
Abstract
Proteins have a significant role in animals and human health. Interactions among proteins are complex and large. Proteins separations are challenging process in molecular biology. Computational tools help to simulate the analysis in order to reduce the training data into small testing data. Large proteins have been mapped using self-organizing maps (SOMs). Neural network based SOMs has a significant role in reducing the irregular shapes of proteins interactions. Iterative checking enables the organizations of all proteins. In next stage, particle swarm intelligence is applied to classify the proteins’ families. In the current work, secondary (Two dimensional) and tertiary proteins (Three dimensional) proteins have been grouped. Two dimensional proteins contain fewer hydro-carbons than three dimensional proteins. For faster analysis, the angles of the proteins are taken into account. The SOMs is compared with Bounding Box approach. In final, the experimental evolutions show that swarm intelligence achieved faster processing through enabling less memory consumptions and time. Since PSO combines proteins datasets in fuzzy values, the compactness or integration of similar proteins are strong. On the other hand, Bounding Box uses the Crisp value. Therefore, it needs more space to organize the whole data. Without SOMs, swarm intelligence also results are poor due to the excessive time consuming and required storage area. Moreover, for almost all classification and clustering tools, it is observed that the overall classification task becomes slow, time consuming, space consuming and also less sensitive because of noises, irrelevant data in input datasets. Thus, the proposed SOM based PSO approach achieved less time consuming with efficient classification into secondary and tertiary proteins.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In the current epoch, large number of biological PROTEINS and DNA (Deoxyribonucleic acid) datasets are build. The main obstacle facing the biologists is to discover manipulating knowledge from such complicated datasets. Development of highly scalable computational and quantitative approaches in order to keep pace with the rapid biotechnology improvements and to discover the inside of the complex biological datasets became essential. Bioinformatics and computational biology is the field for knowledge discovery by manipulating large biological data using computer science, computer architecture and information technology. It requires highly observed and sensitive components to deal with such kinds of huge data. The technologies advancement for sorting or searching DNA sequences indicates improvement. Recently, data mining and bioinformatics assist the researchers to interpret the huge amount of data. A vast number of developed algorithms lead to more reliable and supportive bioinformatics. Recently, a quite challenging for the researchers is to handle data partitioning and current transaction system [1] as well as language modeling [2] due to the huge number of parameters and high dimensions. However, these challenging issues achieve key knowledge for using various bioinformatics algorithms, including data partitioning [1], gene co-expression networks [3] or gene expression graph (GEG) based classifier [4] to separate the genes. These genes are responsible for various dangerous diseases including cancer. The bioinformatics development allows the detection of cleft in organs including lungs, pancreas, kidneys, salivary and mammary glands because of branching morphogenesis process [5]. However, the imperfect/noisy datasets cause the main obstacle to classify the real-world data set [6] as they may the weaken system processing speed along with increasing complexity [7]. Thus, bioinformatics, information technology, computer science and architecture become significant to resolve such drawback and broad range of problems in different fields [8] as well as to detect symptoms of crucial diseases, such as dyslexia, the FURIA classification algorithm [9]. Further, in the field of bioinformatics, computational biology classification of positive unlabeled data [10], and improvement of mine organization rules on various data [11] paves bioinformatics a step ahead to solve difficult problems that was hard to solve yet.
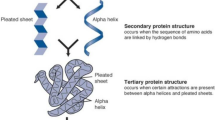
Over the past decade, researchers are interested with genomics and proteomics analysis and classification. They developed different approaches for classification techniques of biological data and for using computational data on biology. Generally, proteins are structural and functional unit of human cell. Proteins are the key feature for performing thousands of reactions while constituting human cell. Most of the time transferring the molecules through the plasma membrane, which plays the main role to create the protective wall around the human cell, is completed by membrane proteins. Approximately 25–75% of the membrane mass consists of proteins [12]. In addition, proteins are very important for being enzymes and for helping in execute necessary reactions that required for creating the human cell. Structurally, proteins have three kinds, namely: primary, secondary, and tertiary, as illustrated in Fig. 1. Various proteins are separated because of their simple pursuit on the human body, such as keratin, elastin, and collagen, which are significant types of support proteins.

