
Abstract
It is a clinically well-established fact that patients with diabetes have very poor stroke outcomes. Yet, the underlying mechanisms remain largely unknown. Our previous studies showed that male diabetic animals show greater hemorrhagic transformation (HT), profound loss of cerebral vasculature in the recovery period, and poor sensorimotor and cognitive outcomes after ischemic stroke. This study aimed to determine the impact of iron chelation with deferoxamine (DFX) on (1) cerebral vascularization patterns and (2) functional outcomes after stroke in control and diabetic rats. After 8 weeks of type 2 diabetes induced by a combination of high-fat diet and low-dose streptozotocin, male control and diabetic animals were subjected to thromboembolic middle cerebral artery occlusion (MCAO) and randomized to vehicle, DFX, or tPA/DFX and followed for 14 days with behavioral tests. Vascular indices (vascular volume and surface area), neurovascular remodeling (AQP4 polarity), and microglia activation were measured. Brain microvascular endothelial cells (BMVEC) from control and diabetic animals were evaluated for the impact of DFX on ferroptotic cell death. DFX treatment prevented vasoregression and microglia activation while improving AQP4 polarity as well as blood-brain barrier permeability by day 14 in diabetic rats. These pathological changes were associated with improvement of functional outcomes. In control rats, DFX did not have an effect. Iron increased markers of ferroptosis and lipid reactive oxygen species (ROS) to a greater extent in BMVECs from diabetic animals, and this was prevented by DFX. These results strongly suggest that (1) HT impacts post-stroke vascularization patterns and recovery responses in diabetes, (2) treatment of bleeding with iron chelation has differential effects on outcomes in comorbid disease conditions, and (3) iron chelation and possibly inhibition of ferroptosis may provide a novel disease-modifying therapeutic strategy in the prevention of post-stroke cognitive impairment in diabetes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Individuals with diabetes not only are at a two- to sixfold-higher risk for having acute ischemic stroke but also suffer from unfavorable functional outcomes and poor recovery after ischemic brain injury for reasons that are not completely understood [1, 2]. Secondary bleeding into the brain known as hemorrhagic transformation (HT) occurs more commonly in diabetes, especially with the use of tissue plasminogen activator (tPA), the only pharmacological therapy for ischemic stroke [3,4,5]. The role and mechanisms by which increased HT contributes to poor recovery in diabetes are unknown. In general, space-occupying, also referred to as symptomatic, bleeding into the brain is believed to worsen outcomes by displacing the brain tissue whereas asymptomatic petechial hemorrhage is considered benign in acute ischemic stroke patients [6, 7]. However, evidence suggests increased odds of poor outcomes in asymptomatic HT as well [8]. In light of the growing evidence that iron accumulation in the brain parenchyma contributes to the pathophysiology of intracerebral hemorrhage (ICH) as well as neurodegenerative diseases including Parkinson’s disease (PD) and Alzheimer’s disease (AD) [9,10,11], the first goal of the current study was to determine the impact of iron chelation on ischemic stroke recovery in control and diabetic rats. The working hypotheses were that excess iron resulting from greater HT in diabetes worsens recovery and iron chelation with deferoxamine (DFX) after thromboembolic stroke, treated with or without tPA, will improve long-term sensorimotor and cognitive function in diabetic rats.
Stroke research on solely neuroprotection strategies has failed clinically, leading to support for the development of the neurovascular unit concept, which emphasizes the interdependence of brain cells and blood vessels for proper brain function [12, 13]. It is likely that the vasculature where the ischemic insult starts has to be closely studied for the development of successful therapeutics for acute protection as well as chronic restoration. We have shown that diabetes promotes excessive pathological neovascularization in the brain [14, 15] and an ischemic injury layered on this pathology causes greater HT and vascular rarefaction that is associated with poor recovery [2, 16,17,18]. Accordingly, the second goal of this study was to determine the impact of iron chelation on cerebral vascularization after stroke in control and diabetic rats. The working hypothesis was that DFX treatment will prevent cerebral vasoregression and neurovascular remodeling after stroke in diabetes and this will be associated with improved functional outcomes. In addition, in vitro studies will investigate iron-induced ferroptotic endothelial cell death as the underlying mechanism of vasoregression after stroke in diabetic conditions.
Research Design and Methods
Study Design and Animal Groups
All ischemic stroke experiments were performed on male Wistar rats procured from Envigo, Indianapolis, IN. Control rats (10 to 12 weeks old) were procured 14 days prior to stroke surgery. For the diabetic arm, animals were procured at 4 weeks of age and diabetes was induced and monitored as described below. All the animals were within the 12- to 14-week age range at the time of stroke surgery. In vivo studies were conducted at the Augusta University in Augusta, GA, before the investigators relocated to the Medical University of South Carolina in Charleston, SC. The animals were housed at the Augusta University animal care facility that is approved by the American Association for Accreditation of Laboratory Animal Care. All protocols were approved by the institutional animal care and use committee. This study was conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research, and adhered to the current RIGOR guidelines for the translational research [19]. The animals were randomly selected (in a block size of 2 rats per cage) for the embolic MCAO with or without tPA or DFX treatment. All behavioral testing and data analyses were conducted in a blinded manner. The animals were fed standard rat chow or high-fat diet (45% fat content) for the control and diabetic groups, respectively, were given tap water ad libitum, and were maintained at 12 h light/dark cycle (6 a.m./6 p.m.). Study design, number of animals that entered and finished the study, mortality rate, and endpoints measured are illustrated in Fig. 1. Details of procedures are described below.

Schematic timeline and description of the experimental design with study groups (C = control; D = diabetes), number of animals, mortality rate, and endpoints measured
Induction of Diabetes
Animals were procured at 4 weeks of age and were immediately put on a high-fat diet (45% fat; Research Diets, New Brunswick, NJ). A low dose of streptozotocin (35 mg/kg body weight; STZ; Cayman Chemicals, Ann Arbor, MI) was injected intraperitoneally at 6 weeks of age. If blood glucose was not above 150 mg/dL 5 days post-injection, a second small dose (20 mg/kg) was administered. The animals were monitored for another 7–8 weeks. Blood glucose levels were measured before noon twice a week from tail vein samples using a commercially available glucometer (Freestyle, Abbott Diabetes Care, Inc.; Alameda, CA). Control rats were maintained on regular chow. Blood glucose levels significantly increased within 3 days of STZ injection in the diabetic group and remained significantly elevated compared to those of the control animals (Table 1). In the first 5 days of the post-operative period, blood glucose and body weight were monitored daily.
Thromboembolic Stroke Surgery
The method of clot preparation was adapted from earlier reports and modified to increase clot stability as we reported [2]. Briefly, arterial blood from a donor rat was supplemented with human fibrinogen (2 mg/mL) and immediately withdrawn into 20 cm of polyethylene (PE)-50 tubing to clot at room temperature for 6 h and subsequently kept at 4 °C for 18 h. The PE-50 tube containing the clot was cut into 5-cm-long pieces. The clots were then transferred to a Petri dish containing sterilized saline and left for further retraction at room temperature for 4 h. A single 4 ± 0.5-cm-long clot was transferred to a PTFE Sub-Lite catheter (Braintree Scientific Inc., Braintree, MA) for surgery.
