
Abstract
A disabling consequence of stroke is cognitive impairment, occurring in 12%–48% of patients, for which there is no therapy. A critical barrier is the lack of understanding of how post-stroke cognitive impairment (PSCI) develops. While 70% of stroke victims present with comorbid diseases such as diabetes and hypertension, the limited use of comorbid disease models in preclinical research further contributes to this lack of progress. To this end, we used a translational model of diabetes to study the development of PSCI. In addition, we evaluated the application of compound 21 (C21), an angiotensin II Type 2 receptor agonist, for the treatment of PSCI by blinding the treatment assignment, setting strict inclusion criteria, and implementing a delayed administration time point. Diabetes was induced by a high-fat diet (HFD) and low-dose streptozotocin (STZ) combination. Control and diabetic rats were subjected to 1 h middle cerebral artery occlusion (MCAO) or sham surgery. Adhesive removal task (ART) and two-trial Y-maze were utilized to test sensorimotor and cognitive function. Three days post-stroke, rats that met the inclusion criteria were administered C21 or vehicle in drinking water at a dose of 0.12 mg/kg/day for 8 weeks. Samples from freshly harvested brains were analyzed by flow cytometry and immunohistochemistry (IHC). Diabetes exacerbated the development of PSCI and increased inflammation and demyelination. Delayed administration of C21 3 days post-stroke reduced mortality and improved sensorimotor and cognitive deficits. It also reduced inflammation and demyelination through modulation of the M1:M2 ratio in the diabetic animals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is a leading cause of long-term disability worldwide, with the onset of cognitive impairment being a frequent contributor [1]. Post-stroke cognitive impairment (PSCI) is a type of vascular contribution to cognitive impairment and dementia (VCID), which is characterized by the progressive worsening of cognition, with 25–30% of stroke survivors developing it within the first 3 months after stroke [2]. With a recent increase in recanalization procedures, more patients are surviving stroke events. In the USA, stroke-related deaths showed a steady decline each year, with a greater decline observed among people aged ≥ 65 (by 54.1%) than their younger counterparts (by 45.9–53.6%) [3]. In 2018, the time by which eligible patients can be selected for recanalization therapy was extended to 24 h after initial stroke symptoms. This has and will continue to lead to more stroke victims receiving life-saving recanalization therapies. As a result, more surviving patients may also develop long-term disabilities, such as PSCI, creating an urgent need to discover possible therapeutic interventions. Unfortunately, poor understanding of how PSCI develops, coupled with the incomplete modeling of PSCI in the laboratory setting, poses a barrier for the development of new treatments. Our lab has shown that, although there are acute cognitive deficits after stroke, young healthy male rats recover within 2 weeks [3, 4]. Diabetic rats, however, remain cognitively impaired at 2 weeks, making the first goal of this paper to discover the long-term effect of diabetes after stroke, in a clinically translational model of PSCI.
More than 40% of stroke victims present with pre-existing diabetes at the time of stroke. In fact, the risk of stroke is up to 2-6 fold greater in diabetic patients [5, 6]. Diabetes not only increases the risk of stroke but also the severity of cognitive impairment [4]. In a large clinical trial, the Rotterdam study, diabetes was associated with a twofold increased prevalence of vascular cognitive impairment (VCI) [5, 7]. Cognitive impairment after stroke is greatly understudied, partly due to the fact that the majority of laboratories utilize young and otherwise healthy male animals. Since the majority of patients present at the time of stroke with comorbid vascular risk factors, studying PSCI in a comorbid disease model is absolutely essential [5]. Utilizing a clinically relevant high-fat diet (HFD)/low-dose streptozotocin (STZ) model, we evaluated the impact of diabetes on long-term cognition up to 8 weeks after a stroke.
Neuroinflammation has been linked to the development of cognitive impairment. Increased microglia activation, reactive astrocytes, and white matter damage have all been associated with the development of cognitive impairment [8,9,10]. Diabetes, as a chronic inflammatory disease also exhibits these same features [4, 11]. Our lab has shown that diabetes increases the number of activated microglia as well as the amount of pro-inflammatory immune cells within both the blood and the brain of HFD/STZ animals [4, 12]. Although it is known that diabetes perpetuates neuroinflammation, it is not yet fully understood how diabetes impacts the microglia phenotype or chronic stroke recovery in an animal model. Microglia are the resident immune cells of the brain and have a major role in modulating neuroinflammation. Upon activation, they exist as two broadly classified phenotypes referred to as “M1” or “M2” [13]. Although M2 consists of many sub-types, overall, they promote immune suppression, injury resolution, and participate in phagocytosis and matrix maintenance [13]. M1, on the other hand, promotes the release of pro-inflammatory cytokines and recruitment of peripheral inflammatory cells [13]. Microglia have a relatively low turnover rate making them susceptible to pro-inflammatory effects of age, injury, or stress, making the second goal of this study to discover how diabetes impacts the M1:M2 ratio and inflammation within the diabetic brain, and how these alterations impact cognition.
After establishing a clinically translational model of PSCI and elucidating the key pathogenic mechanisms contributing to its development, we evaluated a potential therapeutic for disease intervention. In a blinded manner we evaluated the impact of a delayed administration of a small molecule currently in clinical trials, compound 21 (C21). Our lab and others have shown that the neuroprotective effects of angiotensin receptor blockers (ARBs) are mediated through the blockage of the angiotensin II type I receptor (AT1R) which leads to the activation of the angiotensin type 2 receptor (AT2R), due to the increased amount of unbound Ang II able to bind to the AT2R [13, 14]. C21 is a selective AT2R agonist and has been shown by multiple laboratories to ameliorate ischemic damage in different models of stroke [13, 15,16,17,18,19,20,21,22,23]. Our lab has shown that C21, is neurovascular protective and improves stroke outcome, including prevention of cognitive impairment [13, 15]. It has recently been shown to mediate some of its neuroprotective effects through increases in brain-derived neurotrophic factor (BDNF) after stroke [13, 24]. We also reported that C21 downregulates activated microglia after stroke in hypertensive animals, suggesting that therapeutic intervention with delayed administration of C21 post-stroke in diabetic rats may modulate microglia polarization to alleviate cognitive deficits [14, 18]. Taken together, this led us to believe that therapeutic intervention with delayed administration of C21 post-stroke in diabetic rats may modulate microglia polarization to prevent cognitive deficits. The third goal of this study was to evaluate the mechanism by which it exerts its effect.
