
Abstract
Kumamoto is known as the largest groundwater city in Japan. Geochemical modeling (saturation indices and mineral stability diagram) was applied in this area for better understanding a hydrogeochemical evolution in volcanic aquifers in regional scale with additional constraint from stable isotopic dataset. Geochemical evolutionary model was interpreted along the water flow dynamics. In total 136 water samples were collected from wells, springs, and rivers in and around the major groundwater flow lines for geochemical analyses. Our results indicated that plagioclase is the major weathering reactant in aquifers with secondary important weathering minerals of pyroxenes. These reactions facilitate current hydrochemical signatures and produce secondary minerals of kaolinite or halloysite and smectite in later stage. Observed hydrogeochemical processes can be distinguished into two distinct criteria along flow regime. The first processes are material loads from the surface (mixing of contaminants and river waters) and initial stage of silicate weathering, resulting in the formation of Ca-HCO3, Ca-NO3–HCO3, and Ca–Mg–Na-HCO3 type waters. These processes prevail in aquifers at the recharge to lateral flow zones with relatively shorter residence time of < ca. 40 years. The second processes are the precipitation of clay minerals, i.e., smectite, and cation exchange reaction of Ca2+ and Mg2+ to Na+ in downslope aquifers (Na-HCO3 type) with relatively longer groundwater residence time of > 55 years. Microbiological reduction reactions dominate over these areas and salinization occurs at the coast that changes aquifer waters to be Na-Cl type. Proposed models and approach shown in this study may be useful and applicable in interpreting systems in other volcanic aquifer systems at similar climate conditions and for sustainable water resources management.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
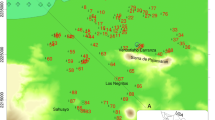
Kumamoto is the largest groundwater user area in Japan (Fig. 1). About one million people in and around the area completely depend on groundwater for their all purposes (e.g., Taniguchi et al. 2019). The area comprises Kumamoto City and its surrounding districts and towns, extended to the western flank of Aso caldera mountains in central Kyushu (Fig. 1), which is one of the largest volcanic calderas in Japan. The abundant groundwater resources in this area are a blessing of combination of both high local precipitations and highly permeable surface geological structures that comprise of Quaternary pyroclastic flow deposits of the Aso volcanic eruptions. Since the people in Kumamoto relies on groundwater for drinking purpose, its quality is one of major concerns to the local citizens and policy makers in a management point of view. However, the groundwater quality is becoming a serious issue in this area due to some geogenic (arsenic and fluoride) (Hossain et al. 2016a, b) and anthropogenic (nitrate and volatile organic compounds) contaminations (Kumamoto Prefecture and Kumamto City 2005; Kumamoto City Waterworks and Sewerage Bureau 2008; Tomiie et al. 2009; Hosono et al. 2013, 2014; Zeng et al. 2016; Okumura et al. 2018).

Location of the study area showing major geographic features, groundwater region and sampling locations along major groundwater flow lines A–A′ and B–B′, which hydrogeologic sections are shown in Fig. 3. Division of flow zones are shown along each arrow. Solid contour lines represent groundwater potential for high water season during September and October (m, above sea level, after Kagabu et al. 2017). Green and yellow squares represent sampling location of spring waters from Aso caldera watershed and Kumamoto area, respectively. Yellow and white stars indicate sampling location of river waters from Shira and Midori Rivers, respectively
In general, chemical evolution of groundwater and thus its quality is greatly controlled by the geochemical weathering processes (Hem 1989; Nordstrom et al. 1989; Drever 1997). Improved knowledge on these processes could help to understand a whole hydrochemical system of an area and for effective management and proper utilization of the water resources (Fisher and Mullican 1997; Cloutier et al. 2006). However, such attempt has not yet been made in the Kumamoto area although flow dynamics of groundwaters and their contamination processes are well studied. Therefore, the primary objective of this study is to depict these principle geochemical processes that control the chemical evolution of groundwaters along their flow systems. After a pioneering work of Garrels and Mackenzie (1967), a good number of researchers explained geochemical evolution of aquifer waters using major ion chemistry and mineral weathering reactions with mass balance approach in different hydrogeological systems (White et al. 1980, 1998; Wood and Low 1986; Thomas et al. 1996 and references therein). However, studies for carbonate-free, silicate dominated volcanic rock aquifers are still in limited numbers (e.g., Join et al. 1997; Nesbitt and Wilson 1992; Koh et al. 2016). Moreover, in most of the previous studies detailed flow dynamics were unclear in regional scale (Kenoyer and Bowser 1992). Due to the ambiguity in groundwater flow delineation, it was somehow difficult to observe geochemical evolutional characteristics precisely.
Kumamoto basin is volcanic terrains composed of pyroclastic flow deposits with coastal vicinity. The recharge and discharge areas of groundwaters with their flow paths are well defined based on hydrogeological assessments (e.g., Taniguchi et al. 2003; Ono et al. 2013; Kagabu et al. 2017; Okumura et al. 2018) and their residence times are well characterized using 85Kr groundwater age tracer (Kagabu et al. 2017). In addition, the effects of material loads from recharge areas and anion reduction processes due to microbiological succession on groundwater chemistry are well documented along flow paths using multiple isotope ratios (δ13C, δ15N and δ34S) (Hosono et al. 2013, 2014), trace elements (Hossain et al. 2016a, b), and PCR analysis (Zeng et al. 2016), which information allow us to depict a combined hydro-bio-geo-chemical evolution of groundwaters from recharge areas to the coast in regional scale. This particular integrated study is associated with systematic use of the major ion chemistry, stable isotope ratios, and geochemical modeling with available mineralogical data to construct a model showing hydro-bio-geochemical evolution in volcanic aquifer systems in a context of regional groundwater flow systems with their known residence time.
Study area
Topography, climate, geology and mineralogy
Kumamoto area is located in the central part of Kyushu Island in southern Japan with the longitude between 130°30′ and 131°05′ E and the latitude between 32°40′ and 33°00′ N (Fig. 1). The area is consisting of the western mountain feet of the Aso caldera mountains, surrounding plateaus and hilly mountains (expressed as ‘highlands’), and Kumamoto plain. The study area, termed as ‘Kumamoto groundwater region’ (Hosono et al. 2013) occupies 945 km2 in an area, lying between the Shira River watershed to the north and Midori River watershed to the south, the mountain foot of western caldera somma to the east and Kinpo Mountain (665 m) and Ariake Sea to the west (Fig. 1). The topography generally inclines from north-east to the south-west. Gradients are drastically changed near the Ezu Lake (Fig. 1), which is spring fed lake as described later. Three major highlands were formed during the Aso volcanic activity with altitude around 100–200 m. Among them Kikuchi and Takayubaru highlands are situated northeastern and eastern side of the study area, respectively, whereas Ueki highland is in the north. Kumamoto area has humid, subtropical climate. Average annual temperature and precipitation of Kumamoto are 16.9 °C and 1986 mm during 1981–2010, respectively (https://www.jma.go.jp/jma/menu/report.html). Around 40% of total precipitation occurs during summer rainy season in June–July.