Male rats were randomly assigned in a block size of 2 rats per cage to 6 different groups: control vehicle, control + DFX, control + tPA/DFX, diabetes vehicle, diabetes + DFX, and diabetes + tPA/DFX. The animals were anesthetized with 5% isoflurane and maintained with 2.0% isoflurane in 70% N2 and 30% O2 using a face mask. The PTFE Sub-Lite catheter containing the clot was inserted up to the origin of MCA through the stump of external carotid artery (ECA), and the clot was gently injected with 100 μL of the sterile saline. The catheter was removed immediately after embolization. Laser Doppler imaging with a scanning system (PIM3, Perimed, North Royalton, OH) was used to confirm a successful occlusion and ensure similar levels of cerebral blood flow (CBF) reduction in all groups 10 min after emboli insertion. DFX (100 mg/kg, ip) or vehicle was given every 12 h for 7 days starting 3 h after the surgery. Our pilot studies showed that HT starts by 2 h after reperfusion. In the tPA groups, the animals received 1 mg/kg tPA (Cathflo Activase (Alteplase), Genentech) intravenously infused over 20 min through the femoral vein starting 90 min after cerebral ischemia induction.
Assessment of Neurological Deficits
Neurobehavioral tests were recorded and scored in a blinded fashion as described before [2, 20]. The animals were handled for 5–7 days prior to surgery in rooms where the tests were to be carried out. Tests included Bederson score, beam walk, adhesive removal test (ART), and Novel Object Recognition (NOR) before and after stroke at days 3, 7, and 14. Mortality occurred on days 1–3. The scores of these animals were not included in the analyses.
The Bederson score for each rat was obtained by using 4 parameters which include (a) observation of spontaneous ipsilateral circling (2 for no circling, 1 for partial circling, and 0 for continuous circling), (b) contralateral hind limb and (c) forelimb retraction which measures the ability of the animal to replace the limb after it is displaced laterally by 2 to 3 cm (2 for immediate replacement, 1 for replacement after minutes, and 0 for no replacement), and (d) resistance to push (1 or 0, depending on whether the animal is able to resist pushing or not). A maximum score of 7 was allotted to a normal rat. Beam walking ability was graded based on a 7-point scale method [21]. The composite neurological score was reported as the sum of the Bederson score and the beam walking score on a 1–14 scale. ART was carried out as previously described with modification [2]. Contact and removal latency of the adhesive paper dot was recorded. For each day, the average was taken from 3 trials with a maximum removal latency of 180 s per trial.
Cognition was measured through the NOR task [14, 20]. The animals were habituated to the test apparatus for 4 days prior to baseline testing for 10 min. On the day of testing, the animal had 3 phases: acclimation, familiarization, and novel. The acclimation phase allowed the animal to habituate to the box each day without any objects present for 5 min. During the familiarization phase, or A/A session, the animals were allowed to explore two identical objects for 5 min. The rats were placed back into their home cages for a 60-min delay, followed by a 5-min novel phase (A/B session) in which the familiar object from the A/A session was paired with a novel object. Objects were placed equidistant from the walls, with 20 cm between the two objects. The rats were placed in the center of the box for each session. Between each phase, objects and the testing apparatus were cleaned with 20% ethanol. The time spent exploring each object was recorded. The recognition index (RI; total time (s) spent exploring the novel object (TN) divided by the total time spent exploring both novel and familiar (TF) objects: RI = TN/(TN + TF)) and discrimination index 2 (D2 = (TN − TF)/(TN + TF)) were calculated. The NOR test was administered at baseline and day 14 after stroke surgery. The following inclusion criteria were used: RI must be between 0.40 and 0.60 in the A/A session and the animals must explore objects a minimum of 30 s within the 5-min trial.
Assessment of Vascularization
To visualize cerebral vessels, the rats were injected with 500 μL of 50 mg/mL FITC-dextran (MW, 2,000,000; Sigma-Aldrich, St. Louis, MO) under anesthesia using isoflurane via the jugular vein before euthanasia as we previously reported [14, 15, 22]. FITC was perfused throughout the animal for a minimum of 10 min. Prior to sacrifice, plasma was obtained for metabolic analysis. Brains were stored in 4% paraformaldehyde for 24–48 h followed by 30% sucrose in PBS. Brains were sectioned using a cryostat, and confocal images were obtained by a Zeiss 760 confocal microscope using 100-μm sections. Specifically, Z-stacked images were obtained from regions of interest in the cortex and striatum (bregma − 1 to + 1). This region was chosen because of proximity to the MCA and its branches that supply the frontal motor cortex, and this is the region where we have previously found hemorrhage in diabetic animals [14, 15, 22]. Images from 3 regions of interest in the ipsilateral cortical and striatal areas in three different sections were captured. The mean value from these images was calculated, and each animal had a total of nine unique images analyzed from the cerebrovasculature. The Z-stack referenced an image size of 1.984 μm, 512 × 512 pixels, and × 20 magnification. Raw images were then imported into Volocity 6.0 (Improvision, Lexington, MA) where they were 3D reconstructed to determine the surface area and vascular volume. Percent vascular volume references the vascular volume ratio in comparison to total volume. Surface area represented the absolute surface area of the total vasculature. Branch density was calculated with FIJI by creating a binary image, skeletonizing this image, and averaging the number of branches over the longest/shortest path multiplied by 100%. For each image, 8–10 measurements were averaged after being sorted by longest/shortest path. Identical threshold parameters were used across all the sections.
Assessment of BBB Permeability
Immunohistochemistry for IgG with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) on 20-μm sections was used to measure BBB permeability [23]. For each rat, three 20-μm sections that were 200 μm apart were averaged together. Briefly, free-floating sections had exogenous peroxidases quenched with 3% H2O2 in methanol for 3 min. After blocking for an hour, the primary IgG antibody in blocking solution (1:250; BD Bioscience 559073) was incubated overnight at 4 °C. Sections were rocked at room temperature for 30 min the next day. After 3 washes, sections were incubated in the biotinylated secondary antibody provided in the Vectastain ABC kit for 1 h. DAB was used as the peroxidase substrate solution. For each animal, three sections were stained and imaged with a × 20 objective using a Zeiss Axioplan 2 Imaging (Carl Zeiss Micro-imaging, Thornwood, NY). MetaMorph Image Analysis Software (Molecular Devices, LLC; San Jose, CA) was used to analyze staining intensity in three ROIs in the cortex and striatum by determining the percent threshold area. Identical threshold parameters were used across all sections. Threshold was averaged for each animal ipsilaterally. Results were expressed as IgG threshold.
Assessment of Neurovascular Remodeling
Free-floating 20-μm cryostat sections were incubated with aquaporin-4 (AQP4) (1:250; Santa Cruz sc-32739) antibody at 4 °C overnight to examine AQP4 polarity as an index of neurovascular remodeling [23]. Following the primary incubation, sections were incubated with a fluorescent-conjugated secondary antibody (1:1000; Thermofisher) for 1 h at room temperature. Z-stacked images were captured for AQP4 and FITC-filled vessels for 10 slices. These images were then used to measure unpolarized AQP4 using Fiji software. The histogram was analyzed and final quantification was calculated by reporting the number of pixels at the 255-value over total number of pixels on the image: (\( \frac{\#\mathrm{of}\ \mathrm{pixels}\ \mathrm{at}\ 255}{\left(\#\mathrm{of}\ \mathrm{pixels}\ \mathrm{at}\ 0\right)+\left(\#\mathrm{of}\ \mathrm{pixels}\ \mathrm{at}\ 255\right)}\Big)\ast 100 \).
