hasC, encoding UDP-glucose pyrophosphorylase from B. subtilis WB600, in Wm. We found that the titer of strain WmB was higher than that of WmA, and it could produce 1.82 g HA/L (Fig. 3a). The HA titer, thus, improved by 103.19% compared to strain WA. Next, we measured growth condition of the three strains. It was found that the growth rate and bacterial concentration of WmB was, respectively, the fastest and highest (Fig. 3b). This may be due to the synthesis of cell walls involved in UDP-glucose 6-dehydrogenase (encoded by hasB) and UTP-glucose-1-P uridylyltransferase (encoded by hasC), further increasing the bacterial concentration.
Titer and MW of HA produced by strain WmB at different growth temperatures
To research the relationship of HA characteristics and the growth temperature of the producing bacteria, we cultured strain WmB in LB with tetracycline at 27, 32, 37, 42, and 47 °C. The cells grew best at 32 °C, and grew poorly at 47 °C (Fig. 4a). The strain was not viable at 50 °C. At 32 °C, the cell density of WmB reached OD600 20.58 in fermentation medium (Fig. 4a) and the HA titer reached 3.65 g/L (Fig. 4b). The average value of HA titer/OD600 is shown in Fig. 4c. To eliminate the effect of bacterial concentration on HA titers, we got the HA yield per hour of the cells using the formula of HA titer/(OD600*time), obtaining the effect of temperature on the titers. It can be seen from Fig. 4c that, as the culture time increases, the unit yield decreases, which may be due to the stable growth period of the bacteria. In each stage, the unit yield is the highest at 32 °C, and the lowest at 47 °C. Briefly, temperature has a significant impact on HA production. The better the growth, the higher the HA titers. Notably, the MW of HA produced by WmB reached 6.973 MDa at 47 °C. This value is the highest observed MW of HA produced by B. subtilis to date. At 32 °C, the MW was as low as 0.392 MDa (Fig. 4d). Thus, WmB can produce both HMW and MMW HA at different temperatures.

a Growth curves, b HA titers, c average HA titer measured every 6 h, and d HA MWs of WmB in 5 L fermenters at different temperatures (27, 32, 37, 42, and 47 °C); 37 °C was used as a control temperature. Data are average of three independent experiments and error bars represent ± SD (*p < 0.05; **p < 0.01; ***p < 0.001)
Relative transcription of clsA in strain WmB at different temperatures
Temperature can make a difference to the cell membrane, e.g., in the fluidity, permeability, and so on. We hypothesized that the content and distribution of CL was affected by the culture temperature and that, in turn, caused the observed temperature-dependent difference in the MW of the HA that was produced (Westbrook et al. 2018). Therefore, we assayed the transcription level of clsA, which encodes cardiolipin synthase, by real-time quantitative reverse transcription PCR (qRT-PCR). Our control (reference) sample was WmB cultured at 32 °C.
The results showed that the relative transcription at 27, 37, 42, and 47 °C is lower than at 32 °C (Fig. 5). The expression level of clsA was consistent with the HA titer. The MW of the HA produced increased when the mRNA level of clsA decreased. These results are consistent with the previous research (Westbrook et al. 2018).

The relative transcription of clsA in WmB at 27, 32, 37, 42, and 47 °C Relative transcription (i.e., relative to the transcription of internal control 16S) was normalized to the values obtained from cultures of WmB at 32 °C. 37 °C was used as a control temperature. Data are average of three independent experiments and error bars represent ± SD (*p < 0.05; **p < 0.01; ***p < 0.001)
We found that the MW of HA produced by B. subtilis strain WmB is related to the culture temperature. This could be due to temperature effects on cell growth, i.e., the better the bacteria grow, the faster the synthetic HA chain is released, resulting in shorter chain length and, thus, lower MW. At a culture temperature of 32 °C, which is optimal for the growth of strain WB600 (Fig. 4a), the CL content might increase and, thus, the release force for the HA chain quickly exceeds the retention force. At 47 °C, the strain grows slowly, and it takes longer for the HA chain to be released. Alternatively, temperature might influence the content and distribution of CL, affecting the function of Class I HAS. Purified SeHAS and SpHAS reached their highest enzyme activity in vitro when CL, rather than other lipids, was added (Triscott and Rijn 1986; Tlapak-Simmons et al. 1999). CL may assist HAS in the formation of pores in the cell membrane through which the growing HA chain is translocated, to some extent (Tlapak-Simmons et al. 1998), and its interaction with HA may contribute to the change of chain length during the polymerization of HA. Altered CL availability in the cell membrane of HA-producing B. subtilis could enable more of the total expressed enzymes per cell to achieve diverse activity (i.e., a diverse polymerization rate) and distinct functionality in the retention or release and translocation of the growing HA chain, resulting in the production of different MWs of HA.