Methods
Animal Model
Male Wistar rats (Envigo RMS, Inc., Indianapolis, IN) were housed in the animal care facility at Augusta University, which is approved by the American Association for Accreditation of Laboratory Animal Care. All experiments were conducted in accordance with the National Institute of Health (NIH) guidelines for the care and use of animals in research. Furthermore, all protocols were approved by the institutional animal care and use committee.
Middle Cerebral Artery Occlusion (MCAO) Surgery
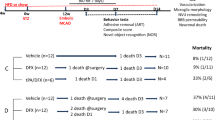
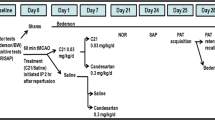
Male control and diabetic animals were subjected to transient focal cerebral ischemia (60 min MCAO) or sham surgery at 12–15 weeks of age using 4–0 silicon-coated nylon suture (Doccol 403756 or 403534), depending on the rat size. The animals that weighed 350 –425 g received the 403756 suture, while animals weighing 300–350 g received the 403534. This was optimized prior to the start of the study to result in similar infarct sizes across weight ranges. The animals were anesthetized using 2–5% isoflurane, a ventral mid-line neck incision was made, the right common carotid artery (CCA) was exposed and lightly tied, and the external carotid artery (ECA) was ligated and cut. The suture was marked at 1.8 and 2 mm, then advanced from a nick at the ECA into the internal carotid artery (ICA) until positioned between the 1.8- and 2-mm marks, indicating the branching of the MCA. The suture was tied in place for the duration of the occlusion and the animals were allowed to recover from anesthesia. At the end of the 60-min occlusion time, the animals were re-anesthetized, the suture was removed for reperfusion and the small nick at the ECA was permanently ligated. In sham surgeries, the CCA was isolated and the ECA was cut and ligated without insertion of the suture. Although there was early mortality in some groups, if an animal that met the pre-set inclusion criteria lived past 4 weeks, they were included in the behavioral analyses (Tables 1 and 2). This led to a total of six groups: control sham (C sham, N = 4), diabetic sham (D sham, N = 5), control MCAO vehicle treatment (C veh N = 5), diabetic MCAO vehicle treatment (D veh, N = 7), control MCAO C21 treatment (C C21, N = 5), and diabetic MCAO C21 treatment (D C21, N = 6). Additional animals (N = 4) were used for lesion examples (Fig S1). In the post-operative period, blood glucose (BG) was monitored daily for 7 days and then weekly until 8 weeks.
Treatment and Behavioral Assessments
Dose and Timing Justification
We chose day 3 to start administering C21 because it is well out of the neuroprotective window and acute infarct evolution is likely to be complete. We chose an 8-week follow-up in an attempt to capture the progressive development of PSCI over time. The oral C21 dose of 0.12 mg/kg was calculated based on an oral bioavailability of 0.25, so is equivalent to the intravenous (IV) dose of 0.03 mg/kg [15].
Blinding and Randomization
The treatment and vehicle were prepared by an individual not involved in the surgery or assessments and labeled as group A and group B. Each animal was numbered before baseline behavioral assessments were taken. After MCAO surgery, the animals that met the pre-set inclusion criteria were assigned to groups A and B using a random number generator. All behavioral and histological assessments were coded and conducted by a blinded investigator.
Inclusion Criteria
In order to ensure inclusion of animals with a significant degree of ischemic injury, we implemented strict inclusion criteria. The inclusion criteria included assessment of sensorimotor function and weight loss. Three days after MCAO, animals underwent the adhesive removal task (ART). Animals that had an ART above 30 s and had more than 11% weight loss were randomly assigned into C21 treatment and vehicle treatment groups.
Assessment of Sensorimotor Function
Sensorimotor function was evaluated by composite neurological score and ART as previously described [25]. The composite score was comprised of the beam walk and Modified-Bederson score and was taken at baseline, day 3 and weeks 1, 2, 4, and 8 post-stroke. The Modified-Bederson score was comprised of circling bias, forelimb retraction, hind-limb retraction, and resistance to push. Circling bias measures spontaneous ipsilateral circling 2 (best) to 0 (worst). For forelimb and hindlimb retraction, each limb was displaced laterally and the ability to recover the replaced limb was measured 1 (best) to 0 (worst). Resistance to push was scored as 1 (resistance) to 0 (no resistance). The animals were trained on the beam walk for 4 days prior to baseline assessments. Animals were given a score from 7 (best) to 1 (worst) based on their balance on a horizontal beam as described previously [26]. For the ART, the animals were trained for 4 days and then baseline measurements were recorded prior to stroke. Three days post-stroke, measurements were recorded to determine eligibility for inclusion in the study. If the rat was included in the study, then subsequent ART measurements were recorded at weeks 1, 2, 4, and 8 post-stroke. ART was carried out as previously described with modification [27]. Contact and removal latency of the adhesive paper dot was recorded and the average was taken from three trials with a maximum removal latency of 180 s per trial.
Assessment of Cognitive Function
Cognition was assessed by the two-trial Y-maze. The animals were trained 4 days prior to baseline testing. Testing was conducted at baseline prior to stroke, followed by weeks 1, 2, 4, and 8 post-stroke. The two-trial Y-maze was used to examine spatial memory. In the first trial, animals were allowed to freely explore two open arms for 10 min. The animal was returned to its home cage for a 15-min delay. In the second trial, animals were allowed to explore all three arms of the Y-maze apparatus freely for 3 min. The total time spent in each arm was recorded. Results were expressed as % time spent in the novel arm (time in novel arm divided by total time in all arms × 100). Preference for the novel arm was interpreted as intact spatial memory.
Euthanasia, Specimen Collection, and Molecular Techniques
Animals were euthanized 8 weeks after stroke or sham surgery using isoflurane overdose and cardiac puncture. Sections of the prefrontal cortex (PFC) and the hippocampus (Hipp) were taken for flow cytometry and immunocytochemistry.