Land-use in the study area is shown in Hosono et al. (2013). Briefly, the western mountain feet of the Aso caldera mountains are covered by forest and grassland. Vegetable and livestock farming are the main land-use in the Ueki, Kukuchi, and Takayubaru highlands. On the contrary, paddy is widely distributed at lowland areas along rivers and plains.
In the Kumamoto groundwater region, the Aso volcanic rocks of Quaternary age and Holocene alluvium deposits overlie hydrogeological basement of the Paleozoic to Mesozoic metamorphic rocks and Tertiary to Quaternary Pre-Aso volcanic rocks (Figs. 2, 3). These metamorphic rocks are exposed only in the southeastern and northwest-western parts of the Kumamoto area (Fig. 2). The Pre-Aso volcanic rocks, which are composed of intermediate to acidic lavas and tuff breccia, present around the Mt. Kinpo, Mt. Tatsuta and Aso caldera somma (Watanabe 1978). The Aso volcanic rocks that form the main body of the Quaternary deposits in Kumamoto widely present between the Kinpo Mountain and western flank of Aso caldera mountains (Fig. 2). Aso volcanic rocks are classified into 4 units by 4 gigantic eruption cycles: Aso-1 (eruption volume > 30 km3, eruptive age 370–270 ka), Aso-2 (> 25 km3, 170 ka), Aso-3 (> 40 km3, 100 ka) and Aso-4 (> 80 km3, 80 ka) (Aramaki 1984) (Fig. 3). There were many minor eruptions occurring in between these large eruption cycles (Ono et al. 1977; Matsumoto et al. 1991). Detailed geology and eruption history in and around caldera are summarized, e.g., in Ono et al. (1977), Kamata (1997), Watanabe (1978), and Ono and Watanabe (1985). The Aso volcanic products are generally permeable due to its porous structures. On the other hand, impermeable clay layers of lacustrine sediments are widely distributed in between Aso-3 and Aso-4 units (Kumamoto Prefecture and Kumamoto City 1995) (Fig. 3). In addition, a thick clay layer of marine origin of the last regression time, Ariake clay layer is deposited over the Kumamoto plain area, overlying the Aso-4 pyroclastic flow deposits at elevation generally lower than 5 m in the west of Ezu Lake in the coastal side (Figs. 2, 3). Beside this, fluvial alluvium sediments are distributed along the rivers at elevation higher than 5 m.

Surface geology map of the study area after Kumamoto Prefecture Geological Map Compilation Committee Report (2008). Dotted line in the figure shows elevation line of ca. 5 m (above sea level). Ariake clay sediments are expected to be distribute at elevation lower than ca. 5 m (see text for details)

Mineralogy of the Kumamoto area has been studied by various researchers (Watanabe 1978, 1979; Ono and Watanabe 1985; Hunter 1998; Kaneko et al. 2007; Miyoshi et al. 2009). In general, plagioclase, clinopyroxene, orthopyroxene, and titanomagnetite are the major phenocrysts in Aso volcanic rocks (Hunter 1998). Dominant secondary clay minerals are kaolinite, illite and smectite with some other alteration products. Detailed information for mineral abundances, compositions and major rock type for each unit of the Aso volcanic rocks are provided in Table S1. In general, the Aso volcanic rocks mainly consist of pyroclastic fall and flow deposits. Aso-1 to -3 consist of a range of welded to non-welded basaltic to deictic pyroclastic flow materials including lava flows, while Aso-4 consist of airfall, ignimbritic deposits, non-welded to welded dacitic ash and pumice, grading up to basaltic scoria (Lipman 1967; Watanabe 1979; Hunter 1998) and contain large amounts of lithic fragments (Ono et al. 1977). Petrographic analyses of Pre-Aso volcanic rocks have found that most common rock type is pyroxene andesite, tracked by subordinate hornblende andesite, biotite rhyolite, and basaltic to dacitic pyroclastic materials (Hunter 1998; Watanabe 1978, 1979).
Hydrogeology and groundwater flows
According to stratigraphy and subsurface geology of Kumamoto area, groundwater mainly occurs in the aquifers that consist of Aso pyroclastic flow and alluvial sedimentary deposits (Fig. 3). Two main aquifers are delineated in this area based on the lithologic properties and hydrodynamic conditions: first (shallow) and second (deep) aquifers from top to the bottom (Kumamoto prefecture and Kumamoto City 1995). In general, depth of the first aquifer is ca. ~ 90 m from the surface whereas second aquifer is situated at depth of ca. 20–250 m (Kumamoto Prefecture and Kumamoto City 1995). The first aquifer is mainly composed of Aso-4 pyroclastic flow and alluvium sedimentary deposits (Figs. 1, 2, 3). On the other hand, Aso-1 to -3 pyroclastic flow deposits and volcanic flow lavas consist of the second aquifer. An impermeable clay layer called ‘Futa’ and ‘Hanabusa’ layers of lacustrine sediments is distributed between the first and second aquifers (Fig. 3). However, this layer is discontinuous especially under the mid-stream area of the Shira River and partially absent around Ezu Lake area (Figs. 1, 2, 3) (Aramaki 2002). Hence, first aquifer is unconfined aquifer in nature whereas second aquifer can be considered as semi confined to confined aquifers.
With hydrogeological point of view, the groundwater in Kumamoto is thought to be largely recharged through infiltration of precipitation at highlands surrounding the plain areas due to the presence of highly permeable pyroclastic flow of Aso-4 (Kumamoto Prefecture and Kumamoto City 1995; Taniguchi et al. 2003; Okumura et al. 2018). It is also confirmed the water can be recharged by surface waters through both natural percolation and artificial infiltration by rice paddy ponding around mid-stream area of the Shira River (Figs. 1, 3). It is suggested in this area that surface water can directly be transported into the second aquifer as there is no aquiclude present and soil infiltration capacity is very high ~ 500 mm/day (Tanaka et al. 2010). Thus the river loses waters particularly at this area. Presence of the Togawa lava, highly porous andesitic lava of Aso-2, makes the confined aquifer more conductive (Fig. 3). At the end of this lava distribution, spring waters emerged at Ezu Lake with the discharge rate of 1.5–1.8 \(\times \) 108 m3/year (Ono et al. 2013). Waters from both aquifers are discharging in this area (Ono et al. 2011). Other than Ezu Lake, spring waters from mountain aquifers discharge through more than 30 major spring sites at mountain feet (Fig. 1) with the discharge rate ranging from 1.0 \(\times \) 103 to 1.6 \(\times \) 107 m3/year. Estimated hydraulic conductivities are 1.0 \(\times \) 10–5 to 1.0 \(\times \) 10–2 m/s and 1.0 \(\times \) 10–5 to 2.5 \(\times \) 10–1 m/s for the first and second aquifers, respectively (Mori et al. 2016; Hosono et al. 2019).