Graphical representations of proteins types [13]
Consequently, from the last decade there had been various attempts to classify proteins using several effective algorithms. Moreover, proteins identification using supervised learning techniques become a new research trend. Besides the researches have been largely attacked by the vast number of data from which it is required to differentiate primary, secondary and tertiary protein using efficient algorithm. Furthermore, the separation of two-dimensional (2D) and three-dimensional (3D) proteins from huge dataset becomes an emerging task. Recently, the improvement in the field of proteomics is proceeding rapidly, which generates a large amount of biological data sets. In order extract useful information and knowledge from this massive data amount, high performance computers and more innovative software tools become in dispensable to manipulate more efficient and time consuming algorithms. The main contravention for the researchers is to achieve accurate findings using various classification algorithms [14]. The resultant findings need to be refined again to achieve the desired data. In addition, for various complex and complicated datasets it is required to identify the functions of each protein, which is an interesting topic in bioinformatics in recent years [15, 16] as well as the main contribution of the current work.
Basically, identification of proteins from any combined and ever growing set of available 2D or 3D data definitely requires usually efficient, time consuming and automatic clustering algorithms. Therefore, for protein classification several techniques can be used including support vector machine (SVM), Self-organizing maps (SOMs), Particle swarm optimization (PSO), Bounding box algorithms, and sequential minimal optimization (SMO). These algorithms can basically handle complex and complicated huge datasets and discover the required findings. Generally, the classification algorithms can be categorized into: (1) pair-wise sequence algorithm, (2) discriminative classifier, and the (3) generative models for proteins classification.
In the current work, algorithms are developed for superior performance compared to other algorithms, namely the bounding box algorithm, particle swarm optimization (PSO), self-organizing maps (SOMs) and particle swarm optimization (PSO), and PSO centric-SOMs. Those algorithms have been commonly mentioned because of their capabilities of efficiently handling large number of data using low memory in shortest possible time. Therefore, the key contribution of the current work is introducing improvised version of clustering and identification methodology on complex, complicated and large imbalance dataset. In order to reduce the runtime and amount of memory to complete action along with ensuring faster classification than previously opposed algorithms. Since the data size is huge, it takes time to attain concluding results, thus the current work machine based unsupervised learning was involved to handle large data size in limited time. Since single method cannot resolve these issues successfully in all cases, thus a more sensible way is to combine multiple methods [17]. Consequently, the proposed method is more preferable as it is mainly combine two methods. Thus, a new framework was designed to manipulate any size of data within shortest possible time and using low machine memory. A combination of the particle swarm optimization (PSO) with self-organizing map (SOMs) is proposed in the current work, which interpreted better accuracy and less time consumption along with shortest memory loss. Moreover, The SOMs based particle swarm optimization approach is proposed in the present work for better performance compared to the SOMs only as well as other common algorithms, such as bounding box algorithm and the support vector machine. The key advantage of using mapping based algorithm is to omit noises, irreverent data and also diminish higher dimension to lower one for visualization of higher dimensional input datasets. Thereafter, PSO is applied to manipulate the noise free data for detecting secondary and tertiary proteins. Although, there are several alternatives for the optimization algorithms, such as genetic algorithms and cuckoo search algorithm, however the PSO ability regarding interpretation of faster and easier computation, better performance with the number of datasets increasing, working with fuzzy logic has motivated the current work. Additionally, the proposed PSO-centric SOMs is capable of providing best performance on any size of the data set most fluently. Therefore, in the present work, huge and complex datasets were gathered from the National Center for Biotechnology Information (NCBI) database. Basically, several databases related to proteins data are present among which proteins data bank (PDB), structural classification of proteins (SCOP), protein information resource, database of interacting proteins, and the national center for biotechnology information (NCBI). Meanwhile, the present work extracted the datasets from NCBI database because the services of this dataset are compared to other databases. Moreover, NCBI offers a wide range of data with faster analytical tools. In general, NCBI also has the feature of offering a sizeable quantity of information people need to extract and in general it’s free for all. Although, bioinformatics data are increasing in an exponential rate, NCBI helps researchers to limit the search-times and to increase the simplicity of making query by maintaining its services efficiently through its website. Moreover, the pipeline of data extraction from NCBI is easier. Because of this phenomenon, the NCBI database is used for collection of data in this work. Despite of this, datasets from any of proteins database is trustworthy. The actual result never varies too much for collecting data from different databases.
Thereafter, the proposed PSO centric SOMs approach was applied. In general, PSO centric SOMs refers to first step classification of two dimensional (2D) and three dimensional (3D) proteins using SOMs as SOMs groups all the similar data together. Afterward, the PSO is used to enclose the resultant proteins as much as possible consuming less memory along with time complexity. For comparison purpose, various single algorithms were also applied on same datasets, such as bounding box algorithms, PSO alone, SOMs alone and the overall findings were compared, which shows superiority of PSO centric SOMs (Sect. 4). Basically, from the perspective of the current work, different alternatives of SOMs as well as PSO could be used for mapping as well as classification task. Some of those alternatives are ISOMAP [18], manifold alignment [19], supervised kohonen network (SKN) [20], counter propagation artificial neural network [21], support vector machine (SVM) [22] and, principle component analysis (PCA) [23]. Such algorithm can be employed to estimate the SOMs, which proved that the SOMs outperforms all the mapping based algorithms where PSO shows better performance with the increasing number of complex as well as complicated datasets. Also, bounding box algorithm performs well compared to other alternatives but bounding box algorithm seems to be slow with increasing datasets, which is depicted graphically and practically as well in the result section. Based on the performance of all the alternatives here for this work, several approaches are considered for comparison purpose. In general, for almost all classification and clustering tools, it is obvious that the overall classification task becomes slow, time consuming, space consuming and also less sensitive because of noises, irrelevant data in input datasets. Thus, for achieving better identification from these types of datasets it is required to remove the noise form the relevant data to boost the classification process speed for less time and space consuming. For this circumstance, the current work proposed mapping based particle swarm optimization which will initially filter the input data, remove the noises from there and map the data within a certain range. Thereafter, the PSO is used to easily classify secondary (2D) as well as tertiary (3D) proteins from the filtered data. This process outperforms all other previous works along with other mapping approaches which have been illustrated in Sect. 4. Comparisons along with performance evolution of our proposed work with various present algorithms have been shown there theoretically, practically and graphically as well. Therefore, the current work presents several practical experiments conducted to evaluate the performance of the algorithms under study for protein classification.
Generally, the secondary protein, only one angle is generated with the Hydrocarbon. However, the tertiary proteins generate two different angles. In the current work, Otsu method [24–26] is applied to check the angles of both these protein types. Basically, Otsu method helps to convert Secondary and tertiary proteins to binary values form where we get the idea about the angles of the both proteins classes. Essentially, for multidimensional data processing, various algorithms [27], such as Otsu’s method, the adaptive binarization method, Lloyd method, Macqueen method are widely used. However, Otsu’s method is the most preferable for image or multidimensional data processing because of its key feature regarding separating an image into two classes according to threshold which diminish the variance between each class. Additionally, Otsu’s is an automatic threshold selection region based segmentation method along with its simpler way of calculation. Moreover, others algorithms execute with lots of irreverent combinations of input image. Thus, the process becomes slow and it detects less combination taking much time. Consequently, the present work employed Otsu’s method for faster and efficient calculation which affects further mapping based particle swarm optimization process. Therefore, the overall process becomes memory and time efficient. Basically, in bioinformatics, most web based tools allow limited number of inputs and output formats and also the input formats cannot be editable, which is the main obstacle for the researchers to broaden knowledge. Therefore, for better improvement a machine-learning based method is proposed in the current work. In addition, we have built our own database. For further comparative study, both in web based and manually created database were involved and our solution presented the best accuracy in both experiments. The proposed method was compared to other classical methods that work with direct protein and its machine based. Hence, in consequence, the proposed solution demonstrated a great energetic and less time consuming.
The organization of the remaining sections is as follows. Section 2 includes related work to protein classification techniques. Section 3 includes the methodology of the proposed approach for protein classification. Section 4 demonstrates the results in details with extensive discussion. Finally, the conclusion is addressed in Sect. 5.
2 Literature review
Nowadays, researchers are interested in developing new algorithms for protein classification. For stepping forward to discover new algorithms in [28], for protein classification the authors mostly concentrated on chemical reactions and proposed high quality protein classifications with external nucleic acid research (NAR) database. A set of chemicals were applied on proteins that are costly along with high risk for human health, whereas the proposed approach is totally machine based and automated based on unsupervised learning. Therefore, huge number of data can be manipulated using less memory and less time which is impossible following chemical reactions approach. Besides that, from [29] it can be realized that the amino-acid substitution matrices have been used to represent the similarities between motifs as well as to prepossess the protein sequences for further protein classification. Since this approach process 20 amino-acids inside each protein, where the proteins data size is extra-large and complicated it cannot process using short time along with low memory size. Thus, this process was time consuming and used large machine memory space. Consequently, the process cannot be efficient enough. Compared to this work, the proposed method is focused on angels of proteins presenting numerically as it is machine based that is why any size of datasets can be manipulated easily in shortest possible time compared to all the discovered algorithms. In the context of structural descriptor database [30], the web-based tools have been used to predict the function of proteins. Basically, in bioinformatics, most web based tools allow a limited number of inputs and output formats and also the input formats cannot be editable, which is the main obstacle for the researchers to broaden knowledge. Hence, for better improvement, machine-learning based methods can be employed, thus the present proposed algorithm is fully machine-based approach. Moreover, it has practically been discovered that machine-learning dependent algorithms are powerful in developing new fold recognition tools [31].Thus, it was established that using machine learning-based database achieved noticeable performance, time of execution, memory usage than using web based tools for various algorithms.
Attempts have been conducted to explore methods for better detection and prediction of protein functions though they failed sometimes to fulfill the requirement for protein–protein reaction in order to determine aspects of functions like sub-spongy localization, tone down after translation and protein–protein cooperation [32]. In addition, this approach required long time to complete the full process. Since single method cannot resolve these issues successfully in all cases, a more sensible way is to combine multiple methods [17]. For proteins classification, Baugh et al. [33] preferred manual experimental data collecting lots of various proteins with known effects on protein function from multiple organisms and curated structural models for each variant from crystal structures and homology models. Afterward, a single method variant interpretation and prediction using rosetta (VIPUR) has been used to integrate the proteins manually, which was costly, time consuming and unpredictable whereas being machine-based and combination of two strong algorithms, without doubt the PSO centric SOMs illustrates superiority.
For protein classification using multi-class protein structure prediction, the study performed in [34] illustrated superior accuracy, while using data from protein data bank (PDB). In general, this work provided poor accuracy level for outside PDB data. Consequently, this process fully depends on the PDB [35]. The current work is the combination of two classification algorithms and based on unsupervised machine-based approach that is why it depicts the capability of high accuracy from any source of data because of high sensitivity. The authors in [36] have illustrated the SVM, which is a web-server for machine learning, competence to acquire best prediction results from a sequence of proteins for protein classification. For the development of SVM models, sometimes the cost parameter rise very high which is unexpected for getting best performance [36]. Nor only for SVM, but also in present situation, the cost that most of the algorithms demand for classification purpose is extremely high. Besides, since some low sequence similarity proteins appear, then the sequence similarity E-value of this low sequence similar proteins is meaningfully high than the globally accepted value which is 0.05 rather generally it does not happen in PSO-centric SOM. Besides, using SVM allows the verification of the known structured proteins [37], which can be considered as the constraint of the SVM algorithm. On the contrary, the proposed approach avoids this kind of risks because of being unsupervised learning as well as machine-based fastest process. In [38], the SVM method was applied for the prediction of bacterial Hemoglobin-like proteins. The authors backed up for the SVM to identify the desired proteins. In addition, amino acid composition process has been used to deal with the class of proteins. Although, for large number of data like 1 billion proteins, then there is almost 20 amino acid inside each protein, thus, the proposed method needs to compete with 20 billion which is terrible to execute along with wastage of time and memory. The proposed algorithm never go through this type of process as it classifies proteins considering angles of proteins converted to numerical values initially. In [39], a new method called protNN has been implemented, which was quite fast with less time consuming. In the first stage, this protNN took almost 3 h for the classification of each protein against all entire PDB. Moreover, this method worked tremendously for three dimensional (3D) proteins where-as quite low for one dimensional (1D) as well as two dimensional (2D) proteins. From the perspective of the work, a secondary-structure matching (SSM) was used as a new tool for fast protein structure alignment in 3D [40]. The authors fully preferred 3D proteins for classification using SSM algorithms. Traditionally, the secondary-structure protein algorithm is a long-term process as it is mainly illustrated by graphical comparison which is quite hard to implement. It is mostly a structure based algorithm and the methods which are mostly dependent on structures are seen to be approximately 2–4 times slower than the sequence neighborhood based algorithm [41]. Basically, the structural neighborhood based classification of nodes in a network [42] is one of the structural based classification algorithms. The memory requirement and computational complexity of this algorithm is very high [43, 44].Therefore, proposed approach in [39] handled efficiently 3D proteins whereas provided low performances while 1D (primary) or 2D (secondary) proteins but ours is universal for all. It worked tremendously for both 2D as well as 3D proteins. In [45], proteins identification was performed using the SCOP (Structural Classification of Proteins) algorithm [46] for proteins function classification, which is fully manual process. Consequently, the process is long term, time consuming and huge memory allocation method, on the contrary to the proposed approach that illustrates dynamically short time discovery ability along with poor memory consumption. Moreover, various recent work regarding real life applications of different contemporary computing techniques associated with fuzzy logic as well as artificial intelligence techniques have been proposed successfully [27, 47–57].
3 Methodology
In previous research work, bounding box [58–60] was implemented by combining the whole datasets into a specific area and dividing the data into specific order until it reach to a certain length. This process allows managing the datasets easily and in a simple way for faster data classification and grouping. In this research, proteins structures are verified and justified with SOM and particle swarm optimization. The pivotal impact of SOMs in this analysis is to connect all the edges of proteins. Since proteins interactions are complex [61], SOMs neural networks manages all of these interactions. Back propagation facilities permit to handle the deficiency among proteins group verifications. Interactions among proteins are mapped by the neural networks input weights with associated edges. Consequently, particle swarm analysis counts all the datasets of proteins under fuzzy values [61, 62]. These fuzzy values allow combining greater limits of the total proteins with specific hydrocarbons angles associated with nitrogen bonds. One file contains all the proteins as training datasets. Based on the situations, these datasets can be divided into other files. These files are initial database for overall data processing. Some training datasets have been collected from online databases [63–66]. These databases allow collecting any proteins structures for better experimental analyses. There are various options to collect proteins from online databases (Fig. 2). The prime considerations of online databases are variations of input with latest timing.