Flow Cytometry
The PFC and Hipp were isolated through separation of the B and D slices as indicated in Fig. 3A. The tissue was then minced into 1-mm3 pieces and was dissociated using Worthington’s Papain Dissociation kit (catalog number LK003153) with the following modifications: (1) tissue was left in dissociation medium for 15–25 min and (2) oxygen was continuously perfused over (not bubbled within) the solution for the duration of the incubation period [28]. Microglia were isolated as described below.
Myelin Debris Removal and Microglial Isolation
A debris removal step was performed using modified protocols from Miltenyi Biotec’s Myelin Removal Kit (catalog number (Miltenyi Biotec, Germany) and CD11b+ Microbeads (Miltenyi Biotec, Germany). Following dissociation, up to 107 cells were suspended in 200 μL 0.5% BSA in PBS buffer and incubated with 20 μL anti-myelin microbeads for 15 min at 4 °C. The cells were then placed in the mini-MACS magnetic separator column and the clean supernatant was eluted out. The cells were then incubated with 20 uL of CD11b+ beads to isolate the microglia/macrophage population and isolated using the mini-MACS separator once again. CD11b+ cells were then further processed with surface and intracellular microglia makers.
Cellular Staining
Cells were incubated with surface markers against pre-conjugated antibodies CD45-APC (eBioscience, San Diego, CA) and CD86-FITC (BD bioscience, San Jose, CA) for 20 min. Cells were then permeabilized for intracellular staining with a fixation/permeabilization solution kit (eBioscience, San Diego, CA). Cells were separated into two groups and incubated with markers CD206 (Abcam, Cambridge, MA) or TNF-α (BD bioscience, San Jose, CA) and TMEM119 (Novus, Centennial, CO). Secondary antibodies PE (eBioscience, San Diego, CA) and PerCP (BD bioscience, San Jose, CA) were used in both groups. Cells were then washed and analyzed using the Cytoflex (Beckman Coulter, Indianapolis, IN).
Imaging and Analysis
To minimize false-positive events, the number of positive events detected with the negative staining control for each individual channel was subtracted from the number of positive cells stained with corresponding antibodies. Cells expressing a specific marker were reported as a percentage of the number of gated events. Markers are listed in Table 1. Microglia were first identified as CD11b+/CD45+ low. M1 microglia were further identified as CD86+, M2 cells were identified as CD206+. Residential microglia versus infiltrating macrophages were also identified as CD11b+/CD45+ without separation of low versus high. Residential microglia were then further identified as TMEM119+, while infiltrating macrophages were identified as TMEM119−. M1 macrophages were then identified as CD11b+/CD45+/TMEM119−/CD86+/TNF-α+ cells.
Immunohistochemistry (IHC)
Brains were extracted and post-fixed in 4% PFA overnight. The B and D slices were derived according to Fig. 3A. Free-floating 30-μm sections were incubated overnight with anti-IBA-1 (Ionized calcium-binding adaptor molecule 1, 1:500, Wako, Japan) and anti-GFAP (Glial fibrillary acidic protein, 1:400, Sigma-Aldrich, Burlington, MA) for D slice sections containing the Hipp and with anti-MBP (Myelin Basic Protein, 1:100, Abcam, Cambridge, MA) and NF200 (Neurofilament, 1:1000, Abcam, Cambridge, MA) for the B slice containing the PFC. Cells were then incubated with Texas red and Alexa Flour 488-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA) used at 1:200 for 2 h at room temperature. Nuclei were counterstained using Dapi (406-diamidino-2-phenylindole, Roche Basel, Switzerland) and sections were mounted. Imaging was performed using the Keyence Microscope (Itasca, IL) and Z stacked through the 30-um thickness at a 1-um pitch to obtain a complete count of the tissue area for IBA-1 and GFAP quantifications. Sections were derived from a single plane for MBP and NF200 quantifications.
Cell Culture
The direct effect of C21 on microglia polarization was determined in mouse cells (BV 2 line) by flow cytometry–based analysis of polarization markers. Cells were treated with LPS (Lipopolysaccharide, 100 ng/ml) and IFNy (interferon y, 20 ng/ml) to induce activation and M1-like polarization. Cells were either pre-treated with C21 (100 nM), 6 h prior, or post-treated, 6 h post-LPS/IFNy exposure to evaluate whether C21 impacts activation and polarization by preventing (pre-treatment) or reversing (post-treatment) M1-like polarization. The AT2R blocker, PD 123319 (0.1 μM), was used to determine if the C21 effects were mediated through AT2R agonism.
Statistical Analyses
SAS V9.4 (SAS Institute, Inc, Cary, NC) software was used to analyze all behavioral data. Prism 7 (GraphPad) was used to analyze all molecular data. Repeated measures ANOVA was performed for measurements taken across time utilizing the last observation carried forward method for any missing data, with the exception of Fig. 2C and 5H, where only raw data points were reported as observational values. Two-way ANOVAs were used to either compare (Control C vs Diabetes D) × (Sham vs Stroke) or (Control C vs Diabetes D) × (Veh vs C21), as indicated within the figure legends. Student’s t test was used to compare two groups. The degree of significance was marked by the number of symbols: 1 symbol indicates p < 0.05, 2 indicates p < 0.01, and 3 indicates p < 0.001. * is used to symbolize an effect of time, + for an effect diabetes vs control (or LPS/IFNy vs control), and $ for an effect of C21 vs veh as indicated on the figures.
Results
Diabetes Increases Mortality, Worsens Stroke Recovery, and Chronically Upregulates Inflammation
During the acute period of stroke recovery (up to 3 days), control animals exhibited a 25% mortality rate, while the diabetic animals exhibited a 40% mortality rate (Table 2). After inclusion at day 3, none of the control animals experienced early mortality prior to week 8, while an additional 55% of the included diabetic animals experienced early mortality, with the bulk of the deaths occurring around week 4. The inclusion criteria resulted in maximum deficits in ART within both the control and diabetic groups at day 3 that improved with time (Fig. 1A, p < 0.001). However, diabetic animals experienced a prolonged recovery period (Fig. 1A++p < 0.01). The neurological score of the animals displayed a similar trend of prolonged and exacerbated sensorimotor deficits within the diabetic animals. Even though both the control animals and diabetic animals experienced a significant degree of ischemic injury, 3 days after stroke, the diabetic animals had a worse neurological score than the control animals which persisted, resulting in a slower rate of improvement (Fig. 1B, *p < 0.05). Overall, the diabetic animals exhibited worsened stroke recovery as measured by both ART and the neurological score when compared to the control animals.