Comparison with HA production in B. subtilis 168
In recent years, industrial HA production has mainly used microbial cultivation (Liu et al. 2011). The most common “GRAS” production strain is B. subtilis 168. Thus, we also assayed the titer and MW of HA produced by B. subtilis 168. The vectors pHY300plk-hasA and pHY300plk-hasABC were transformed into B. subtilis 168 and the strains obtained were named 1A and 1B, respectively. Strain 1B produced a higher titer than 1A, but a lower titer than strain WmA (Fig. 6). The previous work showed that the HA titer could reach 2.26 g/L in B. subtilis 168 engineered by reducing the expression of pfkA or zwf using Clustered Regularly Interspaced Palindromic Repeats interference technology (Westbrook et al. 2018). However, this is still lower than culture of strain WB600, the parental strain used in the present study, for which a titer of 3.21 g/L (Fig. 6) was obtained in shake flasks. As noted above, we obtained an HA titer of 3.65 g/L from strain WmB in optimum conditions.

The HA titers of Bacillus subtilis 168 and WB600 cultured in shake flasks at 32 °C The strains are 1A (Bacillus subtilis 168, pHY300plk-hasA), 1B (Bacillus subtilis 168, pHY300plk-hasABC), WmA, and WmB, separately. 37 °C was used as a control temperature. Data are average of three independent experiments and error bars represent ± SD (*p < 0.05; **p < 0.01; ***p < 0.001)
Conclusions
To decrease the production of byproducts and difficulties in industrial operations, we chose the protease-deficient B. subtilis strain WB600 as the parental strain for production of HA in this work. Then, we removed the spore formation-controlling gene sigF by markerless deletion to avoid spore formation in nutrient-depleted conditions. We found that the growth conditions influenced the HA titer and MW of the resulting strains. We obtained an HA titer of 3.65 g/L in a 5-L fermenter at 32 °C (Fig. 4b). The highest molecular weight HA yet produced in B. subtilis (up to 6.973 MDa) was obtained at 47 °C, although the titer was low. Thus, we report a new strategy to conveniently produce different molecular weights of HA in industry.
References
Bitter T, Muir HM (1962) A modified uronic acid carbazole reaction. Anal Biochem 4:330–334
Cheng F, Luozhong S, Guo Z, Yu H, Stephanopoulos G (2017) Enhanced biosynthesis of hyaluronic acid using engineered Corynebacterium glutamicum via metabolic pathway regulation. Biotechnol J. https://doi.org/10.1002/biot.201700191
Chien LJ, Lee CK (2007a) Enhanced hyaluronic acid production in Bacillus subtilis by coexpressing bacterial hemoglobin. Biotechnol Prog 23(5):1017–1022
Chien LJ, Lee CK (2007b) Hyaluronic acid production by recombinant Lactococcus lactis. Appl Microbiol Biotechnol 77(2):339–346
Chong B, Blank L, McLaughlin R, Nielsen L (2005) Microbial hyaluronic acid production. Appl Microbiol Biotechnol 66(4):341–351
Dowhan W (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem 66(1):199–232
Errington J (2003) Regulation of endospore formation in Bacillus subtilis. Nat Rev Microbiol 1:117–126
Fabret C, Ehrlich SD, Noirot P (2002) A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol 46(1):25–36
Fu J, Ma XW, Huang CF, Zhao YH, Li M (2012) Folding and convoluting manipulation for the treatment of distal radius fractures: a report of 78 cases. Zhongguo Gu Shang 25(9):755–756
Hilbert DW, Piggot PJ (2004) Compartmentalization of gene expression during Bacillus subtilis spore formation. Microbiol Mol Biol Rev 68(2):234–262
Ishiwa H, Tsuchida N (1986) New shuttle vectors for Escherichia coli and Bacillus subtilis. The nucleotide sequence of pHY300PLK and some properties in relation to transformation. Jpn J Genet 61:515–528