Assessment of Microglia Morphology
Iba1 (1:500; Wako 019-19741) was used to measure reactive microglia/macrophages in the cortex and striatum as we described [23]. Microglia were imaged at × 20 and × 60 magnification using Cytation software. Five representative microglia were isolated from each image in the cortex and striatum and converted to binary followed by skeletonization using Fiji software. The AnalyzeSkeleton plugin was used to analyze the number of microglia process endpoints/cell and summed microglia process length/cell. All data that had 2 or fewer endpoints were discarded, and the sum of all endpoints was used for the number of microglia process endpoints/cell. To determine the summed microglia process length/cell, the sum of all branch lengths was used after branch lengths less than 0.1 were removed. Fiji was also used to measure cell swelling and number of protrusions per cell. Cell swelling was measured using the freehand selection tool to draw around the cell soma. A blinded investigator counted the number of protrusions from the soma of each microglia.
To evaluate the activation status of microglia, 20-μm sections corresponding to the start, middle, and end of the brain region (− 1 to + 1 bregma as in vascularization studies) were processed with antibodies at 1:100 dilution for Iba1 (FUJIFILM Wako Pure Chemical Corporation, 019-19741) as well as proinflammatory markers including TNFα (Novus Biologicals; cat no: NBP2-34539) and CD16/32 (BD Biosciences, 553142) and anti-inflammatory marker Arginase 1 (Proteintech Group Inc., 661291IG). Slides were imaged with a × 20 objective using an Olympus IX73 microscope (Olympus Corporation, Japan).
Assessment of Hippocampal Neuronal Death
Identical threshold parameters were used across all the sections for all the following measurements. Cryostat sections 20 μm in thickness (100 μm apart, 3 sections/rat) were incubated in NeuN (1:500; Abcam) antibody at 4 °C overnight. This was followed by incubation with a fluorescent conjugated (1:1000; Thermofisher Alexa Fluor 647) for 1 h at room temperature. The number of NeuN-positive neurons in the entire CA1 or DG was counted in a × 20 objective area using the multi-point tool in Fiji by a blinded investigator.
Brain Microvascular Endothelial Cell Culture and Treatments
Experiments were performed using brain microvascular endothelial cells (BMVECs) isolated from normal Wistar and type 2 diabetic Goto-Kakizaki (GK) rats as reported previously [14, 16]. Both Wistar and GK cells were grown in endothelial growth media (VEC Technologies, Rensselaer, NY, USA) and plated on flasks coated with 0.1% gelatin. Cells were grown to 80–90% confluency and passages 5–8 were used in current experiments. Cells were serum starved in Dulbecco’s modified Eagle’s medium (DMEM) for 2 h and incubated with/without iron(III) sulfate (0.1 mM; Sigma-Aldrich) or DFX (100 μM; Sigma-Aldrich) or combination of both for 6 h in 2% FBS containing DMEM. Cell lysates were collected and prepared for Western blot analysis, lipid peroxidation measurements, total glutathione measurements, and immunofluorescence measurement.
Cell Viability Assay
Cell viability was measured by RealTime Glo-MT Viability Assay kit (Promega, USA). Briefly, BMVECs were grown to 80–90% confluency in a 96-well plate. Media was replaced with low FBS (2% FBS) containing DMEM and allowed to stabilize for 4 h. Reagents in the kit were prepared according to the manufacturer’s instructions. MT cell viability assay substrate and NanoLuc enzyme were diluted with test compounds (iron(III) sulfate, DFX or combination of both) in media, and cells were treated for 48 h. Luminescence was measured every 2 h up to 48 h after treatment. Relative luminescence unit (RLU) was plotted against time in hours.
Western Blot Analysis
Cell lysates of BMVECs were assessed for ferroptosis markers. Briefly, equivalent amounts of cell lysates of BMVECs (15 μg protein/lane) were loaded onto 10% SDS-PAGE, proteins separated, and proteins transferred to nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin followed by incubation overnight at 4 °C with primary antibody anti-ferritin heavy chain (ab65080, Abcam), anti-ferritin light chain (ab69090, Abcam), anti-ACSL4 (acyl-CoA synthetase long-chain family member 4) (PA5-27137, Invitrogen), and anti-IREB2 (iron responsive element binding protein 2) (ab181153, Abcam) at 1:1000 dilution or anti-β-actin (A2854, Sigma) at 1:3000 dilution. After washing, the membranes were incubated for 1 h at 20 °C with appropriate secondary antibodies (horseradish peroxidase [HRP]-conjugated; dilution 1:3000). Pre-stained molecular weight markers were run in parallel to identify the molecular weight of proteins of interest. For chemiluminescent detection, the membranes were treated with enhanced chemiluminescent reagent and the signals were monitored on FluorChem-E Imager (Protein Simple, San Jose, CA). Relative band intensity was determined by densitometry software and normalized with β-actin protein.
Immunofluorescence
Paraformaldehyde-fixed frozen brain sections or BMVECs grown on slides were subsequently washed with TBS followed by treatment with 0.2% Triton X-100 for 3 min. After washing, samples were blocked by 5% BSA for 1 h at room temperature. Slides were then incubated with primary antibodies like anti-IREB2 (iron responsive element binding protein 2) (ab181153, Abcam), anti-4 hydroxynonenal (anti-4HNE) (ab46545, Abcam), anti-TLR4 (toll-like receptor 4) (ab22048, Abcam), and anti-citrate synthase (14309, Cell Signaling) at a 1:100 dilution in 0.2% BSA at 4 °C overnight. Cells were washed and incubated with AlexaFlour 488 conjugated secondary antibody (Jackson Immuno Research Laboratories, Inc., West Grove, PA) at a 1:400 dilution at room temperature for 1 h. Negative control slides were incubated with 0.2% BSA in place of the primary antibody. Slides were imaged on an Axiovert 200 microscope (Carl Zeiss MicroImaging, Thornwood, NY).
Lipid Peroxidation (MDA) Assay
Cell lysates collected after the respective treatments were used to measure the MDA formation by TBARS assay kit (#10009055, Cayman Chemicals) following the manufacturer’s instruction. Results are normalized with the total protein in the cell lysate and expressed as micromolars MDA/milligram protein.
Glutathione Assay
Cell lysates were collected after the respective treatments were used to measure the total glutathione using a glutathione assay kit (#703002, Cayman Chemicals) following the manufacturer’s instruction. Results are normalized with the total protein in the cell lysate and expressed as total GSH/milligram protein.