There are two major geographically distinct groundwater flow systems prevailing in this area (Hosono et al. 2013, 2014 and see groundwater flow simulation results shown in Fig. S1). First one is A–A′ flow that is the most significant flow system from the perspective of groundwater volume as well as city water supply (Figs. 1, 3). This flow line can be divided into three different zones: recharge and mixing zones (A-1), lateral flow to discharge zone (A-2) and stagnant zone (A-3) (Hosono et al. 2013, 2014). The water is recharged by precipitation mainly through Kikuchi and Takayubaru highlands (Taniguchi et al. 2003; Kagabu et al. 2017; Okumura et al. 2018) and also through surface water to the second aquifer in the mid-stream area of the Shira River as mentioned previously. A groundwater residence time was determined by applying 85Kr groundwater age tracer based on piston flow model and exponential piston flow model for specific samples (Kagabu et al. 2017). According to this report, its residence time in A-1 zone was identified as younger than 20 years. The flow path A-2 is the most major flow line over the studied region. Recharged water from A-1 zone is laterally flowing through highly porous Togawa lava formation towards the discharge zone (Ono et al. 2013) with mean flow velocity of ~ 40 m/day (Kumamoto Prefecture and Kumamoto City 1995). The 85Kr age of this groundwater at the end of A-2 is estimated as ca. 40 years (Kagabu et al. 2017). Remaining flow towards coastal plain presents in A-3 zone. This groundwater is almost stagnant with its age determined as > 55 years (Kagabu et al. 2017).
The B–B′ flow line is geographically separated from the major flow line A–A′ and can be divided into three zones (Figs. 1, 3). The size of groundwater systems in this flow line is volumetrically minor compared to that in A–A′ (Fig. S1) but it’s also important for water supply in the vicinity areas. The waters are recharged by precipitation through Ueki highland in the recharge zone (B-1). Waters in this zone generally show younger groundwater age (> 20 years; Kagabu et al. 2017) and this feature is similar to that found in A-1 zone. Then waters pass through basement uplifting area towards the entrance of Kumamoto plain, which area we defined as B-2 zone. The groundwater residence time in this zone show relatively old (> 55 years), due to attenuation in groundwater flow velocity (Kagabu et al. 2017). B-3 is south-westward flow to the coast. Most of groundwaters in A–A′ line emerge at Ezu Lake; those flows of B–B′ line extend towards coast (Fig. S1). In both A-3 and B-3, i.e., the stagnant zones, groundwater residence time is estimated as > 55 years (Kagabu et al. 2017).
As mentioned before the groundwaters can be mixed with surface water in mid-stream of the Shira River where downward fluxes predominate. In addition, mixing of waters from other sources may be occurred in midsection of the major groundwater flow lines including contribution of mountain aquifer waters and water percolations from the first aquifer to deeper second aquifer at where aquicludes between these two aquifers are not distributed. However, in the lateral flow and stagnant zones active groundwater interactions are not expected across two different aquifers. Rather, groundwaters in these areas flow along major flow lines without significant additional water mixing in general that are controlled by regional geographical and geological conditions. Groundwater geochemical evolutions are assessed qualitatively along these well known flow structures.
Methods
Sampling
Groundwater samples were collected from 95 wells along the two major flow lines A–A′ and B–B′, both for the first (well depths 4–80 m) and second aquifers (well depths 44–211 m) (Fig. 1). 58 samples were collected from the second aquifer while 37 samples were collected from the first aquifer. From the flow line point of view, 64 wells are distributed along the A–A′ flow line and 31 wells are distributed along the B–B′ flow line. Most of the wells are observation wells governed and managed by Ministry of Land, Infrastructure and Transport of Japan, Kumamoto Prefectural Government and Kumamoto City. Because of city water supply, most of the observation wells are located along A–A′ flow line and B–B′ flow line has limited number of wells. Besides these observation wells, 35 samples were collected from private wells to complement geographical sampling distributions which are used for domestic purposes and owned by local peoples. Most observation wells have single screen at levels 5–10 m higher than the well depth with its length of ca. 5–10 m, but some wells have 2 or 3 screens in the different depths. Private wells for the second aquifer follow the same well structures but those for the first aquifer are dug wells without screens.
Samples from private wells were collected during July 2011 while all other samples were collected during October–November 2011. Beside these, a total of 30 spring and 11 river samples were also collected (Fig. 1) at around the study area during the same periods. 8 springs are located in the plateaus of Kikuchi and Takayubaru highlands, while 6 springs are situated around Kinpo Mountain. Other 7 spring samples were collected within the Aso caldera watershed. 7 river samples were collected from Shira River from upstream to the downstream and 4 samples were collected from Midori River. Groundwater samples were collected using either existing electric pump or portable pump (MP1, Grundfos, Denmark). During pumping groundwater was continuously monitored with field meter (HORIBA D-54, Japan) for temperature, electrical conductivity (EC), pH, dissolved oxygen (DO), and oxidation reduction potential (ORP). Sample was collected according to standard protocol (Wood 1981), when all the physical parameters were almost at constant values generally in 30–60 min after flushing out of stagnant water in the boreholes.
Laboratory analysis
All samples were filtered through 0.2 μm cellulose acetate filters and collected in a pre-rinsed high-density polyethylene bottles without any headspace. Samples for cations and trace metals were collected in a 100 ml bottle and immediately acidified with 6 N ultrapure HNO3 to produce pH < 2 for stabilization of trace metals. Samples for alkalinity, anion and dissolved silica (SiO2) measurement were collected into a separate 100 ml bottles without preservation. Samples for stable isotopic analysis (δ18O and δ2H) were collected in a 50 ml bottle. The samples were stored in a cooler box at a temperature of < 4 °C and transferred to the laboratory and stored in a refrigerator (< 4 °C) before analysis (usually < 1 week).
All inorganic and stable isotope ratios analyses were carried out at the Hydrology Laboratory of Kumamoto University, Japan following standard analytical procedures (APHA-AWWA-WEF 2012). Alkalinity was measured within the same day of sampling by volumetric titration using 0.05 N H2SO4 and bromocresol green methyl red indicator. The redox potential values in this study have not been corrected to the standard hydrogen electrode, hence, can be used as relative values. Major cations (Na+, K+, Ca2+ and Mg2+) and anions (Cl−, SO42− and NO3−) were measured by ion chromatography (Compact IC 761, Metrohm, Switzerland) within few days of sampling. The analytical precision of major ion analysis is around 3%. Dissolved SiO2 was measured by molybdenum ammonium colorimetric method at a wavelength of 410 nm with a microplate spectrophotometer (Multiskan™ GO, Thermo Fisher Scientific). The accuracy of the chemical analysis was validated by calculating ion balance errors when ion concentration expressed in meq/L (Appelo and Postma 2005) and it was better than ± 5% for all samples except 7 samples in which it was around 6–7%. Trace metals were analyzed by ICP-MS (NexION 300, Perkin-Elmer Co., Ltd, USA).