Structure of the complete adopted design and analysis
3.1 Self-organizing maps
In various aspects of computer science, lower time consumption and higher efficiency are essential requirements [67–69]. All contended learning techniques, such as the SOMs that aim to learn rapidly and helps to work reliably. Such efficient SOMs algorithms have several real time and practical applications in the field of bioinformatics including data mining, speech analysis, and medical diagnostics. The SOMs-based algorithms are efficient to cluster biological data which diminish input space and dimensions. These algorithms are considered a vital biological tool with high sensibility that allow it to control frequent input of data easily rather than other algorithms. The SOMs can classify the data properties and can group the similar data together, thus it can be used with biological components, such as proteins, DNA, and neurons as well as it can introduce new ways to relate new datasets. Thus, these algorithms can serve as a bunch analyzing tool of high dimensional data. The SOMs concept is shown in Fig. 3.

Before applying SOMs (a) and after applying SOMs (b)
Figure 3a displays the phase before applying self-organizing feature map, where the input data is in high dimension and it takes a lot of space along with unclassified data. Figure 3b represents the phase after applying the SOMs, where the colors are now in the right position as they should be just like the yellow colors are on the bottom of right side, and the black colors are in the top of right side. The other colors also classified with their similar pattern together. In Fig. 3b, the dimension is also less than that illustrated in Fig. 3a, which was hard to preview because of high dimensional data, while the SOMs has shorten the dimension, thus it become visible. The SOMs network architecture is structured as illustrated in Fig. 4 for a 3 × 3 size 2D network.

A simple Kohonen network
Generally, in every network, there is numerous numbers of nodes, which depends on the architecture size. In the case of 3 × 3 size network of 2D architecture, there are nine nodes. Each node is directly associated with the input layer to place the input as demonstrated in Fig. 4. Each and every node consists of two appointed co-ordinates \(\left( {x, \, y} \right)\), which contains the data input vector. The dimension and type of the vector data are mostly similar to the ones of the corresponding node. Basically, similar data indicates that if the training data contains vector \(A\) of \(N\) dimension, which is represented by \(A\, = \,[A_{1} ,\,A_{2} ,\,A_{3} , \ldots A_{n} ]\), then the node will also contain the same weight vector \(S\) of dimension \(N\), which is expressed as: \(S\, = \,[S_{1} ,\,S_{2} ,\,S_{3} , \ldots ,S_{n} ]\). The SOMs algorithm deals with several datasets by arranging them in secondary or 2D rectangular or hexagonal grids to form architecture of input space \(W \in R^{\text{n}}\), where the common algorithm steps are as follows:

The preceding SOM algorithm steps can be used to solve any classification problem co-efficiently. Since there are four types of proteins, namely: primary, secondary, tertiary and quaternary, the protein classification using the SOM is as follows:
-
The protein data is used as input to the SOM, and then automatically classify them and bound similar data together with specific space. As the input is taken the structure of input data as illustrated in Fig. 5.
Fig. 5 
SOM applied proteins structure
-
All the nodes are initialized using initializing statement that is given by: \(\sum_{i\, = \,0}^{n} W_{i} \, = \,init\,(W_{i} )\).
-
For the input, measure \(D\) represented in Eq. (2) that represents the difference between the nodes initialized protein and the user input protein.
-
Determine the BMU, which is the input protein which is the most similar to the nodes protein in that constant time.
-
Find the radius of the neighborhood of BMU protein using the Gaussian neighborhood exponential decay function given in Eq. (3). Furthermore, the SOMs algorithms exponential decay function has been applied to calculate the neighborhood size. In lieu of this exponential decay function, radioactive decay function is used to calculate the size of the neighborhood as well. Nonetheless, for simplicity exponential decay function is used, where the datasets may increase exponentially and for manipulation of those datasets the decay function has been used which performs well for large and complicated datasets. Better manipulation of this function will work faster in case of mapping and that must affect the results in the final stage in a positive manner.
-
Although, in first stage the radius was large, and large proteins were detected within the radius, but after necessary iteration only one protein will remain and that protein will be the only neighborhood. So, now the neighborhood protein and its radius expansion of neighborhood are known. Thus, it is simple to easily iterate through all the nodes for detecting which proteins are within the range of neighborhood.
-
Now, Eq. (4) is applied to update the detected protein.
-
Currently, all the obtained proteins that completed the above mentioned procedure are altered depending on their distance and similarities with the neighborhood radius and then altered automatically. This procedure illustrates the steps of the SOM for protein classification.

Basically, the SOMs is preferable due to its elevated features regarding vector projection, vector quantization, reduction of input data set’s dimension along with visualization of those datasets, and faster clustering with less space and memory consumption. In addition, increasing input datasets size decreases the percentage error compared to other clustering methods. Moreover, other alternative algorithms of SOMs, such as ISOMAP [18], and Manifold alignment [19] are mostly based on assumptions as well as probabilities of various parameters associated with those methods. Alongside, SOMs lead to higher sensitivity for clustering and projection purpose. Also, manipulation of input data under SOMs is unsupervised and automatic, while counter propagation artificial neural network (CP-ANN) [21], and supervised kohonen networks (SKN) [20] are based on supervised learning. This is the key advantage for SOMs because it can automatically adjust its parameters for any kinds of input data and no further supervision is necessary. Self-organizing map also supports parallel computing using less inter-neuron contact. Its interpretation of faster mapping, easier computation, manipulation of large and complex data consuming less space and time outperforms other mapping and clustering algorithms, which is illustrated in results section of this work.
3.2 Particle swarm optimization
From the perspective of computer science and engineering, PSO is an optimization process for solving the problem under concern in cyclic order as well as for establishing improved view of previously given solution compared to a given quality [70]. PSO is an intelligent optimization process to find the parameters that provide the maximum value and is easy to use and implement dynamically to any necessary aspects using a few numbers of parameters. This algorithm was inspired from the behavior of animals, such as fish schooling or birds flocking and the evolutionary computational fields like the genetic algorithms.
PSO can be easily used without any disturbances or impersonations regarding the problem that need to be optimized and can explore very large space of region. As illustrated in Fig. 6, the PSO algorithm is initialized randomly using the common built in function rand (.). Afterward, the maximum or minimum value of the given function is determined. Meanwhile, all the particles in the PSO algorithm are always trying to find to the best known position and also being guided by the best known local particle’s position. Whenever the best position is gained by a particle, then the local best known position is updated depending on the particles [71]. The PSO algorithm aims to bind all the given particles within the optima of a specific dimensional space. The velocity and position of individual particles are assigned randomly as follows considering ‘n’ particles in a vector form as \(V\, = \,[V_{1} ,\,V_{2} ,\,V_{3} , \ldots ,V_{n} ]\) in the vector space. Initially, pseudorandom numbers are used to initialize the velocity and position vector of each particle. Here, all the position vectors are dihedral angles which are considered as phi and psi. Rather than that the velocity vectors step forward to get best known position by changing these angles. Therefore, these angles enriches the flexibility of obtaining global best known position. Based on the global best known position and velocity, each and every particle turns and updates their velocities along with positions to cover the optima in the content of time. The following equation is used to update the velocity:

Overview of particle swarm optimization
Here, \(v_{i} (t + 1)\) is the new updated velocity of each particle, \(v_{i} (t)\) is the particle vector before update, \(p_{i} (t)\) is the position of each particle, \(p_{i}^{best}\) is the kn own best position, and \(p_{i}^{gbest}\) is the global known best position. In addition,\(m_{1}\) and \(m_{2}\) are the weights of each particle’s personal best known position and global best position. In addition to the velocity update, another update of each particle position in every iteration is required to cover the optima of the desired position:
where,\(p_{i} (t + 1)\), is the updated position for all individual particles, \(p_{i} (t)\) is the previous position of that instant updated particle, and \(v_{i} (t + 1)\) is the updated velocity of that particle. The theoretical representation of the PSO algorithm is illustrated in Fig. 7.

Theoretical representation of particle swarm optimization
Figure 7 illustrates that if \(f(p_{i} )> f(p_{i} (t + 1))\), then the best known position is automatically assigned to \(p_{i} \,({\text{i}} . {\text{e}} .\,\,P_{i} \, = \,p_{i} (t + 1))\), while if \(f(p_{i} ) < f(g)\), and then \(g\) is the best solution. In order to represent the geometrical analysis for the PSO algorithm, a 2-dimensional space is considered for experimental purpose where the particles are moving with their initial velocity and positions.
From each iteration, the velocity, positions as well as values of \(G_{best}\) and \(P_{best}\) are changed to converge to the best optimum position:
Thus, the particles will reach the \(G_{best}\) to obtain optimum coverage. The nominated PSO parameters’ values [72] are as follows:
-
1.
The theoretical range of the particles is 25–40, which is large enough to get perfect result. Sometimes for better results more number of particles is used.
-
2.
There is a limit of changing the velocity and position for every particle. This criterion is used as stopping criterion.
-
3.
There is a conceptual limit of the weight coefficients \(m_{1}\) and \(m_{2}\) which are generally within [0, 2].
-
4.
The stopping criterion depends on the problem to be optimized, but it is terminated when no improvement occurs over some consecutive iterations, then the algorithm stops using:
$$T_{norm} \, = \,\frac{{T_{\hbox{max} } }}{diameter\,(S)}\,$$(4)where, \(T_{\hbox{max} }\) is the maximum radius and \(diametr(S)\) is the initial swarm’s diameter. In addition,
$$T_{\hbox{max} } \, = \,||\,P_{m} \, - \,G_{best} \,||\;{\text{with}}\;||\,P_{m} \, - \,G_{best} \,||\, \supseteq \,|\,P_{i} \, - \,G_{best} \,||,{\text{ as}}\quad i\, = \,1,\,2,\,3, \ldots ,n$$(5)
The pseudo code for the PSO algorithm is as follows.