Data Analysis
Power analysis was made at alpha = 0.05. Based on the hemoglobin index data for HT in our past studies (control 1.5 ± 3.0 and diabetic 13 ± 4.0, mean ± SD), a sample size of 8/group was predicted to provide at least 85% power to detect the effect of disease on bleeding and 40% extra were added due to the increased mortality with diabetes (n = 12/group). Neurobehavioral tests (Bederson’s and ART) were analyzed using a 2 day × 2 disease × 2 TRT (DFX vs. VEH or tPA/DFX) repeated-measures ANOVA on day 3 and day 14 data. Cognition data (RI and D2) were analyzed as change from baseline at day 14 using a 2 disease × 2 TRT (DFX vs. VEH or tPA/DFX) ANOVA. Baseline data were compared for disease differences using a t test. Vascularization, BBB permeability, neurovascular remodeling, microglia morphology, and neuronal death were analyzed using a 2 disease × 2 TRT (DFX vs. VEH) ANOVA. Area under the curve of cell viability from 0 to 48 h and data from BMVEC cell lysates were analyzed within Wistar or GK lines using a 2 DFX (yes vs. no) × 2 Iron (yes vs. no) ANOVA. A Tukey’s test was used to determine differences for significant interactions for all analyses. Significance was determined at a type I error rate of 5%. NCSS 2007 (NCSS, LLC, Kaysville, UT) was used to calculate area under the curve for cell viability. SAS© 9.4 (SAS Institute Inc., Cary, NC) was used for all analyses.
Results
Iron Chelation Improves Sensorimotor and Cognitive Outcomes in Diabetic but Not in Control Rats: Interaction with tPA
Ischemic injury induced by thromboembolic occlusion of MCA resulted in greater mortality in diabetic rats which occurred in days 1–3 (Fig. 1). Motor function measured by a composite score indicated greater deficits in diabetic rats. While control rats showed spontaneous recovery, deficits in diabetic rats remained consistent. DFX treatment did not have an effect in the control group whereas it improved motor deficits in diabetic animals resulting in a treatment and disease interaction by days 3 and 14 (Fig. 2a). Fine sensorimotor skills measured by ART also showed that the diabetic cohort suffers to a greater extent than the controls. Interestingly, deficits remained greater in the DFX-treated control rats compared to the vehicle group by day 14 whereas there was a steady decline in the diabetic animals (Fig. 2b). Cognitive function as measured by NOR (RI and d2) showed differences at baseline. Data analyzed as change from baseline showed no significant decline in the control group whereas the diabetic vehicle group showed further decline and DFX treatment prevented this by day 14 (Fig. 2c, d).

DFX improved functional outcomes in diabetic but not control rats. Sensorimotor deficits were measured by composite score (a) and ART (b). DFX improved the composite score in diabetic animals starting at day 3. Interestingly, fine motor skills measured by ART worsened by DFX treatment in the control group by day 14. c Recognition index, a measure of working memory, was measured by novel object recognition test. RI was lower in diabetic animals before stroke and showed a further decline by day 14. DFX prevented this response. Results are shown as scattered graphs with individual data points and SEM
We next compared functional outcomes in control and diabetic animals treated with DFX or tPA plus DFX. There was no difference in composite scores among the four groups by day 14 (Fig. 3a, b). For RI and d2, there was no difference for cognitive decline among the groups indicating tPA neither worsened nor improved DFX effects (Fig. 3c, d).

tPA/DFX combination showed differential effects on functional outcomes. While the addition of tPA did not have an effect on motor outcomes measured by the composite score (a), it improved fine sensorimotor skills (b) in both control and diabetic animals and worsened cognitive deficits in the diabetic group (c, d). Results are shown as scattered graphs with individual data points and SEM
Deferoxamine Treatment Has a Differential Effect on Cerebrovascularization in Control and Diabetic Rats
As we have shown previously, vehicle-treated diabetic rats had lower vascular indices including vascular volume, surface area, and branch density in the ipsilateral cortex as compared to the control group (Fig. 4a–d). DFX treatment increased these indices in diabetic rats while mediating a decrease in control animals resulting in a significant disease by treatment interaction. We obtained similar results in the ipsilateral striatum (Supplemental Fig. 1A-C).

DFX has a differential effect on post-stroke vascularization in control and diabetic animals. a Representative images of FITC-filled vasculature acquired from the ipsilateral cortex. Vascular volume (b) and surface area (c) were measured using Volocity software while branch density (d) was quantified with Fiji. Two-way ANOVA indicated a disease and treatment interaction such that DFX improved/preserved these indices in diabetic animals but caused a decline in the controls. High mortality in the diabetic groups and poor FITC staining in some specimens resulted in uneven numbers of animals per group. Results are shown as scattered graphs with individual data points and SEM
Deferoxamine Treatment Attenuates Post-Stroke Neurovascular Remodeling in Diabetic Rats
Since cerebral vasculature is in close contact with astrocytes within the neurovascular unit, we next measured AQP4 polarity as an index of neurovascular remodeling. There was a significant increase in unpolarized AQP4 in the diabetic group suggesting that AQP4, typically located on astrocytic end feet, was redistributed. There was a trend for an interaction (p = 0.058) such that DFX treatment prevented this in diabetic but not control rats in the cortex (Fig. 5a). In the striatum, diabetes increased remodeling but DFX had no effect (Fig. 5b).

DFX prevents NVU remodeling in the cortex of diabetic animals. Unpolarized AQP4 was quantified as a measure of NVU remodeling in the cortex (a) and striatum (b). Representative images of AQP4 staining with FITC-filled vasculature are shown on the top panels. Cumulative data show that DFX prevents the disruption in AQP4 polarity. Results are shown as scattered graphs with individual data points and SEM
Deferoxamine Treatment Attenuates Post-Stroke Microglial Activation in Diabetic Rats
Given that microglia are the resident immune cells of the brain, we assessed microglia morphology as increased cell body size and/or decreased protrusions, endpoints, and branch length indicate microglia activation. There was significantly increased Iba1 staining in the prefrontal cortex of diabetic animals which was reduced by DFX (Fig. 6a). Cell body size was increased in the diabetic groups with no treatment effect (Fig. 6b). Number of protrusions was lower in samples from diabetic rats, and DFX treatment had no effect in controls but reversed this in diabetes resulting in an interaction (Fig. 6c). A similar effect was observed for branch length (Fig. 6e) while it was only a trend for number of endpoints (p = 0.068, Fig. 6d). In the striatum, both control and diabetic animals showed significant Iba1 staining and DFX prevented this response. In contrast to the cortex, DFX was able to increase the number of microglia processes and branch length in both control and diabetic animals (Supplemental Fig. 2). In a subset of samples, the microglial phenotype was evaluated by immunohistochemistry. Brain sections from diabetic animals showed greater staining for proinflammatory markers TNFα and CD16/32 that colocalized with Iba1. DFX treatment prevented the increase in proinflammatory mediators in diabetes while promoting an increase in anti-inflammatory arginase 1 especially in control and animals (Supplemental Fig. 3).

DFX prevents microglia activation in the cortex after stroke to a greater extent in diabetic animals. a Representative images of Iba1 staining in the cortex. Microglia morphology, summarized in panels b–e, demonstrate that activated microglia phenotype with lower endpoints and branch length is more prominent in the diabetes vehicle group and DFX prevents these changes in microglia morphology. Results are shown as scattered graphs with individual data points and SEM
Deferoxamine Treatment Reduces Diabetes-Mediated Increase in BBB Permeability
IgG staining, a measure of increased permeability, was greater in diabetic animals than in control rats and this was more pronounced in the striatum than in the cortex (Fig. 7a, b). DFX was effective in improving BBB integrity in the cortex in both groups, but in the striatum, the effect was greater in diabetes.