The stable isotope ratios of water molecule (δ18O and δD) were measured by an isotope ratio mass spectrometer (Delta V Advantage, Thermo Fisher Scientific, USA). Samples were prepared using H2O-CO2 equilibrium method for δ18O and H2O-H2 equilibrium followed by chromium reduction method for δD. Isotopic ratios are expressed as relative differences (δ values) from Vienna standard mean ocean water (V-SMOW) in parts per mill (‰) (Craig 1961) which are represented by δ (‰) = (Rsample/RV-SMOW – 1) \(\times \) 1000 and R is the ratio of 2H/1H or 18O/16O in either sample or standard. Based on replicate measurements of standards and samples, the analytical precisions for δ18O and δD were better than ± 0.05 and ± 0.5‰, respectively. All analytical results are provided in Tables S2 and S3. Note that δ18O and δD data except for river water samples from Midori River were presented in the recent article (Hosono et al. 2020) that discussed coseismic hydrological change mechanisms after the 2016 Kumamoto earthquake, which research aim was different from our current study.
To identify type of fluid-mineral reactions, the geochemical computer code PHREEQCI (version 2, Charlton and Parkhurst 2002), a complete Windows-based graphical user interface to the geochemical computer program PHREEQC (version 2, Parkhurst and Appelo 1999) operating with accompanying WATEQ4F database, was used to calculate mineral saturation indices and aqueous speciation.
Results and discussion
Origin of groundwater
Figure 4 shows the variation of δ18O and δD compositions of different water samples collected from the study area. Such diagram can provide valuable information on the origins of groundwater (e.g., Gat and Gonfiantini 1981). The most groundwaters show compositions close to the summer local meteoric water line (LMWL) indicating their meteoric origin and implying that summer rainfall is the dominant source of the groundwaters. Kudo et al. (2013) studied in a forested catchment in western slope of the Aso caldera mountains and inferred that major recharge occurs during the summer rainfall, corresponding to our results. In detail, regional aquifer waters fall on compositional areas within certain ranges (Fig. 4) reflecting mixing of waters with different origins.

Bivariate plot of δD and δ18O for river, spring and aquifer waters collected from the study area. Local meteoric lines and compositions of soil pore waters are after Okumura et al. (2018). The global meteoric water line is after Gat and Gonfiantini (1981). Average compositions of 100 core soil waters (10 m deep, 10 cm interval) are shown for four soil cores (Okumura et al. 2018), which sampling locations are shown in Fig. 1
On Fig. 4, pore water samples collected from unsaturated soils in recharge areas are plotted on the compositional area to the right of local summer meteoric line, which isotopic shift can be explained by evaporation at soil surface before infiltration (Okumura et al. 2018). In contrast, spring waters collected outside from Aso caldera show compositions that are on the left of this line. Furthermore, all river waters and spring waters within Aso caldera watershed show most depleted δ18O and δD compositions reflecting their recharge sources with relatively higher elevation. Groundwater samples generally show intermediate compositions among these three end-components, soil pore waters, surrounding mountain aquifer waters, and the Shira River waters (Fig. 4). It is thus reasonable to think that aquifer waters are generally originated from summer rainfall and through multiple recharge pathways, soil water infiltrations, mixing with mountain aquifer waters, and surface water percolations (Hosono et al. 2020).
At a scale of flow line, a major focus of this study, there is no significant difference in terms of range of isotopic compositions between A–A′ and B–B′ lines (Tables S2 and S3): average values of δ18O and δD for A–A′ and B–B′ flows are − 7.1 and − 46.3‰ and − 7.0 and − 46.9‰, respectively. This similarity could be attributed to common origins for both regional flow systems. However, δ18O and δD values of the water samples change along the flow lines that was typically found for A–A′ flow line at mid-stream area of the Shira River in A-1 zone and some part of deep aquifer in A-2 zone and sporadically for groundwaters in B–B’ line. This is due to a mixing process of waters from different origins in the midsection of entire flow lines as discussed in later section.
Hydrochemical characteristics
The principal characteristics of geochemical compositions for river, spring and aquifer waters, in terms of both concentrations and stable isotopes, are listed in Tables S2 and S3, where groundwater results are arranged in order of distance from recharge through discharge to stagnant zones (Fig. 1). Groundwater temperatures ranged from 18.0 to 25.0 °C in A–A′ flow and 17.4 to 25.3 °C in B–B′ flow lines. On the other hand, spring water temperatures are lower than those of groundwaters with a range of 13.5–21.0 °C. An average temperature of river waters was 22.4 °C. Piper diagrams (Piper 1944) show that groundwaters from the study area have a wide range of chemical compositions (Fig. S2). All waters from the study area are broadly classified into four dominant and two mixed water types (Table S2) including Ca-HCO3, Ca-SO4, Na-HCO3, Na-Cl, Ca–Mg–Na-HCO3 (mixed cation) and Ca–(Na)-NO3–HCO3 (mixed anion) types. In general, groundwater chemistry of the first and second aquifers is similar each other (Fig. S2 and Tables S4 and S5); however, it differs along the flow paths.
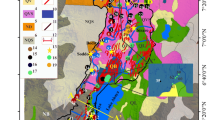
Spatial distribution of hydrochemistry types in the study area is shown in Fig. 5. In the A–A′ flow line groundwaters in the A-1 zone are mainly of Ca-HCO3 type. In general, this type of waters is considered as juvenile waters in groundwater evolution model which are at their early stage of geochemical evolution (e.g., Plummer et al. 1990; Edmunds and Smedley 2000). Groundwaters with rapid circulation process and short residence time, which have not experienced a prolonged water–rock interaction, may also have similar characteristics (e.g., McKenzie et al. 2001). However, groundwaters in active flow line A-2 are mainly of Ca–Mg–Na-HCO3 type, and in the discharge area (A-3), at the end of A–A′ flow line, they became Na-HCO3 type.

Map showing spatial distribution of groundwater types expressed by stiff diagrams
Similarly, groundwaters in the B–B′ flow line begin as Ca-HCO3 type in recharge zone (B-1) followed by Ca–Na-HCO3 type, then flows as Na-HCO3 type in B-2 and B-3 zones (Fig. 5 and Table S2). However, the last three samples in B–B′ flow line are Na-Cl type (Table S2; K16, K35 and K15) which are situated in a reclaimed land at the end of the flows near the coast (Figs. 1, 2) with TDS values ranging 500 to 2,230 mg/L. It has been revealed from nitrate tracers that effect of contaminations through surface soil water infiltrations predominates in recharge areas (Fig. 5, shown as “nitrate dominant” type water) and is limited in lateral flow and discharge zones for both flow lines (Hosono et al. 2013). Thus, observed hydrochemical evolutions from Ca-HCO3 type in recharge areas to other chemistry types in downstream areas are results of several possible processes including silicate mineral weathering, cation exchange, anaerobic degradation of organic matters, deep fluid contributions, and mixing of saline waters.
Water discharging from springs as well as groundwaters in and around recharge zones (A-1 and B-1) are characterized by low electrical conductivities (~ 180 μS/cm) and a predominating Ca-(Mg)-(Na)-HCO3 water type (Tables S2 and S3). However, some waters are enriched in NO3−, due to contaminations from farms, crop lands and orchards in the Ueki, Kikuchi and Takayubaru highlands (Fig. 5). On the contrary, spring waters in Aso Mountain area are dominated with Ca-SO4–(HCO3) type. High sulfate in spring and groundwaters in the Aso area are mainly of hydrothermal origin (Hosono et al. 2018). Since the Aso springs are the main source of Shira River head water, sulfate is one of the dominant anions in this river apart from HCO3− (Ca-SO4 type) though the Midori River is Ca-HCO3 type water.