Diabetes worsened stroke-mediated functional deficits up to 8 weeks after recovery. A Sensorimotor deficits were measured by adhesive removal task (ART) which indicated that diabetes prolonged the recovery after a stroke. Repeated measures ANOVA, (Control C vs Diabetes D) *** main effect of time (p < 0.001), ++ main effect of diabetes (p < 0.01). B Diabetic animals experienced greater deficits after a stroke compared to sham as measured by their neurological score. Repeated measures ANOVA, (Control C vs Diabetes D) Interaction p < 0.05. (n = 5–7/group)
Similar to the worsened sensorimotor deficits, diabetic animals also exhibited exacerbated cognitive deficits. Both the control and diabetic animals exhibited a progressive decline after stroke, but the diabetic animals exhibited a consistently steeper decline than control animals (Fig. 2A, p < 0.001 time, 0.01 diabetes). Eight weeks after stroke, the diabetic animals were significantly more impaired than control animals, with or without stroke. This indicates that diabetes chronically impairs cognition after a stroke (Fig. 2B, p < 0.05). In fact, when the baseline of each animal was compared to their cognition at week 8, the control animals declined by an average of 10.6% after a stroke, while the diabetic animals declined by an average of 27.5%, indicating that the presence of diabetes more than doubled the decline of cognition chronically after a stroke (Fig. 2B, p < 0.05). The threshold of cognitive impairment was set as scoring below the percent chance (33%) that the animal would explore the novel arm for an equal time of the three total arms. Through this method, we found that although 0% of the control sham developed cognitive impairment which increased only to 20% after a stroke, 67% of the diabetic sham group developed cognitive impairment, which increased to 100% if they experienced a stroke (Fig. 2C).

Diabetes exacerbated the development of cognitive Impairment. A 2 trial Y-maze was used to examine cognition. Although both control and diabetic animals experienced an overall progressive decline in cognition after stroke, the diabetic animals experience a steeper decline than the control animals. Repeated measures ANOVA (Control C vs Diabetes D), *** main effect of time (p < 0.001), ++ main effect of diabetes (p < 0.01). B Diabetes with or without the comorbid event of a stroke resulted in a net decline of cognition long-term. Two-way ANOVA, (Control C vs Diabetes D) (Sham × Stroke) * main effect of diabetes (p < 0.05). C 8 weeks after a stroke 100% of diabetic and only 20% of the control animals were classified as “cognitively impaired”. (n = 5–7/group)
Since diabetes is well known to perpetuate a chronically elevated inflammatory state systemically, we evaluated the effects of diabetes on inflammation within the central nervous system (CNS) as an explanation for the exacerbated stroke deficits observed within these animals. We acquired images throughout the B slice (Fig. 3A) from structures of the Prefrontal Cortex (PFC) as well as the Limbic System, as these structures are essential to cognition. We acquired images from the organum vasculosum of the lamina terminalis (OVLT) since this structure is without a blood brain barrier (BBB) and allows access to components of systemic circulation, such as immune cells. We observed a drastic upregulation of IBA-1+ cells (microglia and macrophages) and GFAP+ (astrocytes) cells within each of these structures compared to the control animals (Fig. 3B–Ep < 0.05, 0.01). We observed a trend in the increased presence of IBA-1+ cells that failed to reach significance in diabetic sham animals compared to control sham animals in the absence of a stroke (Fig S2). We then used flow cytometry to validate this in both the B and D slices and noted an increase in the number of both microglia and macrophages (Fig. 3A). Upon further investigation, we discovered that the increase derived not from an increase in microglia but in macrophages (Fig. 3H–J, p < 0.01 PFC, 0.05 Hipp). Utilizing IHC, we then evaluated the D slice, particularly the hippocampus, another area of the brain essential for learning and memory, for consequences of chronic inflammation such as demyelination (Fig. 3F). Diabetic animals experienced significantly reduced myelination, or demyelination, compared to control animals (Fig. 3G, p < 0.01). This suggests that the chronic inflammation observed in diabetes may contribute to the chronic impairment of cognition observed through resultant demyelination of areas responsible for learning and memory, such as the hippocampus.

Diabetes chronically upregulated inflammation after a stroke. A 30-μm sections from the B slice were co-stained with IBA-1 and GFAP as indicated in A. B 10× images were taken from the structures within the PFC, limbic system, and OVLT as indicated with squares in A. Images derived from control stroke (B) and a diabetic stroke groups (D) were Z stacked and quantified throughout the layer. 8 weeks after stroke, diabetic animals exhibited a chronic upregulation of C IBA-1+ cell, and of E GFAP+ cells compared to their control counter parts Student’s t test, (Control stroke C vs Diabetic stroke D) + compared to control stroke (p < 0.05). E) Within the slice B histology GFAP+ cells were also upregulated. Student’s t test, (Control stroke C vs Diabetic stroke D) ++ compared to control stroke (p < 0.01). F 30-μ m sections from the D slice were co-stained with MBP and NF200. Images were taken from the hippocampus as depicted in (A) and (F) and indicate that the diabetic animals also experienced a greater degree of demyelination (G) as measured by the ratio of MBP:NF200 axons, Student’s t test (Control stroke C vs Diabetic stroke D) ++ compared to control stroke (p < 0.01). Flow cytometric analyses were performed on the B and D slices containing the PFC and Hipp, respectively, as indicated in Table 1. This also showed an upregulation of microglia and macrophages (H), with the upregulation deriving from an increase in the macrophage (I) but not microglia population (J). Student’s t test, (Control stroke C vs Diabetic stroke D) ++ and + compared to control stroke (p < 0.01, 0.05). (n = 5–7/group)
Delayed Administration of C21 Reduces Mortality and Improves Stroke Recovery
The delayed administration of C21 resulted in a reduced mortality without impacting blood glucose (BG) (Fig. S3), whereas the vehicle-treated diabetic animals experienced an additional 55% mortality (Table 3). Delayed C21 administration was also associated with improvements in sensorimotor abilities, as measured by ART, as early as week 1 in both control and diabetic animals (Fig. 4A and B, p < 0.05, 0.001). There was no significant difference in body weight between the C21 and vehicle treatments (Fig. S4).