Stroke causes a greater disruption of BBB integrity in the striatum of diabetic animals. Representative images of IgG staining in the cortex (a) and striatum (b) are given on top. Cumulative data in the graphs show that DFX treatment prevented increased BBB permeability in the diabetic group. Results are shown as scattered graphs with individual data points and SEM
Deferoxamine Treatment Attenuates Stroke-Induced Remote Neuronal Damage in the Hippocampus in Diabetes
The hippocampus is highly sensitive to ischemia, and even remote injury can cause neuronal loss. The number of neurons in the CA1 and DG region was significantly lower in the diabetic animals as we have previously shown [20, 23]. Interestingly, DFX treatment increased neuronal loss in the CA1 region of control animals with no effect in diabetic groups (Supplemental Fig. 4).
Deferoxamine Prevents Iron-Mediated Decrease in Cell Viability and Lowers Ferroptosis Markers to a Greater Extent in Brain Microvascular Endothelial Cells Isolated from Diabetic Animals
We have previously reported that post-stroke cerebral vasoregression is associated with endothelial cell death in GK rats, a spontaneous and lean model of type 2 diabetes [15]. Ferroptosis is a recently discovered iron-dependent cell death pathway and gaining increasing attention in neurodegenerative disorders [24, 25]. Thus, we evaluated two markers of ferroptosis, IREB2 and citrate synthase (CS), by IHC in post-stroke day 14 brain sections from control and diabetic rats. There was greater staining for each protein in diabetic animals, and this was prominent around the vasculature (Supplemental Fig. 5).
BMVECs isolated from control Wistar and GK diabetic rats were used to further evaluate the effect of iron on ferroptosis in BMVECs. Ferroptosis which may contribute to the greater vasoregression was observed in diabetic animals. Neither DFX alone nor iron alone had an effect in Wistar cells but iron plus DFX-treated cells had lower ACSL4 levels (Fig. 8a). In GK cells, iron treatment significantly increased ACSL4 protein, which was attenuated by DFX. While IREB2 was not significantly increased (p = 0.13), DFX lowered it to control levels (Fig. 8b). Since ferroptosis is an oxidative form of regulated cell death featuring glutathione (GSH) depletion and lipid peroxidation (Fig. 8c), we next measured GSH and lipid peroxidation (LPO). Iron treatment lowered GSH only in the GK cells, and DFX prevented this in iron- but not vehicle-treated cells (p = 0.029). While we did not do a statistical comparison of Wistar versus GK cells, baseline (control vehicle) LPO levels appeared to be higher in GK cells. Iron increased LPO in both control Wistar and diabetic GK cells. DFX prevented this effect only in iron-treated GK cells. Furthermore, iron treatment increased 4-hydroxynonenal (4-HNE), a product of lipid peroxidation, to a greater extent in GK cells, and DFX prevented this effect (Supplemental Fig. 6). Along with these changes in ferroptotic pathway, iron treatment reduced cell viability (AUC, area under the curve) significantly in GK cells. There was an interaction such that DFX treatment prevented this decrease in iron but not vehicle (control)-treated cells (Fig. 8). In Wistar cells, iron did not have an effect. Similar to GK cells, DFX treatment improved viability only in iron-treated cells.

DFX treatment improves survival of BMVECs isolated from diabetic animals. BMVECs isolated from control (a) and diabetic GK (b) rats were stimulated with iron in the absence and presence of DFX, and markers of ferroptosis (depicted in bold in panel c) were measured. Iron increased IREB2 and ACSL4 proteins as well as lipid peroxidation (LPO) while causing a slight decrease in glutathione (GSH) in GK cells. In control cells, only LPO was elevated. DFX prevented the increases in these markers of ferroptosis in GK cells and improved viability. Representative immunoblots of IREB2 and ACSL4 as well as 4HNE IHC are given in Supplemental Fig. 5. Results are shown as mean and SEM of n = 4–6 per group
Discussion
Based upon our previous findings that (1) there is significant loss of cerebrovasculature after stroke in diabetic animals and (2) vasoregression correlates with the degree of vascular injury, i.e., presence of hemorrhagic transformation, and poor functional outcomes, the objective of this study was to investigate the effect of free iron on long-term ischemic stroke outcomes in a comorbid disease model. Our current novel findings provide evidence that iron chelation with deferoxamine started after ischemia/reperfusion injury induced by thromboembolic stroke would prevent vascular drop out, improve neurovascular integrity, and improve functional outcomes in diabetic but not control animals. Furthermore, in vitro studies suggest ferroptotic endothelial cell death contributes to vascular loss that can be targeted by iron chelation.
Diabetes not only is a risk factor for occurrence [26, 27] but also increases stroke-related mortality and rate of recurrent stroke [28, 29]. It is estimated that more than 30% of patients with stroke have diabetes and these patients suffer from a greater risk of HT, an important complication of ischemic stroke, as well as increased mortality and slower recovery [30,31,32,33]. Clinical studies suggest that diabetes also contributes to cognitive decline and dementia, which are amplified after stroke, further compromising functional outcomes [34,35,36]. The underlying reasons contributing to poor outcomes in diabetes are far from understood. Proposed mechanisms so far focused on the impact of acute injury including greater infarcts, rapid progression of infarction, increased edema, and HT. With respect to HT, it is believed that space-occupying symptomatic bleeding into the brain worsens stroke outcomes by displacing the brain tissue whereas asymptomatic petechial hemorrhage is nonthreatening [6, 7]. However, it is increasingly recognized that blood products, especially iron, can be detrimental and play an important role in neurodegenerative diseases including PD and AD [10, 11, 37, 38]. ICH studies also provided evidence that iron can be detrimental for repair and restoration [9]. Collectively, it has been suggested that iron chelation can improve outcomes in these diseases. While the results of the phase 2 i-DEF trial failed to show any potential beneficial effect of DFX in patients with ICH based on 90-day modified Rankin scores to move forward to a phase 3 trial, long-term cognitive outcomes remain unknown [39]. As recently reviewed, a conservative iron chelation therapy with DFX or deferiprone (DFP) is a viable and attractive target in neurodegenerative diseases including post-stroke cognitive impairment and HT [10, 37] but remains to be tested. Given that there is greater HT in diabetes, we tested the effect of DFX on post-stroke outcomes and the current study provides novel information that when initiated after spontaneous or tPA-mediated reperfusion following thromboembolic stroke in diabetic rats, DFX treatment improves sensorimotor outcomes and prevents further deterioration of cognitive deficits. We used a thromboembolic model of stroke so we can evaluate tPA interactions with iron chelation, which remains to be the only pharmacological therapy for ischemic stroke. This is important as recently demonstrated by the failure of erythropoetin (EPO) in a clinical trial [40]. While single use of EPO in suture occlusion models of stroke was protective, the combination of EPO with tPA worsened the outcome in the embolic model of stroke [40, 41]. In the current study, we did not include a tPA-alone group as our goal was to assess the interaction of tPA with DFX and as such compared only DFX alone or tPA/DFX-treated control and diabetic animals. Our data show that the combination treatment is equally effective if not better in improving sensorimotor deficits, but working memory function appears to be worsened in the tPA/DFX diabetic group. We acknowledge that this result may be due to the small number of animals as a result of greater stroke mortality in the diabetic group. However, a previous study showed that tPA caused greater disruption of the BBB integrity and failed to improve functional outcome in diabetic rats [42]. The same study also reported that an amplified inflammatory response may contribute to the failed protective effects of tPA treatment in this cohort [42]. Studies by us and others showed that interventions that improve functional stroke outcome including cognitive deficits in diabetic models are associated with enhanced anti-inflammatory microglia phenotype [43,44,45]. In this context, there is a need to further investigate the neuroinflammatory response after DFX/tPA treatment.