Changes in each measurement parameter along the flow lines
The trend of chemical constituents as well as isotopes in the Kumamoto groundwaters follows a progressive sequence of hydrochemical evolution. Here, changes of each measurement parameter along the flow lines A–A′ and B–B′ are described and possible causes to explain these changes are documented based on the results from this study and taking previous reports on anaerobic microbiological reactions deduced by multiple isotope ratios (δ13C, δ15N and δ34S) (Hosono et al. 2013, 2014), trace metals (Hossain et al. 2016a, b), and PCR analysis (Zeng et al. 2016) for anions.
The depletion in both δ18O and δD of groundwater at last half part of A-1 zone (Fig. 6) implied that waters were largely recharged from isotopically depleted Shira River while rapid increase in both compositions in the entrance of A-2 indicate predominant contribution of mountain aquifer waters (Fig. 4). Then slight decrease in both isotope ratios afterwards implies increasing contribution of the mixing of Shira River water along the flow. The effect of river water infiltration on hydrochemistry was most effectively traced in SO42− (Fig. 6): it increased in the end of A-1 zone due to strong mixing and after the sudden depression due to the mixing with diluted mountain waters then increased again along the flow in A-2 zone with additional mixing with high SO42− river waters, corresponding well with isotopic results. Similar depleted isotopic and high SO42− anomalies are detected locally at the end of B-2 zone along B–B′ flow near the Shira River, suggesting the occurrence of the same river water infiltration in this limited location. Another isotopic anomaly, an enriched δ18O compared to relatively depleted δD (Fig. 6), confirmed in A-2 zone and some spots on B–B′ flow line, possibly corresponds to soil porewaters (Fig. 4). This may be attributed to surface soil water percolations at local scale via preferential pathways (Kawagoshi et al. 2019); however, simultaneous anomalies were not detected in other geochemical parameters.


Changes in measurement parameters along groundwater flow A–A′ and B–B′. X axis represents the relative distance of sampling location from principal recharge area (Table S2). \({\text{Na}}_{{{\text{corr}}}}^{ + }\) means sodium corrected with input of marine salts or atmospheric spray and is calculated by the formula Na − (Cl− × 10,600/19,000)
A very homogeneous δ34SSO4 feature (8–10‰) was reported through A–A′ flow line (Hosono et al. 2014) except mixing zone and in the end of stagnant zone where anaerobic sulfate reduction occurs with some fractionated δ34SSO4 of up to 16‰. The source of SO42− in the recharge waters is a mixture of surface water (Shira River water), ecosystem-recycled sulfur near surface and of anthropogenic (chemical fertilizers) origins (Hosono et al. 2014) and we think an another SO42− source input is not prominent through the pathways. Cl− followed almost steady pattern (Fig. 6). Similarly, HCO3− showed constant pattern through the flow (ranging ca. 70–100 mg/L) except one sample from the A-3 zone with its value > 200 mg/L (Fig. 6). In the same A–A′ flow line, homogeneous δ13CDIC compositions (− 21 to − 17‰) were confirmed for majority waters except at the end of stagnant zone where methanogenesis reaction occurs with increased HCO3− and fractionated δ13CDIC of up to − 14‰ (Hosono et al. 2014). Observed δ13CDIC signature for majority waters indicates their overall origin that is ecosystem-recycled CO2 with relatively young fresh mineral materials (e.g., Schulte et al. 2011), but contribution from deep seated and volcanically originated CO2 (e.g., Yamada et al. 2011; Koh et al. 2017) is negligible. Nitrate concentrations (Fig. 6; ~ 50 mg/L, originated from chemical fertilizers applied in the recharge zone; Hosono et al. 2013) are the highest in the A-1 recharge zone but decreased gradually toward the end of A–A′ flow. Significant δ15NNO3 isotopic fractionation was not found through the main flow path except in the end of stagnant zone in A-3 where denitrification occurs with slightly fractionated δ15NNO3 of up to 10‰ (Hosono et al. 2013). NO3− decline along A–A′ main flow path seems to be caused by dilution with both mountain aquifer and Shira River waters. Anion patterns observed in this study can reasonably be explained with a help of previously proposed information on their origins and processes.
In B–B′ flow line, nitrate concentrations sharply decreased along the flow (Fig. 6). It has been confirmed in the same flow line that high NO3− (~ 60 mg/L) in the recharge zone is attributed to nitrogen loads from farm lands and it attenuated by denitrification reaction progressed along the flow path (Hosono et al. 2013; Zeng et al. 2016). The same authors confirmed that decreased SO42− through B-1 to B-2 zone (Fig. 6) is associated with anaerobic sulfate reduction, in contrast, HCO3− increased along the flow B–B′ line (Fig. 6), which can be interpreted as it was caused by microbiological respiration through denitrification, sulfate reduction and methanogenesis. More significant anaerobic microbiological activities observed along B–B′ flow line may be facilitated by relatively higher dissolved organic carbon content in the aquifer (Hosono et al. 2015) and/or slower flow rate than A–A′ groundwater flow (Hosono et al. 2014). Cl− concentrations are generally constant in these zones. However, both Cl− and SO42− concentrations increased in stagnant zone (B-3) resulting from mixing of pore waters of marine origin hosted in the Ariake clay sediments (Hosono et al. 2014; Hossain et al. 2016a, b). Exceptionally, Cl− increased sharply (both up to 70 mg/L) in the end of B-1 zone (Fig. 6). The highest groundwater temperature anomaly (~ 23 °C) was depicted only in the end of B-1 zone over the study area (AIST 2014) and many hot spring facilities are developed in this spot. We thus assume that localized deep fluid seeps in this location, which possibility is supported by simultaneous increase in Na+ (Fig. 6).
For cations, significant concentration changes are observed partly associated with the degree of water mineralization along the both flow lines (Fig. 6). For instance, Na+ shows a gradual increasing tendency along the B–B′ flow with the median values of 19, 23 and 121 mg/L in B-1, B-2 and B-3 zones, respectively. Na+ concentrations are relatively constant in A-1 and A-2 zones while little increased at the end of A-3 zone for A–A′ line. Ca2+ shows steady state concentrations in initial two zones of both A–A′ and B–B′ flow lines and decreasing trends in the final part of the both flow lines except last three samples in B–B′ flow. Mg2+ generally follows similar trend with Ca2+ except in A-3 zone where it shows reverse trend. Roughly but gradual decreasing in SiO2 along both flow lines (Fig. 6) is probably due to precipitation of silica phases along the flow lines. At the end, K+ showed increasing concentrations in the recharge areas (Fig. 6), possibly due to the impact of both mineral weathering and dissolution of agrochemicals applied.