Generally, the PSO algorithm can be used to automatically classify biological large number of data for example protein classifications. Moreover, the PSO refers to efficient classification as well as clustering tools compared to all the alternative algorithms that are used for classification of various bioinformatics components, such as DNA, and Proteins. It is practically examined that genetic algorithm (GA) is perfect alternative of PSO algorithm and the main differences between these two algorithms are computational efficiency as well as effectiveness of less space consumption. Thus, the PSO algorithm outperforms the GA; even they are quite similar from the perspective of using a combination of deterministic and probabilistic procedure in each iteration. However, PSO is better than genetic algorithm in terms of efficiency as well as space purpose.
3.3 PSO-centric SOMs
Generally, the classification of 2D (secondary) and 3D (tertiary) proteins are much more complicated as it requires more sensitive, less time consuming, and less memory consumption algorithms. It is practically proved that one single algorithm cannot deal appropriately with this situation (Sect. 4). Therefore, a reliable and fast way to gain better performance is required. Thus, combining two bioinformatics algorithms can outperform all the methods discovered previously mentioned, such as the PSO, SOMs, and Bounding Box algorithms. Therefore, for achieving superior approach, the current work introduces a combination of the PSO and SOMs algorithms that performs efficiently. Here, the proposed PSO centric SOMs illustrate the effectiveness of both SOMs and PSO on vast complex and complicated datasets. The SOMs usually diminishes the input space as well as dimensions and represents lower preview facility of higher dimensional data. It has also capability of differentiating all the similar data and group them like 2D and 3D proteins. Therefore, in the first phase, SOMs are applied on the proteins datasets to diminish the dimensions and visualize them as well as differentiating 2D (secondary), 3D (tertiary) and noise data, thereafter, the PSO algorithm is applied. Generally, the PSO algorithm manipulates dynamically movable data using fuzzy logic. It usually follows a dynamic process to bind all the similar data within a certain limit. After the execution of SOMs, the grouped proteins dynamically move around. Afterwards, for acquiring best performance and not to let the movable proteins out of range, immediately the PSO algorithm is applied. This clarifies the reason of the superiority of the proposed PSO-centric SOMs compared to the other algorithms. The overall process is illustrated in Fig. 8.

Overall PSO-Centric SOMs process over a higher dimensional and complex dataset
In Fig. 8, various colors indicate different properties of proteins like 1D (primary), 2D (secondary), and 3D (tertiary).
3.4 Datasets collection process
In the current work, the real-world datasets of Secondary (two dimensional) and tertiary (three dimensional) proteins were excerpted from the NCBI database. In general, for exploration purpose the overall operation of the data excerption is a mobilized process which in greater terms helps gathering data from Google. Usually, for extracting data there are some steps which are mandatory to follow. Therefore, initially, the user logs into Google using authorized entry towards the world class 2D experiment as well as to the 3D proteins’ datasets. Afterward, those datasets are excerpted from the NCBI database proteins part, which contains various types of primary, secondary, and tertiary proteins along with different properties of proteins. Moreover, all the datasets are public and downloadable. Thereafter, the necessary file format is selected to gather the desired proteins and mark them all to send to a particular file as well as for downloading. Then, the datasets will be trained using various bioinformatics algorithms for extraction of pure and noise-free data because the noise full data slow down the overall process. Till now and then, enormous number of researchers, biologists, academician, and trainers are interested with finding an efficient and faster process for detecting the secondary and tertiary proteins from a large number of datasets. Moreover, automatic and unsupervised identification procedure helps a lot for efficient classification of desired proteins. Especially, large proteins datasets along with high frequency make a barrier to the way of desired exploration. The overall collection process for the dataset is illustrated in Fig. 9.

Data collection overall process
4 Results and discussion
In the present work, huge and complex datasets were gathered from National Center for Biotechnology Information (NCBI) database to evaluate the proposed PSO centric SOMs approach. In general, PSO centric SOMs ensure faster identification of both secondary (2D) and tertiary (3D) proteins. Initially, the SOMs manipulate the input data and for efficiency of upcoming level it removes noises, irreverent data and maps the whole data within a certain range so that the PSO algorithm can be applied easily. Afterward, PSO is used to enclose and group similar resultant proteins as much as possible consuming less memory along with consuming less time. For comparison purpose, various single algorithms, such as the bounding box algorithms, PSO alone, SOMs alone have been applied on same datasets and the overall findings have been compared, which showed the superiority of PSO centric SOMs (Sect. 4). All the findings of those algorithms along with PSO centric SOMs have been illustrated graphically and mathematically in the following results section. Moreover, the performance evolution of those algorithms based on their findings has been depicted in the bottom of the result section. Therefore, the current work included several practical experiments to evaluate the performance of many algorithms for proteins classification. Various complex and large datasets have been used for the comparative purpose with the findings of proposed approach. The PSO-centric SOMs algorithm is opted out for its noticeable capability of time saving, high performance in memory reduction, and faster processing. Other applied algorithms are capable of reaching concluding points slower compared to the PSO-centric SOMs. A comparison among findings of various algorithms in terms of the time, memory usage, and possible number of secondary (2D) and tertiary (3D)proteins have been described in details to the following sections (Sects. 4.1–4.7.4). These evaluation parameters associated with 2D and 3D proteins have been represented for bounding box algorithm, PSO, and PSO-centric SOMs using 3D graph. In addition, the comparisons of different algorithms have been illustrated using multiple 3D graphs. In order to implement the proposed approach, Java programming language along with NetBeans platform has been used for this work. Java development Kit (JDK) version 1.7 has been adopted to compile the overall process. Since the adopted JDK system has independent platform of its own, thus it consumes less space and perform efficiency for classification. The overall configuration of the system to execute those algorithms includes 8 GB of RAM, 1000 GB HDD, Core i5 Processor with Windows 7. Moreover, the system can be easily adjusted with Linux, Vista and Windows 10 also. In the proposed work, the Bounding box algorithm, Self-organizing maps algorithm, particle swarm optimization algorithm with the help of java Netbeans IDE along with the features described above to convert it to source code. For obtaining accurate result the output has been evaluated for each and every specific input vectors many times. Similar tasks have been executed for every algorithm and stored the output for that particular algorithm. After that, to evaluate the correctness of the present work contribution, an online database is considered for comparison evaluation. That is how we had created our own database. The comparisons, performance evaluation, findings of various algorithms are illustrated graphically, mathematically and theoretically in the following Sects. (4.1–4.7.4). These evaluation parameters associated with 2D and 3D proteins have been represented for bounding box algorithm, particle swarm optimization, and PSO-centric SOMs using 3D graph. In addition, the comparisons of different algorithms have been illustrated using multiple 3D graphs. In order to implement the proposed approach, Java programming language along with NetBeans platform has been used for this work. Java development Kit (JDK) version 1.7 has been adopted to compile the overall process. Since the adopted JDK system has independent platform of its own, thus it consumes less space and perform efficiency for classification. Moreover, java programming language along with netbeans IDE is used to implement the proposed algorithms in the current work. Python, C++ and C language can be used for this purpose. Basically, java is used for its built-in key feature of Java Virtual Machine, which is language independent. On the contrary, python interpreter is language dependent, which is more sophisticated than JVM. Additionally, java programming language guarantees accurate and faster manipulation of input data. This will definitely bring a positive effect on the findings of the proposed approach. The overall configuration of the system to execute those algorithms includes 8 GB of RAM, 1000 GB HDD, Core i5 Processor with Windows 7. Moreover, the system can be easily adjusted with Linux, Vista and Windows 10 also. We have executed Bounding box algorithm, Self-organizing maps algorithm, particle swarm optimization algorithm with the help of java Netbeans IDE along with the features described above to convert it to source code. For obtaining accurate result the output has been evaluated for each and every specific input vectors many times. Similar tasks have been executed for every algorithm and stored the output for that particular algorithm. After that, to evaluate the correctness of our contribution, we have considered online database to compare with ours and outcome of both seems to be similar and accurate. That is how we had created our own database. The comparisons, performance evaluation, findings of various algorithms are illustrated graphically, mathematically and theoretically in the following Sects. (4.1–4.7.4).
4.1 Bounding box algorithm for proteins separations
Bounding box approach [59, 60] is data processing mechanisms that allow all the datasets under specific sizes. This process is helpful for limited datasets. In this process, whole DNA sequences are grouped into row and column modules where each row contains eighty DNA base pairs and columns contains sixty DNA base pairs. A significant step of Bounding Box is that it sub divide the whole datasets until it reach a satisfied DNA segments. The number of proteins in various dimensions using Bounding box algorithm for proteins separation was determined as reported in Table 1, where the significant values are in bold. The algorithm returned different values for different sizes of data in 2D space as well as 3D space.
Table 1 reports that for 100 MB proteins data, 77 two-dimensional proteins along with 4 three-dimensional proteins were obtained. In the case of simplicity, the constant experimental number of proteins were considered as 1000 proteins of various types, then exactly \(\frac{number\,of\,2D\,proteins\, \times \,\,100}{total\,\,number\,of\,proteins}\) = \(\frac{77\, \times \,\,100}{1000}\) = 7.7% of 2D proteins and \(\frac{number\,of\,3D\,proteins\, \times \,\,100}{total\,\,number\,of\,proteins}\) = \(\frac{4 \times \,\,100}{1000}\) = 0.4% of 3D proteins were obtained. Respectively, the same procedure is perceived for the remaining data as resumed by third and fifth columns in Table 1. From these results, it could be realized that the possibilities of getting 2D and 3D data from same number of proteins is in increasing order. The determination of only 2D protein is clear in the case of the 900 MB having 134.3% for 2D protein, and 11.5% for 3D protein as well as with the 950 MB having 132.1% for 2D protein, and 12.5% for 3D protein.
However, using the bounding box algorithm, the differences of resultant data for 2D and 3D proteins were (10.3 − 7.7)% = 2.6%, and (0.5 − 0.4)% = 0.1% for 100 and 150 MB data, respectively. The differences seem to be huge with the increasing size of data to be experimented. For more simplicity, the differences and details relationships between the experimented and resultant data is exhibited are Fig. 10. In Fig. 10, X-axis shows the proteins data size, Y-axis exhibits the findings of both 2D and 3D proteins and Z-axis shows the number of difference between the 2D and 3D findings compared to data size.