Delayed administration of C21 improved functional deficits. C21 administration improved functional deficits in A control animals and B diabetic animals as measured via ART. Repeated measures ANOVA, (Control C veh vs C C21) and (Diabetic D veh vs D C21) *** main effect of time (p < 0.001), + and +++ main effect of C21 (p < 0.05 C veh, 0.001 D veh). (n = 5–7/group)
The delayed administration of C21 also preserved brain tissue volume within the control and diabetic animals after a stroke (Fig. 5C, p < 0.05). C21 administration additionally improved myelination within the diabetic animals, but not the control animals (p < 0.05). This translated to the observed improvements in the cognitive deficits experienced by the diabetic animals and alleviated both the acute and chronic deficits in cognition observed in the diabetic animals, resulting in a dimorphic recovery from stroke (Fig. 5F, Interaction p < 0.05). When the baseline of each animal was compared to their cognition at week 8, C21 administration not only prevented the 27.5% decline in diabetic animals, but actually resulted in a 5.6% increase from baseline after a stroke (Fig. 5G, p < 0.01). When the threshold of cognitive impairment was set as scoring below the % chance (0.33) that the animal would explore the novel arm for an equal time of the three total arms, we discovered that C21 treatment reduced the development of cognitive impairment from 100 to 0%. By week 8, none of the diabetic animals that received C21 were cognitively impaired (Fig. 5H).

Delayed administration of C21 after a stroke reduced brain atrophy in both control and diabetic animals but only improved cognitive deficits in diabetic animals. A Depicts an image of the B slice of a vehicle-treated control and B C21-treated control 8 weeks after stroke. C C21 treatment preserved brain volume in both control and diabetic animals. Student’s t test, (Diabetes D Veh vs Diabetes D C21) and (Control C Ceh vs Control C C21) $ compared to Veh (p < 0.05). D It also drastically improved the myelination within the hippocampus from the D slice of the diabetic animals only. Student’s t test, (Diabetes D Veh vs Diabetes D C21) $ compared to D Veh (p < 0.05). E Although C21 treatment did not significantly improve cognition in control animals, it did significantly improve cognition in diabetic animals and prevented the progressive decline in cognition (F). Repeated measures ANOVA, (Control C vs Diabetes D) Interaction p < 0.05. G C21 treatment actually resulted in a net improvement of cognition from baseline to 8 weeks after a stroke in diabetic animals. Student’s t test, (Diabetes D veh vs Diabetes D C21) $$ compared to D Veh (p < 0.01). H C21 resulted in 0 animals classified as “cognitively impaired” while the veh treated animals developed cognitive impairment at a rate of 100% (n = 3–4/group for C and 5–7/group for D–H). Representative images from diabetic animals are not shown due to the extensive tissue damage in sections of vehicle-treated animals
C21 Modulates the Polarization of Microglia to Downregulate Inflammation in Diabetes
C21 administration did not reduce the number of GFAP+ cell nor IBA-1+ cells in the diabetic cohort (Fig. 6A and B). Additionally, it did not impact the percentage of microglia which were activated (Fig. 6C). However, within the activated microglia population, it actually shifted the M1:M2 ratio toward a more anti-inflammatory profile. Within the ipsilateral hemispheres containing both the PFC and hippocampus, the M1:M2 ratio was upregulated within diabetic animals after a stroke compared to sham, but downregulated with C21 administration when evaluated with flow cytometry (Fig. 6E, p < 0.01, 0.05). There was not a significant difference between any of the groups within the contralateral hemisphere (Fig. 6E).

C21 may exert its beneficial effects on stroke recovery and cognition through modulation of the M1:M2 ratio in diabetic animals. Treatment with C21 did not impact the amount of A GFAP+ cells or B IBA+ cells that were stained histologically within the brain of the diabetic animals. C It also did not impact the percentage of activated microglia, as measured by flow cytometry as indicated in Table 1. Flow cytometric analyses were conducted as illustrated in D, and indicated that C21 did however reduce the M1:M2 ratio within the ipsilateral hemisphere (E) without altering the contralateral hemisphere. Student’s t test, (Diabetes D veh vs Diabetes D sham) and (Diabetes D veh vs Diabetes D C21) ++ vs sham (p < 0.01) and $ compared to D veh (p < 0.05). (n = 5–7/group)
We then evaluated the direct impact of C21 on microglia in vitro using a BV2 mouse cell line. The M1:M2 ratio was evaluated as illustrated in Fig. 7A. We discovered that LPS/IFNy exposure did polarize the cells to a M1 phenotype and away from a M2 phenotype, as expected (Fig. 7B–D). Both the pre- and post-treatment with C21 polarized the cells toward a M2 phenotype to ultimately decrease the M1:M2 ratio (Fig. 7B–D). Post-treatment C21 was administered 6 h after LPS/IFNy exposure, yet it was still able to prevent the inflammatory effects of LPS/IFNy exposure, highlighting the potency of this therapeutic application on microglia polarization. Interestingly, when the AT2R was blocked with PD, the effects were not completely reversed, suggesting that perhaps the microglia polarization effects of C21 are, at least partly, independent of the AT2R.