Overall, it can be summarized that groundwater characteristics along A–A′ flow line are mainly of aerobic environment and mixed cation (Ca2+, Mg2+ and Na+) dominated with no drastic variations along the flow except in A-1 zone where groundwater chemistry is heterogeneous due to mixing of different source waters. In contrast, B–B′ flow can be characterized evolving as Na-dominated, Ca-poor, high pH and high alkalinity groundwater along the flow line with strong reducing activity. In contrast to the variations in the groundwater chemistry along flow directions, no obvious gap was observed between first and second aquifers.
Mineralization processes
Alumino-silicate minerals are the most reported rock-forming minerals in the study filed (Table S1). On Fig. S3, most data tend to plot within the domain of global average of silicate weathering, suggesting that incongruent dissolution or leaching of the siliceous volcanic rocks is the major controlling process responsible for the formation of groundwater chemistry. Additional analysis using mass-balance calculations were provided in supplementary material (see ‘mass-balance calculations’). The extent of weathering and leaching depends on the chemical and mineralogical compositions of the host rocks. Following Herczeg (2001) and Appelo and Postma (2005), incongruent dissolution reactions of silicate weathering in the study area can be hypothesized as follows that are applicable for entire aquifers:
Weathering rate of plagioclase and mafic minerals is significantly faster than that of quartz and K-feldspar (White et al. 1998). Hence, if plagioclase has a larger modal abundance than mafic minerals, groundwater chemical compositions can largely be explained by plagioclase weathering and leaching tends to be occurred in order of Ca2+ > Na+ > Mg2+ (Mukherjee et al. 2009) as the reaction proceeds. Therefore, Ca-HCO3 and Ca-Mg-Na-HCO3 would be the major groundwater type in such area. According to Garrels and Mackenzie (1967), hydrochemical evolutions in igneous rocks are dominated by hydrolysis of plagioclase by CO2 and kaolinite is often an early weathering product which was accounted for ca. 80% of the total weathering capacities in Sierra Nevada spring systems. In addition, a presence of any volcanic glasses enhances weathering rate of the rocks in ca. 10 times faster than the crystalline basaltic rocks (e.g., Gislason and Eugster 1987) and Na+ releases preferentially over Ca2+. In the study area, plagioclase phenocryst has the highest modal abundance, around 3–43% (Table S1), followed by pyroxenes (1–25%), and crystalline glass is the most common groundmass (see “Topography, climate, geology and mineralogy” and Table S1).
Enrichment of Na+ is one of the principal geochemical features of the Kumamoto groundwater. These excess Na+ are shown in \({\text{Na}}_{{{\text{corr}}}}^{ + }\)/Ca2+ and \({\text{Na}}_{{{\text{corr}}}}^{ + }\)/Mg2+ changes along flow paths (Fig. 6), which could be explained in two different ways, either weathering of silicate minerals or cation exchange reaction (Maybeck 1987; Plummer et al. 1990). It was proposed that the molar ratio of dissolved Na+/Ca2+ in groundwater falls approximately equal to 1 in the common plagioclase phenocryst dominated volcanic regions (Garrels and Mackenzie 1967). Figure 6 shows all the groundwaters from A–A′ flow line have the molar ratio equal or less than 1 except few samples from A-3 zone with the molar ratio of > 1, which results entirely consistent with congruent dissolution of plagioclase. In the same zones (A-1 and A-2), groundwater chemistry evolved from Ca-HCO3 type to Ca–Mg–Na-HCO3 type along the flow path (Fig. 5) that should be caused by the weathering of minerals containing Mg, i.e., pyroxenes, the second important host minerals over the aquifer systems, in addition to plagioclase weathering.
In the B–B′ flow line, however, the molar ratio \({\text{Na}}_{{{\text{corr}}}}^{ + }\)/Ca2+ for the samples from B-1 and B-2 zones (with pH < 8) was approximately or slightly over than 1. Na+ is accumulating to the groundwater and Ca2+ is being depleted with increasing pH nearly or above 8 (Fig. 6). Observing variations in \({\text{Na}}_{{{\text{corr}}}}^{ + }\)/Ca2+ ratio along the flow lines B–B′ and end of A–A′, we can postulate for another hydrochemical evolution process taking place especially in the stagnant zone. These phenomena can be described in two different ways, addition of Na+ originated other than plagioclase weathering or Ca2+ removal via cation exchange. Dissolution of volcanic glasses that are not described but maybe present in the study field may cause observed Na+ elevations. However, this phenomenon should occur in earlier stage of weathering sequence than plagioclase weathering. Thus former hypothesis is unlikely to explain observed hydrochemical signatures, although we agree the occurrence of volcanic glass dissociation itself but it might have been completed during time elapsing after the last volcanic eruptions.
Figure 7 expresses the influence of cation exchange process. In this figure, concentrations of divalent cations (Ca2+ + Mg2+) that may have been involved in exchange reactions are corrected by equivalent concentrations of associated anions (HCO3− + SO42−). Again, the amount of Na+ is corrected by Cl− to account for Na+ that is solely from the aquifer geology. If cation exchange is a significant controlling process, then the relation between these two sets of parameters should be linear with a slope of ~ − 1 (Fisher and Mullican 1997). Figure 7 shows that samples from A-3 zone and B–B′ flow line (except two high salinity samples) having a trend line (r2 = 0.97) with a slope of − 0.98, which is close to -1, indicating that cation exchange has an important control on those groundwaters. We further confirmed that reverse ion exchange processes would not expect to be occurred based on (Ca2+ + Mg2+)–(Na+ + K+) vs HCO3−–(SO42− + Cl−) classification diagram (Fig. S6). To support cation exchange mechanism, there would need to exist sodium-charged cation exchange media within the rock unit to facilitate the process. It is already described that Kumamoto plain area is covered by marine and flood sediments which partly contains clays with saline pore waters with Na+. Moreover, in B-2 zone aquifers are covered by wet-land composed of clay materials. Both areas are characterized by relatively long (> 55 years) groundwater residence time. It is plausible therefore that Ca2+ and Mg2+ in the water exchanged with previously absorbed Na+ on the surface of the clay minerals (Domenico and Schwartz 1990) with relatively later stage of geochemical evolutions. Several researchers studied similar characteristics in groundwater systems and reported calcite and plagioclase dissolutions followed by cation exchange (Edmunds 1981; Appelo and Postma 2005; Mukherjee et al. 2009).