2D and 3D proteins regions determined by the bounding box algorithm
In Fig. 10, the black and red stairs indicate resultant number of secondary (2D) and tertiary (3D) proteins experimented by bounding box algorithm. The black stair represents the findings number of 2D proteins, whereas the red stair defines findings of 3D proteins. The red stair is quite low than the black which indicates the findings of 2D proteins were higher than to the 3D as the initial points of X-axis and Y-axis were 100 and 0 MB. In addition, the topmost points were 1150 MB and 1800 although the topmost resultant point of 2D proteins was 15. Consequently, the above designed graph represents three dimensional views of findings of experimented data for the bounding box algorithm.
4.2 Self-organizing maps for proteins separations
By applying the SOMs for proteins classification were obtained the values indicated in Table 2 as to 2D and 3D proteins. The SOMs algorithm detected various numbers of 2D as well as 3D proteins.
The data in Table 2 suggests that from the aspects of SOMs algorithm, the difference between the two findings of 2D or 3D protein was quite large for 100 MB size of data as 109 for 2D along with 6 for the 3D were detected. Considering 10,000 proteins as constant experimented value for 100, 150, 200 MB and so on, it was observed that \(\frac{number\,of\,2D\,proteins\, \times \,\,100}{total\,\,number\,of\,proteins}\) = \(\frac{109\, \times \,\,100}{10000}\) = 1.09% for 2D along with \(\frac{number\,of\,3D\,proteins\, \times \,\,100}{total\,\,number\,of\,proteins}\) = \(\frac{6\, \times \,\,100}{10000}\) = 0.06% for 3D. Consequently, applying same process for 150, 200, 650, 800, 1000 MB it could be obtained 1.88, 2.99, 12.99, and 25.67% of 2D proteins as well as 0.09, 0.17, 1.57, 2.43, and 3.78% of 3D proteins. For self-organizing maps the findings were increasing with the increasing rate of data size. Initially, the rising rate was low, but gradually its increment were large to be observed as for the 100 and 150 MB resultant difference of 2D proteins, which were (188 − 109) = 79 in 2D proteins and (9 − 6) = 3 in 3D proteins; whereas when the data size was 950 and 1000 MB, then the difference was 267 for the 2D proteins and 43 for the 3D proteins. The differences seem to be huge with the increasing experimented data size and there is no break of this rising rate of the data results. Furthermore, a graph regarding the relationship, differences, and data sizes is provided in Fig. 11 within which both of black and red staircases perform the role of determined 2D and 3D proteins after applying the SOM algorithm.

2D and3D proteins regions determined by self-organizing maps algorithm
In Fig. 11, the black stairway indicates the 3D number of proteins, while the red stairway represents 2D number of proteins, X-axis shows the data size, Y-axis defines the findings both (2D and 3D), and Z-axis indicates the difference level of 2D and 3D proteins like here black stair is higher than red. From Table 2, it can be realized that the initial value of data size was 100 MB, so in graph the initial level was defined as 100 MB and topmost value was seemed to be 1150 MB, similarly for both findings, the initial value was zero and topmost was defined as 3000 to cover the experimented last value 2654. Finally, the designed graph illustrates three dimensional views of findings of experimented data for self-organizing maps algorithm.
4.3 Bounding box algorithm versus the self-organizing maps
The increasing rate of 2D proteins with the increasing number of input data size for both the bounding box and SOMs algorithms is reported in Table 3.
Table 3 depicts that for 2D proteins, when the data size was100 MB, the number of findings in SOMs was greater than the Bounding Box. For 150 MB data, it was (188 − 103)/100 = 0.85% rise of Bounding box algorithm. Consequently, for 200, 250, 450, 700, 900, and 1150 MB data size, the increasing rate was 1.44, 1.61, 3.51, 5.76, 8.75, and 10.65%, respectively, for the SOMs from the Bounding Box algorithm. Initially, the rising rate was low, but with the increasing amount of input data size, the rate seemed to be large such as from 150 to 200 MB data, the rising rate was(1.41 − 0.85)% = 0.56%, whereas for 450–700 MB data, the rising rate was approximately (5.76 − 3.51)% = 2.25%, which is obviously greater than 0.56%.
The same indications can be perceived from the data in Table 3 for 3D proteins region. Initially, the difference was [(6 − 4)/100 − (9 − 5)/100] which is exactly 0.02% increase of SOM, where for 1150 MB data, the difference seemed to be exactly (421 − 154)/100 − (378 − 141)/100 which is 0.3% increase of SOMs from Bounding Box algorithm for 3D data. Thus, for both the 2D and 3D findings, the SOMs provided better results than the Bounding box algorithm although in initial stage the difference was poor; afterwards, it increased with the increasing data size.
Figure 12 illustrates the varieties of both 2D and 3D findings between SOMs and Bounding Box algorithms. The green staircase represents secondary (2D) SOMs findings, the red one defines findings of Bounding Box algorithm for 2D proteins, and the blue stairway shows the Bounding Box detected tertiary (3D) protein, whereas cyan colored mount is presenting experimented result of SOMs number of 3D proteins. In Fig. 12, X-axis is defined as input data size, Y-axis is indicated as findings of SOMs and Bounding Box algorithm, and the Z-axis shows the experimented results differences between them.

2D and 3D findings after applying both SOMs and bounding box algorithm along with data size
In Fig. 12, Z-axis demonstrates that the green mount and Cyan colored mount are higher for both secondary (2D) and Tertiary (3D) findings than the red and blue one. This happened because of the efficiency, sensitivity, and time-consumption that the SOM depicted much. Although initially it is hard to differentiate as the difference is reduced, but with the context of increasing number of data the perspective or view is changed. Consequently, clear graphical views after rising of data size can be obtained. Consequently, Fig. 12 suggests that for large number of input data the SOM algorithm definitely had better ability than the bounding box algorithm for separating proteins.
4.4 Time comparisons among bounding box, SOMs and PSO centric SOMs
A comparative study for the three algorithms under comparison, namely bounding box algorithm, self-organizing map and PSO Centric-SOMs algorithm, led to the values report in Table 4 in terms of the time consuming in nanoseconds. Generally, the bounding box algorithm is a linear time measurement algorithm, which is simple, efficient, and requires less time consuming. It takes approximately O(n) times to be completed, though the implementation is little bit complicated. Besides, the SOM is also very easy to understand and to implement. It can easily classify any type of data in an effective manner even faster. Also, it can differentiate the similarities and dissimilarities between data fluently. The SOMs is more sensitive, thus it can go through higher dimensional data and evaluate them. The time complexity of the SOMs is quite stable of order O(S2), where S is computational time indicated in Table 4.
The hybrid proposed algorithm (PSO centric-SOMs) (Fig. 2) is basically the combination of two methods, namely the PSO and the SOM. This process was most reactive and also the best less time erosive algorithm. In this proposed solution, the SOMs is applied to classify the data and then PSO encloses the classification results as much as possible in the shortest possible time. The effectiveness of PSO centric with SOMs algorithm can be noticeable from the data in Table 4 regarding the time consuming of both 2D and 3D proteins for Bounding Box algorithm, SOMs algorithm and PSO centric with SOMs algorithm in nanoseconds for specific size of data.
Table 4 establishes that the proposed algorithm was the most proficient compared to the other algorithms. Thus, the SOMs algorithm enhanced the capability and the performance of the PSO algorithm, where the significant values are in bold. Table 4 suggests large difference between the execution time of the different algorithms. For example, in the case of 100 MB proteins data, the Bounding Box algorithm detected the 2D and 3D proteins in 1654 (ns), and the SOM took 1209 (ns), while the proposed PSO centric-SOMs algorithm took 784 (ns). A big difference in the time consuming when using the proposed PSO centric-SOM solution over that required when using the Bounding Box is obtained, where the proposed method was(1654 − 784) = 870 times or \(\frac{(1654\, - \,784)}{1654}\) = approximately 52.59% faster than Bounding Box. In addition, the proposed PSO Centric-SOM was (1209 − 784) = 425 times or \(\frac{(1209\, - \,784)}{1209}\) = approximately 35.15% more efficient than the SOMs algorithm. Similarly, for 200 MB proteins data, the PSO centric-SOM was almost 1823 time or 50.59% faster than the bounding box and 898 times or 33.87% more fluent than the SOM. Consequently, when the increasing data size was up to 500 MB, such as in the case of 650 MB, then the PSO centric-SOM was 13126 times or 56.54% and 8567 times or 45.92% efficient than bounding box and SOM, respectively. Therefore, for 850 MB data, it was 55.52 and 39.41% and for 1150 MB data, the PSO centric with SOM was approximately 62 and 48.49% more fluent than the others.
Furthermore, with the increasing size of data, the working capability of PSO centric with SOMs raised and so the finding time was also raised in a large rate. For PSO centric with SOM, when the data size was 150 MB, the time consuming was 1230(ns). Similarly, for 200, 250, 300, 350, and 400 MB, the execution time was 1753, 2387, 2765, 3365, and 3987 ns, respectively, this suggests increasing rate of times. In addition, it could be established that the SOM was more efficient than the Bounding Box algorithm as in every increasing size of data like 100, 150, 200, 250, 600, 700, and 750 MB, the SOM was respectively 26.90, 30.08, 25.58, 26.69, 26.64, 23.90 and 27.75% faster than the bounding box algorithm.
From the extensive preceding results, it could be established that the PSO centric with SOMs was efficient and less time consuming than both the bounding box algorithm and self-organizing map algorithm. The relations, differences between results, and the time comparisons reported in Table 4 are exhibited in Fig. 13. In Fig. 13, X-axis represents the data size in Megabyte and each unit differences is 20 MB of data so 5 means 5 × 20(MB) = 100 MB, similarly 10 is 200 MB, 15 is 300 MB and so on. Y-axis exhibits the time required by each algorithm compared to a specific size of data.