C21 directly modulated microglia partly independent of AT2R activation. Representative flow cytometry charts in panel A and cumulative data in panel B demonstrate that BV2 cells polarized toward a M1 phenotype with the incubation of both LPS and IFNy and treatment with C21 before or after LPS and IFNy challenge reduced the M1:M2 ratio. Blockade of AT2R with PD123319 did not reverse the effect, suggesting C21 effects may be independent of AT2R activation within microglia cells. Student’s t test (LPS/IFNy vs control), (LPS/IFNy vs LPS/IFNy/C21 pre-tx) and (LPS/IFNy vs LPS/IFNy/C21 post-tx) * compared to control (p < 0.05), $ compared to LPS/IFNy (p < 0.05). C Although C21 did not impact the M1 microglia polarization, it drastically upregulated E D M2 microglia polarization. Student’s t test (LPS/IFNy vs control), (LPS/IFNy vs LPS/IFNy/C21 pre-tx) and (LPS/IFNy vs LPS/IFNy/C21 post-tx) * compared to control (p < 0.01), $$ compared to LPS/IFNy (p < 0.01). (n = 5–6/group)
C21 Exerts Dimorphic Effects on Control and Diabetic Animals
Although delayed administration of C21 improved sensorimotor deficits in control animals and preserved brain volume, it exerted dimorphic effects on the immune cell modulation when compared to its effects in diabetic animals. Within the control cohort, C21 administration increased the presence of IBA-1+ cells within the B slice in regions of the PFC, limbic system, and the OVLT (Fig. 8A, p < 0.01), while it had no impact in diabetes (Fig. 6A–C). Upon further assessment of the residential status of the microglia, C21 administration increased the number of macrophages in the hippocampus, but not the PFC in control animals and tended to decrease them in diabetic animals (Fig. 8C, interaction p < 0.05) exerting a dimorphic effect on each group. This suggested that the increase in IBA-1+ cells may be due to increased infiltration of macrophages. When further assessing the polarization of the macrophages, we found that although C21 increased their presence within the tissue, it actually decreased the percentage of M1 macrophages compared to vehicle-treated control animals (Fig. 8D, p < 0.05).

C21 administration exerted a dimorphic effect in diabetic and control animals. A While C21 did not significantly impact the number of microglia nor the degree of macrophage infiltration in diabetic animals chronically after a stroke, it promoted a dimorphic increase in M2 macrophage infiltration of control animals. A IHC staining with IBA-1 showed an increased amount of IBA1+ cells within C21-treated control animals. Student’s t test (Control C veh vs C C21) $$ compared to C veh. B Flow cytometric analyses indicated a dimorphic interaction of C21 to increase the TMEM119- (macrophage) population within the CD45+ cells of the hipp of control animals, but not of diabetic animals. Two-way ANOVA (Control C veh vs C C21) × (Diabetic D veh vs D C21), interaction p < 0.05. It also trended toward this within the PFC but did not reach significance (B). Although C21 increased macrophage infiltration within the Hipp, within the macrophage population of the control animals C21 reduced the percentage of M1 macrophages within the Hipp. Student’s t test (Control C veh vs Control C C21) $ compared to C veh (p < 0.05). (n = 5–6/group)
Discussion
The observation that the HFD/STZ model of diabetes increased stroke mortality, worsened sensorimotor deficits, and exacerbated cognitive deficits, similar to that observed clinically, validated it as a translational animal model for the study of diabetic stroke and the development of PSCI. Clinically, more than 40% of stroke victims present at the time of stroke with pre-existing diabetes, which also increases the severity of cognitive impairment [7]. Similar to the increased occurrence of cognitive impairment in diabetes reported in the Rotterdam study, in our preclinical model, diabetic animals not only showed lower Y-maze scores indicative of worse cognitive function but also a fivefold increased incidence of cognitive impairment after stroke [5, 7].
Our lab has previously shown worsened cognition and increased inflammation within diabetic rats up to 2 weeks after a stroke, but to the best of our knowledge this is the first study evaluating the long-term effect of diabetes on cognition and inflammation [4]. While a previous study reported impaired cognition 2 months after stroke in a type 1 model of diabetes, inflammation was not assessed in that particular study [29]. It was interesting to note that both diabetic and control animals experienced a similar degree of brain atrophy after a stroke, but only the diabetic animals developed impaired cognition at a significant rate. This may be due to the chronically elevated inflammatory state in diabetic rats, as indicated by the increased number of astrocytes and microglia and by the advanced degree of demyelination. A similar effect of diabetes on microglia activation as well as impaired white matter and cognition has been reported in mice out to 35 days after stroke [11]. However, this is the first study to evaluate out to 60 days and confirm that the elevated inflammatory state produced by diabetes remains chronically elevated after stroke, without any apparent resolution. Although both control and diabetic animals experienced a similar degree of atrophy, the neuronal networks within the remaining tissue of the control animals appear to retain superior functionality and connectivity.
In this study, we noted a large Mtrans phenotype, where the CD206 M2 marker and the CD86 marker often were co-expressed, even when intracellular markers were additionally implemented. Since this Mtrans population has been shown to be detrimental, similar to M1 microglia over-activation, this was gated out by only selecting M2 microglia from the CD86- population [30]. While in this study we did not measure BDNF levels, this leads us to speculate whether the M2 microglia maintain traditional M2 roles such as increased BDNF production, beneficial phagocytosis, and synaptic plasticity in diabetes. Previous studies have reported increased phagocytosis and synaptic stripping by microglia in conjunction with impaired hippocampal function, which may suggest a reprogramming or a deleterious role within a sub-population of M2 microglia in diabetic animals [31].
After a stroke, many animals spontaneously recover. This may be due to the fact that they are not adequately stroked in comparison to the rest of the stroke group. It may also be due to compensatory mechanisms that vary between animals or to surgical variance. In either case, it can dilute or exaggerate the effectiveness of a therapeutic intervention. Our implementation of inclusion criteria served to address this problem and homogenize the stroke volumes in an effort to depict an accurate picture of the therapeutic potential of C21. In an added effort to increase the translatability of our studies, we blinded the experimenters and we implemented a delayed administration of C21, which served two purposes. First, many drugs are evaluated as a pre-treatment and show therapeutic efficacy but in clinical practice therapeutic interventions are rarely implemented in this manner. This ultimately contributes to the failure rate when translating an experimental therapeutic into clinical trials. Secondly, we wanted to elucidate the ability of C21 administration to impact the secondary neurodegenerative processes separate from acute neuroprotection which was reported by us and others [20, 21, 23]. While Bennion and colleagues reported long-term neuroprotection with C21, this effect was seen when C21 was administered 90 min after MCAO in aged rats. The observed preservation of brain tissue (lower brain atrophy) in the control and diabetic groups in our study leads one to question, whether this is due to neuroprotection or prevention of secondary neurodegenerative processes. Since C21 was initiated 3 days after stroke, when the infarct size has completed its evolution, this led us to believe that this may be due to secondary neurodegenerative processes, but further experiments are warranted.