In this study, we focused on the effects of DFX treatment on cerebral vascularization after stroke for several reasons. First, the failure of all the neuroprotective strategies tested to date led to the development of neurovascular niche for functional recovery concept [46]. Angiogenesis is required for neurogenesis and functional recovery to occur [46]. A seminal study demonstrated that angiogenesis is required for neuroblast migration from the subventricular zone, a key area for endogenous neuronal progenitor cells, and inhibition of angiogenesis worsens functional recovery [46]. Stimulation of angiogenesis is being evaluated as a therapeutic modality in stroke [47,48,49]. Second, we showed that in contrast to reparative angiogenesis in control animals, there is cerebrovascular regression associated with poor recovery of motor and cognitive functions after stroke in diabetes and the degree of post-stroke vascular dropout correlates with the occurrence of HT [15, 22]. Other studies also suggest impaired vascular restoration after diabetic stroke [50, 51]. Type 2 diabetic mice subjected to ischemia showed decreased number of microvessels after stroke in the ischemic hemisphere. Another study conducted with diabetic GK rats also showed a reduced rate of angiogenesis 7 days after stroke [52]. However, the impact of HT and iron chelation on vascularization and neurovascular integrity remained unexplored. Our current data show that vascularization indices such as vascular volume, surface area, and branch density are lower in diabetic animals than in control animals subjected to stroke as we have previously reported [14, 22]. Interestingly, DFX restores these indices to control levels in diabetic rats but causes a decrease in control animals. In this study, we did not have a sham group but our previous studies showed that typically control animals show a reparative angiogenic response after stroke when compared to sham animals and this vasoregression occurs in the diabetic cohorts [15]. In this 14-day study, we observed that fine sensorimotor skill deficits were greater in DFX-treated control animals but whether and to what extent this drop in the vascularization impacts chronic outcomes especially post-stroke cognitive impairment needs to be shown in longer-term studies.
There is no doubt that cerebrovascular integrity and function are critical for brain health [12, 13] and neurovascular unit is at the core of this concept. Given that astrocytes wrap around blood vessels and provide critical communication between the vasculature and neurons, we measured AQP4 polarity as an index of NVU remodeling. Our results show that in the diabetic group there is an increase in unpolarized AQP4 suggesting relocation of this protein from astrocytic end feet to body only in the cortex, but not in striatum, and DFX has a modest effect preventing this remodeling. BBB integrity in the cortex is significantly compromised after stroke in both control and diabetes groups and DFX prevents this pathology, more so in diabetic rats. In the striatum, there is increased BBB permeability only in the diabetic group and again DFX is effective in preventing a disruption of barrier function.
Ferroptosis is a recently discovered cell death mechanism that is gaining attention in neurodegenerative diseases and brain injury [11, 24, 25]. This nonapoptotic form of cell death is differentiated by iron-dependent accumulation of ROS leading to lasting lipid damage and membrane permeabilization. This mechanism of cell death is unique that it does not require any transcriptional activation or post-translational modification of any effector molecule but occurs when the cellular antioxidant defense system is overwhelmed [53]. Inhibition of the plasma membrane cystine/glutamate antiporter known as system XC− leads to decreased cysteine levels, which is needed for glutathione biosynthesis. When glutathione levels drop, glutathione peroxidase 4 (GPX4) can no longer function to clear lipid peroxides leading to cell death. This is particularly important for the brain as glutamate, which causes excitotoxic cell death, also inhibits system XC−. Ferroptosis pathways are further enhanced by activation of IREB2 and acyl-CoA synthetase long-chain family member 4 (ACSL4), which facilitates the incorporation of polyunsaturated fatty acids into membrane phospholipid layers rendering cells more ferroptosis susceptible. Given the neurotoxic effects of iron, the possible role of ferroptosis in neurodegenerative diseases and brain injury, from a neuronal perspective, was elegantly discussed in recent review articles [10, 11, 25]. In the current study, given the vasoregression observed in diabetic rats after stroke, we approached ferroptosis from a vascular perspective. While we did not perform cellular localization studies, initial immunohistochemistry for markers of ferroptosis, IREB2, and CS showed a strong vascular localization (Supplemental Fig. 5) prompting us to investigate ferroptosis in vitro using BMVECs isolated from control and diabetic animals. We have observed a robust iron-mediated increase in lipid peroxidation in BMVECs from both control and diabetic animals, but markers of ferroptosis and lipid peroxidation end products were significantly increased in the “diabetic” cells and DFX was effective in improving cell viability in this group. A recent paper also reported iron-mediated ROS-dependent changes in the endothelial cell genome that is associated with senescence and ferroptosis and the DFX-conjugated nanoparticle prevented these responses in vitro as well as in vivo in an ICH model [54]. We recognize the limitations of cell culture and phenotypic changes that can occur when cells are grown in a dish. However, we have shown that these primary cells retain some of the pro-angiogenic properties observed in vivo when used in early passages [16, 55]. Thus, in vitro results support the idea that excessive iron resulting from HT may promote endothelial ferroptosis and vasoregression. Additional studies focusing on the impact of the inhibition of ferroptosis on vascularization patterns as well as long-term sensorimotor and cognitive outcomes after stroke may identify novel therapeutic approaches.
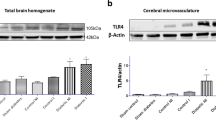
Brain and systemic inflammation contribute to the injury and recovery after stroke. In this regard, microglia have taken a central role as resident immune cells of the brain. Microglia activation is highly relevant to HT as these cells take up the iron from the brain parenchyma. We have observed a robust increase in Iba1-positive cells in the ipsilateral cortex of diabetic animals as well as a decrease in endpoints and branch length suggesting activated microglia morphology. DFX treatment prevented these changes. It is highly possible that chelation of iron prevented the activation of microglia directly, but it is also known that nonapoptotic forms of cell death can trigger sterile inflammation through the release of damage-associated molecular patterns, which are recognized by innate immune receptors including toll-like receptors (TLRs). We have reported that iron increases the expression of TLR4 in brain microvessels as well as in endothelial cells obtained from diabetic animals and iron-mediated endothelial cell death have features of both necroptosis and ferroptosis that can be prevented by the inhibition of TLR4 [56]. In the current study, we observed increased TLR4 signal in the vasculature in diabetic rats (Supplemental Fig. 7). A recent study reported that inflammatory processes following cardiac transplant are initiated through ferroptotic death of graft endothelial cells in a TLR4-dependent manner [57]. It is reasonable to speculate that iron-induced ferroptotic cell death can trigger a vasoneuronal inflammatory loop that leads to chronic inflammation and progressive post-stroke cognitive impairment in diabetes.