Jia Y, Zhu J, Chen X, Tang D, Su D, Yao W, Gao X (2013) Metabolic engineering of Bacillus subtilis for the efficient biosynthesis of uniform hyaluronic acid with controlled molecular weights. Bioresour Technol 132:427–431
Jin P, Kang Z, Yuan P, Du G, Chen J (2016) Production of specific molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab Eng 35:21–30
Kawai F, Hara H, Takamatsu H, Watabe K, Matsumoto K (2006) Cardiolipin enrichment in spore membranes and its involvement in germination of Bacillus subtilis Marburg. Genes Genet Syst 81(2):69–76
Keasling JD (2010) Manufacturing molecules through metabolic engineering. Science 330:1355–1358
Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY (2012) Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol 8:536–546
Liu L, Liu Y, Li J, Du G, Chen J (2011) Microbial production of hyaluronic acid: current state, challenges, and perspectives. Microb Cell Fact 10(1):99
Mao ZC, Chen RR (2007) Recombinant synthesis of hyaluronan by Agrobacterium sp. Biotechnol Prog 23(5):1038–1042
Overkamp W, Kuipers OP (2015) Transcriptional profile of Bacillus subtilis sigF-mutant during vegetative growth. PLoS One 10(10):e0141553
Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, Moorman AFM (2009) Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 37:e45
Salzberg LI, Helmann JD (2008) Phenotypic and transcriptomic characterization of Bacillus subtilis mutants with grossly altered membrane composition. J Bacteriol 190(23):7797–7807
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Harbor
Schallmey M, Singh A, Ward OP (2004) Developments in the use of Bacillus species for industrial production. Can J Microbiol 50(1):1–17
Tlapak-Simmons VL, Kempner ES, Baggenstoss BA, Weigel PH (1998) The active streptococcal hyaluronan synthases (HASs) contain a single HAS monomer and multiple cardiolipin molecules. J Biol Chem 273(40):26100–26109
Tlapak-Simmons VL, Baggenstoss BA, Clyne T, Weigel PH (1999) Purification and lipid dependence of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J Biol Chem 274(7):4239–4245
Triscott MX, van de Rijn I (1986) Solubilization of hyaluronic acid synthetic activity from streptococci and its activation with phospholipids. J Biol Chem 261(13):6004–6009
Weigel PH, Baggenstoss BA (2012) Hyaluronan synthase polymerizing activity and control of product size are discrete enzyme functions that can be uncoupled by mutagenesis of conserved cysteines. Glycobiology 22(10):1302–1310
Westbrook AW, Moo-Young M, Chou CP (2016) Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl Environ Microbiol 82(16):4876–4895
Westbrook AW, Ren X, Moo-Young M, Chou CP (2018) Engineering of cell membrane to enhance heterologous production of hyaluronic acid in Bacillus subtilis. Biotechnol Bioeng 115(1):216–231
Widner B, Behr R, Von Dollen S, Tang M, Heu T, Sloma A, Sternberg D, Deangelis PL, Weigel PH, Brown S (2005) Hyaluronic acid production in Bacillus subtilis. Appl Environ Microbiol 71(7):3747–3752
Wu XC, Lee W, Tran L, Wong SL (1991) Engineering a Bacillus subtilis expression-secretion system with a strain deficient in six extracellular proteases. J Bacteriol 173(16):4952–4958
Yang G, Guo X, Luan Y (2005) The application on different molecular weight of sodium hyaluronate. Food Drug 12:1–3
Yu H, Stephanopoulos G (2008) Metabolic engineering of Escherichia coli for biosynthesis of hyaluronic acid. Metab Eng 10(1):24–32
Acknowledgements
This work was supported by the Tianjin Research Program of Application Foundation (16JCTPJC50100).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Y., Li, G., Zhao, X. et al. Regulation of hyaluronic acid molecular weight and titer by temperature in engineered Bacillus subtilis. 3 Biotech 9, 225 (2019). https://doi.org/10.1007/s13205-019-1749-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-019-1749-x