Interestingly, the delayed administration of C21 did not reduce the number of IBA-1+ cells within the diabetic animals, nor their activation status. This is different than what our lab previously observed in spontaneously hypertensive rats (SHRs), where C21 drastically reduced microglia number and activation [14]. Difference may arise from differences within the comorbid background of diabetes versus hypertension. It may also arise from region of analysis. As determined by flow cytometry, the microglia number was unchanged in the PFC of the diabetic animals but tended to decrease in the hippocampus of diabetic animals. Differences in activation induced by C21 may arise from the use of two completely different methods. In this study, we used flow cytometry and the absence of M1/M2 markers to indicate activation status. Our lab previously utilized morphological analyses such as the transformation index (TI) and circularity index, which may be more indicative of M1-activated microglia (i.e., swollen cell body, shortened processes). This study did, in fact, show a reduction in M1 microglia [14]. Although C21 administration did not alter the number of microglia, it did shift the activated cells to a more anti-inflammatory phenotype. The treatment with C21 appears to work by polarizing the activated microglia toward a M2 phenotype. It is encouraging to observe that both the delayed administration of C21 in vivo, as well as the post-treatment of C21 in injured microglia in vitro resulted in significant improvements. Interestingly, in the absence of injury in vivo and in vitro, C21 did not alter the M1:M2 ratio. In the context of diabetes or when the cells were challenged with LPS and IFNy, C21 markedly reduced the M1:M2 ratio. Studies have shown that M2 microglia and macrophages drive oligodendrocyte differentiation, which could underlie the improved MBP:NF200 ratio observed within the diabetic animals [28].
The quantification of infiltrating macrophages was utilized as a surrogate marker for BBB integrity 8 weeks after stroke. With this surrogate marker, we noted a large increase in infiltrating macrophages when comparing diabetic animals to control animals. This was decreased with C21 treatment in the hippocampi of diabetic animals. Interestingly, control animals treated with C21 experienced a dimorphic upregulation of macrophages, opposite to the downregulation observed in the diabetic animals. Upon further investigation into this phenomenon, it was discovered that even though C21 upregulated the macrophages in general, it drastically reduced the percentage of M1 macrophages within that population. This leads us to believe it is both recruiting macrophages into the control brain and impacting their polarization. Further investigation is warranted to evaluate whether this potential massive recruitment of M2 macrophages observed only in the controls is accompanied by a breakdown of the BBB and whether this recruitment is actually beneficial or detrimental. Since we were interested in long-term protective effects of C21 independent of acute neurovascular protection it confers as we previously reported [15] and started the treatment 3 days after stroke, we did not measure the effect of C21 on acute vascular injury including hemorrhagic transformation and edema after stroke in diabetic animals. Given that diabetes exacerbates vascular injury and impairs vascular recovery [3, 25, 32], additional studies are warranted to evaluate neurovascular protective and restorative effects of C21 in diabetes.
In summary, we found the HFD/STZ model of diabetic to be a highly translational model for the study of diabetic stroke and the development of PSCI, displaying both similar increased mortality, functional deficits, cognitive impairment, and elevated inflammation to that observed clinically. We also found the implementation of the inclusion criteria to be a valuable tool to ensure ischemic injury and homogenization of the acute ischemic damage. C21 is currently in clinical trials for the treatment of pulmonary fibrosis. Since this therapeutic has already been shown to be safe for human administration and granted orphan drug status, it has high repurposing potential. The level of AT2Rs within the brain is a highly debated concept, but our intriguing in vitro results suggest that C21 may additionally act independent of these receptors [13]. Due to the complexity of our design, we included only the results obtained in male animals. Nevertheless, this translational study emphasizes the importance of incorporating disease models into dementia research to more closely mimic the clinical variables present within patient populations.
References
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322.
Levine DA, Galecki AT, Langa KM, Unverzagt FW, Kabeto MU, Giordani B, et al. Trajectory of cognitive decline after incident stroke. JAMA. 2015;314(1):41–51.
Li W, et al. Post-stroke neovascularization and functional outcomes differ in diabetes depending on severity of injury and sex: potential link to hemorrhagic transformation. Exp Neurol. 2018;311:106–14.
Ward R, et al. Post stroke cognitive impairment and hippocampal neurovascular remodeling: the impact of diabetes and sex. Am J Physiol Heart Circ Physiol. 2018.
Hardigan T, Ward R, Ergul A. Cerebrovascular complications of diabetes: focus on cognitive dysfunction. Clin Sci (Lond). 2016;130(20):1807–22.
Ergul A, et al. Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets. 2012;12(2):148–58.
Wang Q, et al. Prediabetes is associated with post-stroke cognitive impairment in ischaemic stroke patients. Brain Res. 2018;1687:137–43.
Felsky D, Roostaei T, Nho K, Risacher SL, Bradshaw EM, Petyuk V, et al. Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat Commun. 2019;10(1):409.
Kalaria RN, Akinyemi R, Ihara M. Stroke injury, cognitive impairment and vascular dementia. Biochim Biophys Acta. 2016;1862(5):915–25.
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672–713.
Ma S, et al. Diabetes mellitus impairs white matter repair and long-term functional deficits after cerebral ischemia. Stroke. 2018;49(10):2453–63.
Ladonya Jackson WL, Abdul Y, Dong G, Baban B, Ergul A. Diabetic stroke promotes a sexually dimorphic expansion of T cells. NeuroMolecular Med. 2019;In Press(71).
Jackson L, et al. Within the brain: the renin angiotensin system. Int J Mol Sci. 2018;19(3).
Ahmed HA, Ishrat T, Pillai B, Fouda AY, Sayed MA, Eldahshan W, et al. RAS modulation prevents progressive cognitive impairment after experimental stroke: a randomized, blinded preclinical trial. J Neuroinflammation. 2018;15(1):229.
Alhusban A, Fouda AY, Bindu Pillai, Ishrat T, Soliman S, Fagan SC. Compound 21 is pro-angiogenic in the brain and results in sustained recovery after ischemic stroke. J Hypertens. 2015;33(1):170–80.
Ahmed HA, et al. Role of angiotensin system modulation on progression of cognitive impairment and brain MRI changes in aged hypertensive animals - a randomized double- blind pre-clinical study. Behav Brain Res. 2018: 346:29–40.