This study is not without limitations. Mortality is high in diabetic animals, and as a result, numbers of animals that completed the study are lower than those of control rats. Animals were randomized to groups. When an interim analysis showed the jump in mortality with tPA, especially in the control tPA/DFX group (8% vehicle vs 33% tPA/DFX, Fig. 1), we stopped the tPA/DFX arm of the study, which resulted in a very low number of animals. Another limitation is the duration of the study. We followed the animals up to 14 days because that was the time used in our previous studies in which we showed cerebral vasoregression. Higher mortality was also a factor in this decision. In other studies, we have shown that comorbid diseases including hypertension and diabetes cause progressive post-stroke cognitive impairment (PSCI) and diabetic animals show further deterioration after 4 weeks when followed up to 8 weeks [58, 59]. While mortality was an issue in diabetic rats, we recognize that longer-term studies are needed to determine the impact of DFX on PSCI. We were not able to determine the infarct size. Due to extensive injury in untreated diabetic animals, brain tissue collected on day 14 was extremely fragile and could not be processed for thin sections needed for histology. Infarct size measurements in this group were not reliable for group comparisons. However, we previously showed that infarct sizes are similar between control and diabetic rats in this model of embolic stroke when measured by TTC on day 3 [2]. Neuroinflammation was assessed by changes in microglia morphology and expression of pro/anti-inflammatory markers by immunohistochemistry. Flow cytometry-based approaches would have been more desirable, but that would have necessitated additional animals. It is increasingly recognized that there is functional and regional microglial heterogeneity [60,61,62]. As such, different microglial subtypes may respond differently to any intervention, especially in combination with tPA, and deserve further exploration. In the current study, we report the results obtained with male animals. We have shown very poor short-term outcomes in diabetic females [17] which necessitated a study of its own in female animals. These results will be published separately. Finally, as discussed in preceding paragraphs, we did not use inhibitors of ferroptosis neither in vivo nor in vitro but rather assessed the impact of DFX on this cell death pathway in the context of diabetes. Nevertheless, our findings provide significant novel information and suggest that therapeutic targets may differ in the presence of comorbidities. Enhanced understanding of the impact of HT on the endogenous reparative/restorative mechanisms of the brain and long-term functional outcomes after ischemic stroke will allow more targeted and effective therapeutic strategies for this debilitating disease and associated complications.
References
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–596.
Li W, Ward R, Valenzuela JP, Dong G, Fagan SC, Ergul A. Diabetes worsens functional outcomes in young female rats: comparison of stroke models, tissue plasminogen activator effects, and sexes. Transl Stroke Res. 2017;8:429–39.
Bruno A, Levine SR, Frankel MR, Brott TG, Lin Y, Tilley BC, et al. Admission glucose level and clinical outcomes in the NINDS rt-PA stroke trial. Neurology. 2002;59(5):669–74.
Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab. 2007;27(3):435–51.
McCormick MT, Muir KW, Gray CS, Walters MR. Management of hyperglycemia in acute stroke: how, when, and for whom? Stroke. 2008;39(7):2177–85.
Larrue V, von Kummer RR, Muller A, Bluhmki E. Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European-Australasian Acute Stroke Study (ECASS II). Stroke. 2001;32(2):438–41.
Yong M, Kaste M. Dynamic of hyperglycemia as a predictor of stroke outcome in the ECASS-II trial. Stroke. 2008;39(10):2749–55.
Park JH, Ko Y, Kim WJ, Jang MS, Yang MH, Han MK, et al. Is asymptomatic hemorrhagic transformation really innocuous? Neurology. 2012;78(6):421–6.
Wilkinson DA, Pandey AS, Thompson BG, Keep RF, Hua Y, Xi G. Injury mechanisms in acute intracerebral hemorrhage. Neuropharmacology. 2018;134(Pt B):240–8.
Morris G, Berk M, Carvalho AF, Maes M, Walker AJ, Puri BK. Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav Brain Res. 2018;341:154–75.
Derry PJ, Hegde ML, Jackson GR, Kayed R, Tour JM, Tsai AL, et al. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog Neurobiol. 2020;184:101716.
Gorelick PB, Furie KL, Iadecola C, Smith EE, Waddy SP, Lloyd-Jones DM, et al. Defining optimal brain health in adults: a presidential advisory from the American Heart Association/American Stroke Association. Stroke. 2017;48(10):e284–303.
Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96(1):17–42.
Prakash R, Johnson M, Fagan SC, Ergul A. Cerebral neovascularization and remodeling patterns in two different models of type 2 diabetes. PLoS One. 2013;8(2):e56264.
Prakash R, Li W, Qu Z, Johnson MA, Fagan SC, Ergul A. Vascularization pattern after ischemic stroke is different in control versus diabetic rats: relevance to stroke recovery. Stroke. 2013;44(10):2875–82.
Abdelsaid M, Prakash R, Li W, Coucha M, Hafez S, Johnson MH, et al. Metformin treatment in the period after stroke prevents nitrative stress and restores angiogenic signaling in the brain in diabetes. Diabetes. 2015;64(5):1804–17.
Li W, Qu Z, Prakash R, Chung C, Ma H, Hoda MN, et al. Comparative analysis of the neurovascular injury and functional outcomes in experimental stroke models in diabetic Goto-Kakizaki rats. Brain Res. 2013;1541:106–14.
Elgebaly MM, Prakash R, Li W, Ogbi S, Johnson MH, Mezzetti EM, et al. Vascular protection in diabetic stroke: role of matrix metalloprotease-dependent vascular remodeling. J Cereb Blood Flow Metab. 2010;30(12):1928–38.
Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR guidelines: escalating STAIR and STEPS for effective translational research. Transl Stroke Res. 2013;4(3):279–85.
Ward R, Valenzuela JP, Li W, Dong G, Fagan SC, Ergul A. Poststroke cognitive impairment and hippocampal neurovascular remodeling: the impact of diabetes and sex. Am J Physiol Heart Circ Physiol. 2018;315(5):H1402–H13.
Kelly-Cobbs AI, Prakash R, Li W, Pillai B, Hafez S, Coucha M, et al. Targets of vascular protection in acute ischemic stroke differ in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2013;304(6):H806–15.
Li W, Valenzuela JP, Ward R, Abdelbary M, Dong G, Fagan SC, et al. Post-stroke neovascularization and functional outcomes differ in diabetes depending on severity of injury and sex: potential link to hemorrhagic transformation. Exp Neurol. 2019;311:106–14.
Ward R, Li W, Abdul Y, Jackson L, Dong G, Jamil S, et al. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res. 2019;142:237–50.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Magtanong L, Dixon SJ. Ferroptosis and brain injury. Dev Neurosci. 2018;40(5–6):382–95.
Folsom AR, Rasmussen ML, Chambless LE, Howard G, Cooper LS, Schmidt MI, et al. Prospective associations of fasting insulin, body fat distribution, and diabetes with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) study investigators. Diabetes Care. 1999;22(7):1077–83.
Stegmayr B, Asplund K. Diabetes as a risk factor for stroke. A population perspective. Diabetologia. 1995;38(9):1061–8.
Kernan WN, Inzucchi SE. Type 2 diabetes mellitus and insulin resistance: stroke prevention and management. Curr Treat Options Neurol. 2004;6(6):443–50.
Luscher TF, Creager MA, Beckman JA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Circulation. 2003;108(13):1655–61.
Nannetti L, Paci M, Baccini M, Rinaldi LA, Taiti PG. Recovery from stroke in patients with diabetes mellitus. J Diabetes Complicat. 2009;23(4):249–54.