Relation between (Ca2+ + Mg2+) – (HCO3− + SO42−) against (Na+ – Cl−) to indicate the cation exchange reactions for groundwater samples collected from A-3 flow zone of the A–A′ line and B-1 to B3 zones of the B–B′ line
Thermodynamic interpretation of water–rock interactions
Saturation indices (SI) can be used to investigate thermodynamic control on groundwater chemistry and to represent the possible reacting mineral phases. It is helpful to evaluate the states of mineral equilibria in the groundwater system and to identify the reacting mineral phases in absence of accurate mineralogical data of the aquifer matrix. The SI of a particular mineral is defined as:
where, IAP is the ion activity product of the mineral–water reaction and KT is the equilibrium solubility constant at a given temperature for particular samples. SI value of a specific mineral nearly zero indicates that the groundwater system is in equilibrium with this specific mineral. Whereas, SI > 0 or SI < 0 specifies groundwater is oversaturated or undersaturated that means minerals favorably precipitate or dissolve, respectively. The average state of saturation of the groundwater in different zones along the flow lines with respect to selective minerals is shown in Table 1. Hydrochemical data except Al3+ shown in Table S2 were used to calculate the SI value. We do not have complete set of Al3+ data for all samples from present study and thus used average Al data as per flow lines from different set of samples as input data (A–A′ first aquifer: 147 µg/L, A–A′ second aquifer: 79 µg/L, B–B′ first aquifer: 51 µg/L, B–B′ second aquifer 47 µg/L). Al3+ has a very low solubility in groundwater and tends to be conserved in solid phase (Freeze and Cherry 1979). There is a possibility of the presence of colloidal particles even though the samples were filtered through 0.2 μm membrane filter. Furthermore, in higher concentrations, Al3+ is sensitive to complexation with other dissolved species especially with organic compounds. A number of samples from Kumamoto plain area contains relatively high Al3+, indicating inorganic conservation of aluminum (Cleaves et al. 1970). Therefore, a part of dissolved aluminum in solution may be organically bounded and should not be included in calculations of saturation index. However, calculated SI values were not significantly affected with a possible uncertainty of these Al3+ concentrations due to their trace concentrations relative to other major ions.
It is clear from Fig. 8 and Table 1 that groundwaters in the Kumamoto basin are undersaturated with respect to reported alumino-silicate minerals. The calculated SI values for albite and anorthite, diopside, and phlogopite (a micaceous mineral chemically similar to biotite) remain strongly negative along the flow lines showing that the groundwater is undersaturated with respect to these minerals (Table 1; Fig. 8). It is notable that albite and anorthite tend to reach saturation along the flow paths at the end of A-3 and in B-3 zones, suggesting that the mineral saturation and subsequent precipitation of secondary minerals, i.e., smectite as explained later, could occur. Like most natural waters (e.g., Hem 1989), groundwaters are oversaturated with quartz (0.05 < SI > 1.05) and undersaturated with amorphous silica (− 1.26 < SI > − 0.27). In addition, they are in equilibrium to little oversaturated condition with respect to cristobalite and chalcedony phases. Thus silica in amorphous form is likely a major contributor in the groundwater systems. All groundwater samples are undersaturated with respect to gypsum (− 4.6 > SI < − 1.98) and halite (− 9.18 > SI < − 4.76) (Table 1). The soluble sulfate and halite minerals are not present in the host rocks. Presence of high concentrations of sulfate in A-1 zone water and Na-Cl type groundwater in B-3 zone are originated by a mixing of Shira River water and high saline pore waters in marine clay sediments, respectively. Groundwater remains close to or somewhat above saturation with respect to gibbsite, whereas, it becomes significantly oversaturated with respect to secondary minerals of weathering products including kaolinite, smectite and illite (Fig. 8; Table 1).

Variations in saturation indices for selected rock-forming minerals and clay minerals along the A–A′ and B–B′ flow lines. Thermodynamic values were calculated by using PHREEQCI with wateq4f.dat data base
Figure 9 shows mineral stability diagrams for the Na2O–Al2O3–SiO2–H2O and CaO–Al2O3–SiO2–H2O systems. These figures were generated by assuming end-member compositions using the equilibrium relationship of Tardy (1971) for standard temperature and pressure (25 °C and 1 atm). The groundwater samples from A–A′ flow were plotted mainly on the kaolinite stability field while some samples fall on boundary filed between kaolinite and smectite. On the contrary, most of the samples from B–B′ flow line was placed in the stability field of smectite and few samples followed boundary area. Groundwaters are oversaturated for both minerals for both flow lines and thus equilibrium with kaolinite or smectite is an important process that controls groundwater chemistry. However, some mineralogical study revealed that halloysite is the major clay mineral formed in resulting from plagioclase weathering in some outcrops of the study field (Aomine and Wada 1962). It is difficult however to assess this issue thermodynamically because of the lack of stability diagram involving halloysite and the result showing groundwaters are generally undersaturated with respect to this type of clay minerals (Table 1). Thus here we only state halloysite presented presumably as a major weathering product under humid and subtropical climate like in Kumamoto.

Stability diagram of a Na2O–Al2O3–SiO2–H2O and b CaO–Al2O3–SiO2–H2O systems showing minerals expected to be in thermodynamic equilibrium with groundwater samples. Dashed lines represent saturation line of quartz and amorphous silica, respectively. Thermodynamic data are taken from Tardy (1971). Legend: A–A′ first aquifer (open triangle), A–A′ second aquifer (solid triangle), B–B′ first aquifer (open circle), and B–B′ second aquifer (solid circle)
It has been reported that availability of excess silica from relatively high rate of chemical weathering of basaltic rocks prevailed in humid tropical climate favors kaolinite stability while sample from granitic area with arid climate tends to be plot in smectite field which reaction occurs in later stage of weathering process with longer water–rock contact time (Appelo and Postma 2005; Stallard and Edmond 1987). It is thus reasonable to think that more stability of A–A′ flow samples in the kaolinite field indicates that groundwater is well drained, whereas, samples plotting in smectite field from B–B′ flow suggest restriction in groundwater flow. This is in good agreement with groundwater flow regime in this area where waters in A-1 and A2 zones have shorter residence time (< 40 years) with active hydrological cycle than B–B′ flow line with longer residence time of > 55 years (Kagabu et al. 2017). The diagrams (Fig. 9) depicted greater maturity of waters in B–B′ than A–A′ lines in hydrochemical evolution. This assumption is supported by simultaneous saturation in plagioclase (Fig. 8) and decline in SiO2 concentrations of matured waters (Fig. 6), which accompany consumption of silica in forming smectite to be precipitated. These results correspond well with recent report in other volcanic areas (e.g., Ide et al. 2018).
Summary and implications
Geochemical weathering processes were studied to describe comprehensive hydrobiogeochemical evolutionary model for volcanic aquifer systems in Kumamoto area along two well-defined groundwater flow lines A–A′ and B–B′ from land to ocean (Fig. 10). Groundwaters in this area are recharged largely by summer precipitation through soil infiltration at plateaus and percolation of waters from surrounding mountains and locally through river waters. Impact of agrochemical applications predominates in recharge areas for both flow paths, most clearly shown by elevated NO3− concentrations (~ 60 mg/L). However, major water type (Ca-HCO3 type) in these areas is controlled by mineral decomposition mainly of plagioclase to form secondary minerals, i.e., kaolinite (however, it is highly probable as it presents as halloysite), through atmospheric CO2 mediated volcanic rock weathering.