Time comparisons among bounding box, SOM and PSO centric SOM algorithms
In Fig. 13, the blue line indicates the time needed for Bounding Box algorithm, for example in the case of 100 MB data, 1656 ns time were required, which is figured in the graph as point (100, 1656). Similarly, the red line shows the time consumed by the SOMs algorithm. The other green line represents the consuming time for PSO Centric-SOMs. From the graph shown, it is clear that the PSO Centric with SOMs was efficient with less time consuming compared to SOMs. Moreover, SOMs required less time consuming compared to the Bounding Box algorithm. Consequently, for increased size proteins data, the proposed PSO Centric with SOMs is efficient, requires less time consuming, and is more capable than SOMs and Bounding Box algorithm. The proposed approach is concerned with the angles and hydrocarbon bonds of the proteins during separations. However, interactions among proteins are not considered. These interactions among proteins are complex and significant for drug design and disease investigation, which can be studied further in the future.
4.5 Particle swarm optimization for proteins classification
Proteins are biologically complicated in nature but execute a number of significant accessories in human body as well as for any kind of living cell. From the last decades, proteins have drawn the attention of both the researchers and biologists to discover the uncovered knowledge. In this current work PSO has been implemented to optimize the input datasets. For optimization purpose, the particle swarm optimization algorithm was used here considering various remarkable benefits in classification:
-
1.
The particle swarm optimization can easily manipulate the dynamic movements of input proteins. While each node of PSO takes proteins as input the proteins are movable. Basically, PSO itself follow dynamic process to bring all the similar proteins within a particular limit. Figure 14 shows dynamic movements of proteins. After a certain time, all the moving proteins will be bound within a limit following fuzzy logic.
Fig. 14 
Dynamic movements of proteins
-
2.
It always helps to step forward to a solution using less number of parameters as well as efficient enough compared to other algorithms. Furthermore, the PSO algorithm is employed to work on fuzzy logic which bind each and every proteins within the limit of fuzzy logic whereas others algorithms works with crisp logic which is almost straightforward.
-
3.
The information sharing system PSO follow quite exceptionally different from others like in PSO the input layers are connected with enormous other nodes. Information regarding various proteins is transferred not only for some exact known values but also each and every possible fraction values of proteins.

Therefore, the basic particle swarm optimization algorithm (PSO) is quite easy to implement and a few parameters are required to be evaluated. It also has no overlapping and mutation calculation along with the ability of better performance.
4.6 Impact self organization mapping on particle swarm optimizations
Mapping plays significant role in large dataset processing. Self-Organization mapping enables whole datasets into specific format with the mapping functions as well as feed forward learning. As a result, SOM centric PSO outperforms only PSO performances in proteins classifications. This research work only considers the number of proteins in both the cases (Table 5), where the significant values are in bold.
Table 5 indicates the differences of resultant findings for both 2D and 3D proteins by PSO algorithm and SOMs based particle swarm optimization (PSO).A large difference is noticeable while using SOMs based PSO and while using only PSO. For the simplicity of manipulation, if we consider the constant experiment data as 10,000 proteins, then from Table 5, we can notice that for 100 MB data, the number of resultant 2D proteins were 109 \(\left( {\frac{109\, \times \,100}{10000}\, = \,1.09\% } \right)\) after demonstrating only PSO, whereas while using SOMs based PSO the finding, was 177, \(\left( {\frac{177\, \times \,100}{10000}\, = \,1.77\% } \right)\) 2D proteins. The difference is approximately (1.77 − 1.09)% = 0.68%. Similarly, for 3D proteins, the PSO algorithm found about 6, \(\left( {\frac{6 \times \,100}{10000}\, = \,0.06\% } \right)\) proteins, meanwhile, SOMs based PSO detected 9, \(\left( {\frac{9 \times \,100}{10000}\, = \,0.09\% } \right)\), led to a difference of about (0.09 − 0.06)% = 0.03%. Following same procedure for 150 MB, 200 MB, 250 MB, 300 MB, the resultant 2D proteins for only PSO were 188,\(\left( {\frac{188\, \times \,100}{10000}\, = \,1.88\% } \right)\), 299 \(\left( {\frac{199\, \times \,100}{10000}\, = \,2.99\% } \right)\), 365 \(\left( {\frac{165\, \times \,100}{10000}\, = \,3.65\% } \right)\), 476 \(\left( {\frac{476\, \times \,100}{10000}\, = \,4.76\% } \right)\). On the contrary, while SOMs based PSO was used, the 2D protein findings were remarkable in amounts and for 150, 200, 250, 300 MB the findings were, respectively, 245 \(\left( {\frac{245\, \times \,100}{10000}\, = \,2.45\% } \right)\), 301 \(\left( {\frac{301\, \times \,100}{10000}\, = \,3.01\% } \right)\), 489 \(\left( {\frac{489 \,\times \,100}{10000}\, = \,4.89\% } \right)\), and 603 \(\left( {\frac{603\, \times \,100}{10000}\, = \,6.03\% } \right)\). The noteworthy differences were about (2.45 − 1.88)% = 0.57%, (3.01 − 2.99)% = 0.02%, (4.89 − 3.65)% = 1.24%, (6.03 − 4.76)% = 1.27%, which represents the effectiveness of proposed algorithm compared to particle swarm optimization (PSO). If we pay attention to 3D findings for 150, 200, 250, 300 MB, we get 0.09, 0.17, 0.23, and 0.36% from PSO alone and also 0.14%, 0.20%, 0.27%, 0.44% from SOMs based PSO, respectively. Therefore, the SOMs based PSO solution led to 0.05, 0.03, 0.04, 0.08% better results for 3D proteins than PSO alone. Increasing data sizes enlarge the effectiveness of SOMs based PSO than PSO alone. If we consider the data size of 950 MB, 1000 MB and 1150 MB, then we get, respectively 23, 25.67, 26.54% of 2D proteins and 3.35, 3.78, 4.21% of 3D proteins while experimenting by PSO alone. Rather than that while using SOMs based PSO on same data set, we get 45.09, 57.80, 70.21% number of 2D proteins and 16.09, 23.21, 32.14% of 3D proteins. Therefore, the difference which is most remarkable was obtained by the proposed algorithm (SOMs based PSO),which was approximately 22.09, 32.13, 43.67% faster in 2D protein classification than PSO, and also 12.74, 19.43, 27.93% faster in 3D protein classification compared to PSO alone. Here, one thing which also draws our attention is the classification process was getting faster with the increasing size of data set for proposed approach, where PSO alone could not maintain that efficiently. Figure 15 illustrates the findings comparison while using SOMs centric PSO and PSO alone. In Fig. 15, X-axis indicates the data size and Y-axis depicts the findings value of both 2D and 3D proteins. Also, the dark salmon curve illustrates the findings of 2D proteins after using SOMs centric PSO, whereas the deep sky blue curve represents the 2D findings after accomplishing PSO alone. Furthermore, the yellow curve is the presenter of 3D findings for SOMs centric PSO and grey curve represents the findings of 3D proteins experimented by PSO alone. After evaluating Fig. 15, it comes to our realization easily that overall performance or classification ability of SOMs centric PSO was (177 − 109) = 68 2D and (9 − 6) = 3, 3D proteins faster than PSO alone which was approximately 38.41% 2D and 33.3% 3D proteins.