Jorgensen H, Nakayama H, Raaschou HO, Olsen TS. Stroke in patients with diabetes. The Copenhagen Stroke Study. Stroke. 1994;25(10):1977–84.
Khatri P, Wechsler LR, Broderick JP. Intracranial hemorrhage associated with revascularization therapies. Stroke. 2007;38(2):431–40.
Mankovsky BN, Ziegler D. Stroke in patients with diabetes mellitus. Diabetes Metab Res Rev. 2004;20(4):268–87.
Megherbi SE, Milan C, Minier D, Couvreur G, Osseby GV, Tilling K, et al. Association between diabetes and stroke subtype on survival and functional outcome 3 months after stroke: data from the European BIOMED Stroke Project. Stroke. 2003;34(3):688–94.
Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70(5):774–80.
Saczynski JS, Jonsdottir MK, Garcia ME, Jonsson PV, Peila R, Eiriksdottir G, et al. Cognitive impairment: an increasingly important complication of type 2 diabetes: the age, gene/environment susceptibility--Reykjavik study. Am J Epidemiol. 2008;168(10):1132–9.
Devos D, Cabantchik ZI, Moreau C, Danel V, Mahoney-Sanchez L, Bouchaoui H, et al. Conservative iron chelation for neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis. J Neural Transm (Vienna). 2020;127(2):189–203.
Yan N, Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front Neurosci. 2019;13:1443.
Selim M, Foster LD, Moy CS, Xi G, Hill MD, Morgenstern LB, et al. Deferoxamine mesylate in patients with intracerebral haemorrhage (i-DEF): a multicentre, randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2019;18(5):428–38.
Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009;40(12):e647–56.
Jia L, Chopp M, Zhang L, Lu M, Zhang Z. Erythropoietin in combination of tissue plasminogen activator exacerbates brain hemorrhage when treatment is initiated 6 hours after stroke. Stroke. 2010;41(9):2071–6.
Ning R, Chopp M, Yan T, Zacharek A, Zhang C, Roberts C, et al. Tissue plasminogen activator treatment of stroke in type-1 diabetes rats. Neurosci. 2012;222:326–32.
Yan T, Venkat P, Chopp M, Zacharek A, Ning R, Cui Y, et al. Neurorestorative therapy of stroke in type 2 diabetes mellitus rats treated with human umbilical cord blood cells. Stroke. 2015;46(9):2599–606.
Jackson L, Dong G, Althomali W, Sayed MA, Eldahshan W, Baban B, et al. Delayed administration of angiotensin II type 2 receptor (AT2R) agonist compound 21 prevents the development of post-stroke cognitive impairment in diabetes through the modulation of microglia polarization. Transl Stroke Res. 2020;11(4):762–75.
Jackson L, Dumanli S, Johnson MH, Fagan SC, Ergul A. Microglia knockdown reduces inflammation and preserves cognition in diabetic animals after experimental stroke. J Neuroinflamm. 2020;17(1):137.
Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26(50):13007–16.
Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11(3):298–308.
Chopp M, Li Y, Zhang J. Plasticity and remodeling of brain. J Neurol Sci. 2008;265(1–2):97–101.
Chopp M, Zhang ZG, Jiang Q. Neurogenesis, angiogenesis, and MRI indices of functional recovery from stroke. Stroke. 2007;38(2 Suppl):827–31.
Sweetnam D, Holmes A, Tennant KA, Zamani A, Walle M, Jones P, et al. Diabetes impairs cortical plasticity and functional recovery following ischemic stroke. J Neurosci. 2012;32(15):5132–43.
Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA. Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab. 2007;27(4):710–8.
Zhu M, Bi X, Jia Q, Shangguan S. The possible mechanism for impaired angiogenesis after transient focal ischemia in type 2 diabetic GK rats: different expressions of angiostatin and vascular endothelial growth factor. Biomedicine & pharmacotherapy. 2010;64(3):208–13.
Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73(11–12):2195–209.
Dharmalingam P, Talakatta G, Mitra J, Wang H, Derry PJ, Nilewski LG, et al. Pervasive genomic damage in experimental intracerebral hemorrhage: therapeutic potential of a mechanistic-based carbon nanoparticle. ACS Nano. 2020;14(3):2827–46.
Prakash R, Somanath PR, El-Remessy AB, Kelly-Cobbs A, Stern JE, Dore-Duffy P, et al. Enhanced cerebral but not peripheral angiogenesis in the Goto-Kakizaki model of type 2 diabetes involves VEGF and peroxynitrite signaling. Diabetes. 2012;61(6):1533–42.
Abdul Y, Abdelsaid M, Li W, Webb RC, Sullivan JC, Dong G, et al. Inhibition of toll-like receptor-4 (TLR-4) improves neurobehavioral outcomes after acute ischemic stroke in diabetic rats: possible role of vascular endothelial TLR-4. Mol Neurobiol. 2019;56(3):1607–17.
Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129(6):2293–304.
Jackson L, Dong G, Althomali W, Sayed MA, Eldahshan W, Baban B, et al. Delayed administration of angiotensin II type 2 receptor (AT2R) agonist compound 21 prevents the development of post-stroke cognitive impairment in diabetes through the modulation of microglia polarization. Transl Stroke Res. 2019.
Ahmed HA, Ishrat T, Pillai B, Fouda AY, Sayed MA, Eldahshan W, et al. RAS modulation prevents progressive cognitive impairment after experimental stroke: a randomized, blinded preclinical trial. J Neuroinflamm. 2018;15(1):229.
Stratoulias V, Venero JL, Tremblay ME, Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38(17):e101997.
Tan YL, Yuan Y, Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. 2020;25(2):351–67.
Dubbelaar ML, Kracht L, Eggen BJL, Boddeke E. The kaleidoscope of microglial phenotypes. Front Immunol. 2018;9:1753.
Acknowledgments
The authors would like to thank the summer medical students David Foreman and Thomas Menk for their assistance with the analysis of vascularization data with the Volocity software.
Funding
This study was supported by Veterans Affairs (VA) Merit Review (BX000347), VA Senior Research Career Scientist Award (IK6 BX004471), National Institute of Health (NIH) R01 NS083559 and R01 NS104573 (multi-PI, Susan C. Fagan as co-PI) to Adviye Ergul; Diabetic Complications Research Consortium DiaComp awards 17AU3831/18AU3903 (DK076169/115255) to Dr. Weiguo Li; Scientist Development Grant (16SDG30270013) and Mercer University seed grant to Dr. Mohammed Abdelsaid; and NIH T32 HL007260 to Victoria Wolf.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Statement on the Welfare of Animals
All rats were housed in the animal care facility at Augusta University, which is approved by the American Association for Accreditation of Laboratory Animal Care. All experiments were conducted in accordance with the National Institutes of Health (NIH) guidelines for the care and use of animals in research. Furthermore, all protocols were approved by the institutional animal care and use committee.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PPTX 86473 kb)
Rights and permissions
About this article

Cite this article
Abdul, Y., Li, W., Ward, R. et al. Deferoxamine Treatment Prevents Post-Stroke Vasoregression and Neurovascular Unit Remodeling Leading to Improved Functional Outcomes in Type 2 Male Diabetic Rats: Role of Endothelial Ferroptosis. Transl. Stroke Res. 12, 615–630 (2021). https://doi.org/10.1007/s12975-020-00844-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-020-00844-7