Fouda AY, Pillai B, Dhandapani KM, Ergul A, Fagan SC. Role of interleukin-10 in the neuroprotective effect of the angiotensin type 2 receptor agonist, compound 21, after ischemia/reperfusion injury. Eur J Pharmacol. 2017;799:128–34.
Ahmed HA, et al. Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals-a randomized double- blind pre-clinical study. Behav Brain Res. 2019;359:560–9.
Ishrat T, et al. Dose-response, therapeutic time-window and tPA-combinatorial efficacy of compound 21: a randomized, blinded preclinical trial in a rat model of thromboembolic stroke. J Cereb Blood Flow Metab. 2018:271678X18764773.
Joseph JP, Mecca AP, Regenhardt RW, Bennion DM, Rodríguez V, Desland F, et al. The angiotensin type 2 receptor agonist compound 21 elicits cerebroprotection in endothelin-1 induced ischemic stroke. Neuropharmacology. 2014;81:134–41.
Bennion DM, et al. Post-stroke angiotensin II type 2 receptor activation provides long-term neuroprotection in aged rats. PLoS One. 2017;12(7):e0180738.
Min LJ, et al. Direct stimulation of angiotensin II type 2 receptor initiated after stroke ameliorates ischemic brain damage. Am J Hypertens. 2014;27(8):1036–44.
Shan BS, et al. Attenuation of stroke damage by angiotensin II type 2 receptor stimulation via peroxisome proliferator-activated receptor-gamma activation. Hypertens Res. 2018;41(10):839–48.
Schwengel K, et al. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J Mol Med (Berl). 2016;94(8):957–66.
Li W, et al. Diabetes worsens functional outcomes in young female rats: comparison of stroke models, tissue plasminogen activator effects, and sexes. Transl Stroke Res. 2017.
Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science. 1982;217(4562):855–7.
Chen J, Li Y, Chopp M. Intracerebral transplantation of bone marrow with BDNF after MCAo in rat. Neuropharmacology. 2000;39(5):711–6.
Holt LM, Olsen ML. Novel applications of magnetic cell sorting to analyze cell-type specific gene and protein expression in the central nervous system. PLoS One. 2016;11(2):e0150290.
Zhang L, Chopp M, Zhang Y, Xiong Y, Li C, Sadry N, et al. Diabetes mellitus impairs cognitive function in middle-aged rats and neurological recovery in middle-aged rats after stroke. Stroke. 2016;47(8):2112–8.
Kumar A, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. Microglial/macrophage polarization dynamics following traumatic brain injury. J Neurotrauma. 2016;33(19):1732–50.
Hao S, et al. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun. 2016;51:230–9.
Ergul A, et al. Impact of comorbidities on acute injury and recovery in preclinical stroke research: focus on hypertension and diabetes. Transl Stroke Res. 2016;7(4):248–60.
Acknowledgments
This study is from the doctoral thesis of Ladonya Jackson, presented to the University of Georgia. The authors would like to thank Dr. Darrell Brann for providing the BV2 cell line and Vicore Pharma for providing C21. We would also like to thank the Electron Microscope and Histology Core at Augusta University for the histological staining of our samples, with a special thanks to Ms. Penny Roon.
Funding
This study was supported by National Institute of Health (NIH) R01 NS104573 to Adviye Ergul and Susan C. Fagan; R01 NS083559, Veterans Affairs (VA) Merit Review (BX000347), VA Senior Research Career Scientist Award (IK BX004471) to Adviye Ergul; and TL1 TR002382 and UL1TR002378 to Ladonya Jackson.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Statement on the Welfare of Animals
All rats were housed in the animal care facility at Augusta University, which is approved by the American Association for Accreditation of Laboratory Animal Care. All experiments were conducted in accordance with the National Institute of Health (NIH) guidelines for the care and use of animals in research. Furthermore, all protocols were approved by the institutional animal care and use committee.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Materials
ESM 1
Fig. S1 The degree of weight loss correlated with lesion size. In order to set inclusion criteria, we first evaluated the correlation between weight loss at day 3 and the infarct size at day 3. We discovered that weight loss correlated with the size of the infarct (R2 = 0.8), Pearson’s Correlation. Since a 10% drop in weight loss only correlated with a 20% infarct size, we required the drop in weight loss of the included animals to be greater than 10%. Fig. S2 In an effort to ensure inclusion of animals with a significant degree of ischemic injury, a strict inclusion criteria was employed. 3 days after the animals underwent MCAO surgery, those that met the weight loss and sensorimotor deficit criteria were included in the study. Sham diabetic animals trended to show increased inflammation. A) 30 um sections from the B slice were co-stained with IBA-1 and GFAP and 10x images were taken from the structures within the prefrontal cortex, limbic system and OVLT. Images derived from control sham (A) and a diabetic sham groups (B) were Z stacked and quantified throughout the layer. Diabetic sham animals trended to exhibit a chronic upregulation of C) IBA-1+ cells (p < 0.07). Student’s t test, (Control sham C vs Diabetic sham D) + compared to control sham (p < 0.05). No difference was found in D) GFAP+ cells compared to their control counterparts. (n = 5/group). Fig. S3 Delayed C21 administration did not impact blood glucose. Blood glucose ranged from 300–350 mg/dL throughout the majority of the study. Fig. S4 Delayed C21 administration did not impact weight loss post-stroke. There was no significant difference in weight with the treatment of C21. Since weight loss is typically maximized by day 3 post-stroke, the delayed administration of C21 was not administered early enough to prevent significant weight loss, and it also did not later contribute to a larger weight gain. Fig. S5 Flow cytometry gating strategy. Strategy showing how cells were gated for flow cytometric analysis to identify infiltrating macrophages. Path A was utilized to differentiate infiltrating cells from resident cells, while path B was utilized to analyze the M1:M2 ratio. (PPTX 711 kb)
Rights and permissions
About this article

Cite this article
Jackson, L., Dong, G., Althomali, W. et al. Delayed Administration of Angiotensin II Type 2 Receptor (AT2R) Agonist Compound 21 Prevents the Development of Post-stroke Cognitive Impairment in Diabetes Through the Modulation of Microglia Polarization. Transl. Stroke Res. 11, 762–775 (2020). https://doi.org/10.1007/s12975-019-00752-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-019-00752-5