Comparison between SOMs based PSO and PSO alone

Relationship between precision and recall for mapping less PSO

Relationship between precision and recall for SOM centric PSO
Therefore, the overall comparison confirms the effectiveness of the proposed approach (SOMs centric PSO) compared to particle swarm optimization (PSO) alone.
4.7 Performance evaluation of SOMs over PSO and SOMs less PSO
F-measure has been calculated to verify the performances of PSO albeit the SOMs and SOMs less PSO. In addition, the specificity and precision have been checked along with recall. There are few parameters that control the overall processing. These are true positive, false positive, false negative and true negative. True positive indicates exact result that should be the real output. True negative indicates the real limitations of the analysis. False positive defines about the wrong predictions during the experiments. False negative means the wrong outputs are showing as correct one:
In addition, more other feasible alternative indicators are sensitivity and accuracy. Specificity measures the desired outcomes and precision computes the exact values. Sensitivity and accuracy will increase the intensity of the outcomes. However, in the present work, only protein structures have been considered. In that case specificity and precision are enough to handle the outcome.
Since the current work focuses on the structures, specificity and precision are sufficient. In future work, the sensitivity can be measured.
4.7.1 1F-measures of SOM less PSO for 3D portions
F-measures reflect the actual facts of the classification for SOM less PSO. F-measure is the ratio between precision and recall. Precision measures the true classification by considering ratio between true classification and summation of true and false classification. The values obtained as to F-measures of SOM less PSO are indicated in Table 6.
4.7.2 Mapping less PSO Specificity Measurements
Basic experimental results must have two parts as target areas and non-target areas. Targeted values are the pivotal part of the research concentration. On the other hand, non-focused points are very important for its presence in total dataset. In recent machine learning analysis, specificity includes both focused and non-focused area. Moreover, the specificity defines to the exact options of identifying the non-focused points accurately from collected dataset. Mathematically, the specificity is calculated using:
From the specificity analysis performed, the values presented in Table 7 were obtained. From these values, it can be easily concluded that SOM less PSO accurately detected the non-3D proteins area parts of the total datasets. Consequently, the specificity of this processing was 1 − 0.23 = 0.77 = 77% accurate for SOM less PSO.
4.7.3 3F-measures of SOM centric PSO for 3D portions
F-measures define the actual outcomes of the classification for SOM centric PSO. This is the ratio between precision and recall. The values obtained as to F-measures of SOM based PSO were the ones presented in Table 8.
4.7.4 Mapping based PSO specificity measurements
General outcomes of the processing contain two parts as target points and non-target points. Targeted parts are the pivotal concentration of the research work. On the other hand, non-values points are also very important for its presence in total dataset. Mathematically, the specificity is calculated using:
From the specificity analysis conducted, the values indicated in Table 9 were obtained. From the obtained values, it can be easily concluded that SOM based PSO accurately detected the 3D proteins area parts of the total datasets. Hence, the specificity of SOM oriented processing was 1 − 0.018 = 0.9918 = 99.18% accurate for SOM based PSO.
5 Conclusion
Machine learning base proteins classifications system help to separate proteins into two different groups. Training datasets were verified with set of mining methods. Self-organizing maps (SOMs) process the whole data with exact shape. It acts as data mapper as well as organizer. Initially, shape the similar data into a certain region by considering relative distances among proteins. Then, shape the proteins into corner according to their angles. Particle Swarm intelligence enables faster processing by associating features with common nature. Experimental result established that swarm bases SOM outperform the swarm less SOMs. The experimental results established that the proposed PSO-centric with SOM approach is faster than the other algorithms process along with less time consumption.
Comparing the proposed algorithm with the findings of [32, 17], it is possible to notice a remarkable improvement by using the proposed algorithm due to the combination of the two methods which strengthened the overall process. The present contribution refers a trifles representation regarding classification of 2D (secondary) and 3D (tertiary) proteins. In addition, quite remarkable findings were noticeable after manipulating the huge datasets using PSO centric SOMs compared to other algorithms. Since computer science generally addresses numerical data, thus for the manipulation purpose, the biological huge datasets were converted to binary values by using the Otsu method [24–26] for further process. For clear visualization and interpretation purpose, the datasets were represented considering the angles of proteins for detection of variation, patters and trends within the datasets. Therefore, all experiments presented in the current article using various single and multi-classification algorithms were efficient achieved using less time, and allocating shortest memory space along with complex, complicated data in sophisticated way. The comparisons have been illustrated in the results section (Sects. 4.1–4.7.4).
The results established that the proposed combination of algorithms detected higher number of secondary and tertiary proteins compared to the findings of bounding box and self-organizing maps alone. The performance evolution also shows better result for PSO centric SOMs rather than others. Meanwhile, the main contribution of the current work is focused on the secondary and tertiary proteins data which refers to the use of only homogeneous datasets for experimental purpose. Therefore, a long term goal of working with both homogeneous along with heterogeneous datasets is recommended in future work. For this consequence, the proposed algorithm needs to be developed little bit more to ensure faster and accurate manipulation of both homogeneous and heterogeneous datasets. Moreover, these works is quite straightforward for imbalance datasets and ignore this type of data while mapping. Thus, the manipulation of imbalance data for achieving more accurate and exact result is recommended as a future work.
References
Turcu A, Palmieri R, Ravindran B, Hirve S (2016) Automated data partitioning for highly scalable and strongly consistent transactions. IEEE Trans Parallel Distrib Syst 27(1):106–118
Chien JT, KuBayesian YC (2016) Recurrent neural network for language modeling. IEEE Trans Neural Netw Learn Syst 27(2):361–374
Deng SP, Zhu L, Huang DS (2016) Predicting hub genes associated with cervical cancer through gene co-expression networks. IEEE/ACM Trans Comput Biol Bioinform 13(1):27–35
Hsieh SY, Chou YC (2016) A Faster cDNA microarray gene expression data classifier for diagnosing diseases. IEEE/ACM Trans Comput Biol Bioinform 13(1):43–54
Dhulekar N, Ray S, Yuan D, Baskaran A, Oztan B, Larsen M, Yene B (2016) Prediction of growth factor-dependent cleft formation during branching morphogenesis using a dynamic graph-based growth model. IEEE/ACM Trans Comput Biol Bioinform 13(2):350–363
Sáez JA, Luengo J, Herrera F (2016) Evaluating the classifier behavior with noisy data considering performance and robustness: the equalized loss of accuracy measure. Neurocomputing 176:26–35
Saez JA, Galar M, Luengo J, Herrera F (2016) INFFC: an iterative class noise filter based on the fusion of classifiers with noise sensitivity control. Inf Fusion 27:505–636
Fdez JA, Alonso JM (2016) A survey of fuzzy systems software: taxonomy, current research trends and prospects. IEEE Trans Fuzzy Syst 24(1):40–56
Palacios A, Sanchez L, Couso I (2016) An extension of the FURIA classification algorithm to low quality data through fuzzy rankings and its application to the early diagnosis of dyslexia. Neurocomputing 176:60–71
González M, Bergmeir C, Triguero I, Rodríguez Y, Benítez JM (2016) On the stopping criteria for k-nearest neighbor in positive unlabeled time series classification problems. Inf Sci 328:42–59
Martin D, Fdez JA, Rosete A, Herrera F (2016) NICGAR: a niching genetic algorithm to mine a diverse set of interesting quantitative association rules. Inf Sci 355–356:208–228
Butt AH, Khan SA, Jamil H, Rasool N, Khan YD (2016) A prediction model for membrane proteins using moments based features. Biomed Res Int 2016:8370132. doi:10.1155/2016/8370132
Vala MHJ, Baxi A (2013) A review on otsu image segmentation algorithm. Int J Adv Res Comput Eng Technol 2(2):387–389 (ISSN: 2278–1323)
Akbal-Delibas B, Farhoodi R, Pomplun M, Haspel N (2016) Accurate refinement of docked protein complexes using evolutionary information and deep learning. J Bioinform Comput Biol 14(3):1642002. doi:10.1142/S0219720016420026
Wang B, Wang M, Jiang Y, Sun D, Xu X (2015) A novel network-based computational method to predict protein phosphorylation on tyrosine sites. J Bioinform Comput Biol 13:1542005. doi:10.1142/S0219720015420056
Wang D, Hou J (2015) Explore the hidden treasure in protein–protein interaction networks—an iterative model for predicting protein functions. J Bioinform Comput Biol 13(5):1550026. doi:10.1142/S0219720015500262
Watson JD, Laskowski RA, Thornton JM (2005) Predicting protein function from sequence and structural data. Curr Opin Struct Biol 15(3):275–284
Tan S, Guan Z, Cai D, Qin X, Bu J, Chen C (2014) Mapping users across networks by manifold alignment on hypergraph. In Proceedings of the twenty-eighth AAAI conference on artificial intelligence (AAAI’14), 159–165
Bangyal W, Jamil A, Shafi I, Abbas Q (2011) propagation network-based approach for contraceptive method choice classification task. J Exp Theor Artif Intell 24(2):211–218
Brereton RG, Lloyda GR (2010) Support vector machines for classification and regression. Analyst. doi:10.1039/B918972F
Iranmanesh A, Fahimi M (2001) Genetic algorithm trained counter-propagation neural net in structural optimization. In: Proceedings of the sixth international conference on Application of artificial intelligence to civil and structural engineering (ICAAICSE ‘01), Topping BHV, Kumar B (Eds.). Civil-Comp Press, pp. 85-86
Bollen J, Van de Sompel H, Hagberg A, Chute R (2009) A principal component analysis of 39 scientific impact measures. PLoS One 4(6):e6022. doi:10.1371/journal.pone.0006022
MacQueen JB (1967) “Some methods for classification and analysis of multivariate observations. In: Proceedings of 5-th Berkeley symposium on mathematical statistics and probability”. Berkeley, University of California Press, 1:281–297
Yuan X, Martínez J-F, Eckert M, López-Santidrián L (2016) An improved Otsu threshold segmentation method for underwater simultaneous localization and mapping-based navigation. Sensors 16(7):1148. doi:10.3390/s16071148
Xu ZB, Chen PJ, Yan SL, Wang TH (2014) Study on Otsu threshold method for image segmentation based on genetic algorithm. Adv Mater Res 999:925–928
Hegde GP, Seetha M, Hegde N (2016) Kernel locality preserving symmetrical weighted fisher discriminant analysis based subspace approach for expression recognition. Int J Eng Sci Technol 19(3):1321–1333. doi:10.1016/j.jestch.2016.03.005
Taormina R, Chau KW (2015) Data-driven input variable selection for rainfall–runoff modeling using binary-coded particle swarm optimization and extreme learning machines. J Hydrol 529:1617–1632
Pedruzzi I, Rivoire C, Auchincloss AH et al (2013) HAMAP in 2013, new developments in the protein family classification and annotation system. Nucleic Acids Res 41(D1):D584–D589. doi:10.1093/nar/gks1157
Maddouri RSM, Nguifo EM (2010) Protein sequences classification by means of feature extraction with substitution matrices. BMC Bioinform 11:175
Bernardes JS, Fernandez JH, Vasconcelos ATR (2008) Structural descriptor database: a new tool for sequence-based functional site prediction. BMC Bioinform 9:492
Yan R-X, Si J-N, Wang C, Zhang Z (2009) DescFold: a web server for protein fold recognition. BMC Bioinform 10:416
Rost B, Liu J, Nair R, Wrzeszczynski KO, Ofran Y (2003) Automatic prediction of protein function. Cell Mol Life Sci. 60(12):2637–2650
Baugh EH, Simmons-Edler R, Müller CL, Alford RF, Volfovsky N, Lash AE, Bonneau R (2016) Robust classification of protein variation using structural modelling and large-scale data integration. Oxf J Sci Math Nucleic Acids Res 44(6):2501–2513
Dinubhai PM, Shah HB (2013) Comparative study of multi-class protein structure prediction using advanced soft computing techniques. Int J Eng Sci Innov Technol 2(2):275–282
Burkhardt K, Schneider B, Ory J (2006) A biocurator perspective: annotation at the research collaboratory for structural bioinformatics protein data bank. PLoS Comput Biol 2(10):e99. doi:10.1371/journal.pcbi.0020099
Li YH, Xu JY, Tao L, Li XF, Li S et al (2016) SVM-Prot 2016: a web-server for machine learning prediction of protein functional families from sequence irrespective of similarity. PLos One 11(8):e0155290. doi:10.1371/journal.pone.0155290
Cai Y-D, Liu X-J, Xu X-B, Zhou G-P (2001) Support vector machines for predicting protein structural class. BMC Bioinform 2:3
Selvaraj MK, Puri M, Dikshit KL, Lefevre C (2016) BacHbpred: support vector machine methods for the prediction of bacterial hemoglobin-like proteins. Adv Bioinform 2016:8150784. doi:10.1155/2016/8150784
Dhifli W, Diallo AB (2016) ProtNN: fast and accurate nearest neighbor protein function prediction based on graph embedding in structural and topological space, Cornell University, pp 1–28
Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect D Biol Crystallogr 60(12):2256–2268
Bhattacharya S, Bhattacharyya C, Chandra NR (2007) Comparison of protein structures by growing neighborhood alignments. BMC Bioinform 8:77. doi:10.1186/1471-2105-8-77
Nandanwar S, Murty MN Structural neighborhood based classification of nodes in a network. In: Proceeding, KDD ‘16 Proceedings of the 22nd ACM SIGKDD international conference on knowledge, discovery and data mining, pp. 1085–1094, ACM New York, NY, USA
Bhatia N, Vandana SSCS (2010) Survey of nearest neighbor techniques. Int J Comput Sci Inf Secur 8:302–305
Desrosiers C, Karypis G (2010) A comprehensive survey of neighborhood-based recommendation methods. In: Ricci F, Rokach L, Shapira B, Kantor PB (eds) Recommender systems handbook. Springer, Boston, pp 107–144. doi:10.1007/978-0-387-85820-3_4
Hadley C, Jones DT (1999) A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Structure 7(9):1099–1112
Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540
Hore S, Chatterjee S, Sarkar S, Dey N, Ashour AS, Balas-Timar D, Balas VE (2016) Neural-based prediction of structural failure of multistoried RC buildings. Struct Eng Mech 58(3):459–473
Zhang J, Chau KW (2009) Multilayer ensemble pruning via novel multi-sub-swarm particle swarm optimization. J UCS 15(4):840–858
Sharma K, Virmani J (2017) A decision support system for classification of normal and medical renal disease using ultrasound images: a decision support system for medical renal diseases. Int J Ambient Comput Intell 8(2):52–69
Wang WC, Chau KW, Xu DM, Chen XY (2015) Improving forecasting accuracy of annual runoff time series using ARIMA based on EEMD decomposition. Water Resour Manag 29(8):2655–2675
Li Z, Shi K, Dey N, Ashour AS, Wang D, Balas VE et al (2017) Rule-based back propagation neural networks for various precision rough set presented KANSEI knowledge prediction: a case study on shoe product form features extraction. Neural Comput Appl 28(3):613–630
Manogaran G, Lopez D (2017) Disease surveillance system for big climate data processing and dengue transmission. Int J Ambient Comput Intell 8(2):88–105
Zhang S, Chau KW (2009) Dimension reduction using semi-supervised locally linear embedding for plant leaf classification. In: International conference on intelligent computing. Springer, Berlin, pp 948–955. doi:10.1007/978-3-642-04070-2_100
Wu CL, Chau KW, Li YS (2009) Methods to improve neural network performance in daily flows prediction. J Hydrol 372(1):80–93
Chau KW, Wu CL (2010) A hybrid model coupled with singular spectrum analysis for daily rainfall prediction. J Hydroinform 12(4):458–473
Wang XZ, He YL, Dong LC, Zhao HY (2011) Particle swarm optimization for determining fuzzy measures from data. Inf Sci 181(19):4230–4252
Wang XZ, Xing HJ, Li Y, Hua Q, Dong CR, Pedrycz W (2015) A study on relationship between generalization abilities and fuzziness of base classifiers in ensemble learning. IEEE Trans Fuzzy Syst 23(5):1638–1654
Nimmy SF, Kamal MS (2015) Next generation sequencing under De-Novo genome assembly. Int Journal of Biomath 8(5):1–29
Kamal MS, Khan MI (2014) performance evaluation of Warshall algorithm and dynamic programming for markov chain in local sequence alignment. Interdiscip Sci Comput Life Sci 7(1):78–81
Kamal MS, Khan MI (2014) An integrated algorithm for local sequence alignment. Netw Model Anal Health Inform Bioinforma 3:1–9. doi:10.1007/s13721-014-0068-8
Chatterjee S, Hore S, Dey N, Chakraborty S, Ashour AS (2016) Dengue fever classification using gene expression data: a PSO based artificial neural network approach. In: 5th International conference on frontiers in intelligent computing: theory and applications, volume: Springer AISC
Wang D, He T, Li Z, Cao L, Dey N, Ashour AS, Balas VE, McCauley P, Lin Y, Xu J, Shi F (2016) Image feature-based affective retrieval employing improved parameter and structure identification of adaptive neuro-fuzzy inference system. Neural Comput Appl. doi:10.1007/s00521-016-2512-4
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M (2016) O. J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14):6614–6624
Tateno Y, Miyazaki S, Ota M, Sugawara H, Gojobori T (2000) DNA Data Bank of Japan (DDBJ) in collaboration with mass sequencing teams. Nucleic Acids Res 28:24–26 (Updated article in this issue: Nucleic Acids Res. (2002), 30, 27–30)
Benson DA, K-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL (2000) GenBank. Nucleic Acids Res 28:15–18
Schmuker M, Schwarte F, Brück A, Proschak E, Tanrikulu Y, Givehchi A, Scheiffele K, Schneider G (2007) SOMMER: self-organising maps for education and research. J Mol Model 13:225–228
Faigl J (2016) An application of self-organizing map for multirobot multigoal path planning with minmax objective. Comput Intell Neurosci 2016:2720630. doi:10.1155/2016/2720630
Muñoz A, Muruzábal J (1998) Self-organizing maps for outlier detection. Neurocomputing 18(1):33–60. doi:10.1016/S0925-2312(97)00068-4
Rini DP, Shamsuddin SM, Yuhaniz SS (2011) Particle swarm optimization: technique, system and challenges. Int J Comput Appl 14(1):19–27
Hu X, Shi Y, Eberhart R (2004) Recent advances in particle swarm. Evol Comput 1:90–97 (CEC2004)
Kohonen T (1995) Self-organizing maps. Springer, New York
Bai Q (2010) Analysis of particle swarm optimization algorithm. Comput Inf Sci 3(1). doi:10.5539/cis.v3n1p180
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kamal, M.S., Sarowar, M.G., Dey, N. et al. Self-organizing mapping based swarm intelligence for secondary and tertiary proteins classification. Int. J. Mach. Learn. & Cyber. 10, 229–252 (2019). https://doi.org/10.1007/s13042-017-0710-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13042-017-0710-8