In the A–A′ flow path, recharged waters flow laterally with enhanced weathering mainly of plagioclase and secondary of pyroxenes but without significant anaerobic microbiological reactions, resulting Ca–Mg–Na-HCO3 type waters that are discharged in the entrance of plain as springs in Ezu Lake. Relatively active groundwater flow with short residence time (< 40 years) facilitates this type of water evolutions. However, some stagnant waters remain in the plain, which are defined as Na-HCO3 type with depleted NO3−, due to combined processes including ion exchange and anaerobic denitrification. At this stage of geochemical evolution, plagioclase decomposition is not important but smectite precipitation predominates in association with SiO2 saturation. This type of waters is formed in reducing environments under inactive flow regime with long water–rock/sediments contact time, at least, over 55 years.
Groundwaters in B–B′ line evolve their geochemical signatures to Na-HCO3 type with depleted SiO2, NO3− and SO42− but elevated HCO3− in the middle of lateral flow zone due to progressive ion exchange reaction, smectite precipitation, and anaerobic microbial succession from denitrification until methanogenesis under extensive anoxic aquifer setting. Attenuation of the active groundwater flow (groundwater residence time > 55 years) in thinning aquifer systems at basement uplifting area enhanced water–rock-microbiological interaction in steady state. Then waters are transported towards the coast maintaining similar hydrobiogeochemical signatures in upgradient, however, with significant contribution of pore waters in marine clay sediments with high salinity. Mixing of these high-salinity and biochemically evolved pore waters of marine origin further alters original water characters to intensify ion exchange reaction that is triggered by clays with large amount of Na+ attachment. Finally, water changes to Na-Cl type due to strongest interaction with sea water components in the coast.
We understand that additional assessments with combined thermodynamics and kinetic control and inverse geochemical modeling may provide further insights into proposed scenario; these tests favorably require more data (e.g., concentrations of gas phases in water such as N2, NH3 and H2S and organic compounds like dissolved organic carbon) and additional analyses (e.g., definition of confined flow systems without any water mixing along the flow line, identification of initial and final groundwater chemistry) and thus could not be included in this study. Nevertheless, this study quantitatively illustrates how groundwater chemistry could evolve along its flow path in volcanic aquifers in regional scale at given time scales in consequence of multiple processes, i.e., different source inputs, water–rock interaction, contaminations and microbial activities. This study can serve as an important basis for understanding groundwater chemical evolutions in active groundwater flow systems, such as volcanic and sedimentary fan aquifers, and for management and protection of groundwater resources.
References
AIST (2014) Water environmental map. no. 7 Kumamoto area. Geological Survey of Japan, National Institute of Advanced Industrial Science and Technology
Aomine S, Wada K (1962) Differential weathering of volcanic ash and pumice, resulting in formation of hydrated halloysite. Am Miner 47(9–10):1024–1048
APHA-AWWA-WEF (2012) Standard methods for the examination of water and waste water, 22nd edn. American Public Health Association, Washington, DC
Appelo CAJ, Postma D (2005) Geochemistry, groundwater and pollution, 2nd edn. CRC Press, Boca Raton
Aramaki S (1984) Formation of the Aira caldera, southern Kyushu, c. 22 000 years ago. J Geophy Res 89:8485–8501
Aramaki S (2002) Studies on the groundwater conservation of KITAAMGI spring water in KASHIMA. Report of research project, Grant-in-Aid for Scientific Research, 12680582
Charlton SR, Parkhurst DL (2002) PhreeqcI–a graphical user interface to the geochemical model PHREEQC: US Geological Survey, Fact Sheet FS-031-02, 2
Cleaves ET, Godfrey AE, Bricker OP (1970) Geochemical balance of a small watershed and its geomorphic implications. Geol Soc Am Bull 81:3015–3032
Cloutier V, Lefebvre R, Savard MM, Bourque É, Therrien R (2006) Hydrogeochemistry and groundwater origin of the Basses-Laurentides sedimentary rock aquifer system, St. Lawrence Lowlands, Québec, Canada. Hydrogeol J 14(4):573–590
Craig H (1961) Isotopic variations in meteoric waters. Science 133:1702–1703
Domenico PA, Schwartz FW (1990) Physical and chemical hydrogeology. Wiley, New York
Drever JI (1997) The geochemistry of natural waters: surface and groundwater environments. Prentice-Hall, Englewood Cliffs
Edmunds WM (1981) Hydrogeochemical investigations. In: Lloyd JW (ed) Case studies in groundwater resources evaluation. Oxford Science Publications, Oxford, pp 87–112
Edmunds WM, Smedley P (2000) Residence time indicators in groundwater: the East Midlands Triassic sandstone aquifer. Appl Geochem 15:737–752
Fisher RS, Mullican WF (1997) Hydrochemical evolution of sodium-sulfate and sodium-chloride groundwater beneath the Northern Chihuahuan Desert, Trans-Pecos, Texas, USA. Hydrogeol J 5(2):4–16
Freeze RA, Cherry JA (1979) Groundwater. Prentice Hall, Englewood Cliffs
Garrels RM, Mackenzie FT (1967) Origin of the chemical composition of some springs and lakes. In: Gould RF (ed) Equilibrium concepts in natural water systems, advances in chemistry series, vol 67. Amer. Chem. Soc, Washington, D.C., pp 222–242
Gat JR, Gonfiantini R (1981) Stable isotope hydrology: deuterium and oxygen-18 in the water cycle. IAEA Technical Report No. 210, p 339
Gislason SR, Eugster HP (1987) Meteoric water–basalt interactions. II: a field study in N.E. Iceland. Geochim Cosmochim Acta 51(10):2841–2855
Hem JD (1989) Study and interpretation of the chemical characteristics of natural water. US Geol. Surv. Water-Supply Paper, 2254
Herczeg A (2001) Can major ion chemistry be used estimate groundwater residence time in basaltic aquifer. In: Cidu R (ed) Proc. 9th internat. symp. water–rock interaction. A.A. Balkema, Rotterdam, pp 529–532
Hosono T, Tokunaga T, Kagabu M, Nakata H, Orishikida T, Lin IT, Shimada J (2013) The use of δ15N and δ18O tracers with an understanding of groundwater flow dynamics for evaluating the origins and attenuation mechanisms of nitrate pollution. Water Res 47:2661–2675
Hosono T, Tokunaga T, Tsusima A, Shimada J (2014) Use of δ13C, δ15N and δ34S to study anaerobic bacterial processes in groundwater flow systems. Water Res 54:284–296
Hosono T, Lin I-T, Alvarez K, Morimura S, Zeng X-X, Mori K, Tawara Y, Matsunaga M, Hossain S, Shimada J (2015) Research trend and direction for groundwater nitrate pollution study: an insight from Kumamoto area. J Groundw Hydrol 57(4):439–465 (in Japanese)
Hosono T, Hartmann J, Louvat P, Amann T, Washington KE, West AJ, Okamura K, Böttcher ME, Gaillardet J (2018) Earthquake-induced structural deformations enhance long-term solute fluxes from active volcanic systems. Sci Rep 8:14809
Hosono T, Yamada C, Shibata T, Tawara Y, Wang C-Y, Manga M, Rahman ATMS, Shimada J (2019) Coseismic groundwater drawdown along crustal ruptures during the 2016 Mw 7.0 Kumamoto earthquake. Water Resour Res 55(7):5891–5903
Hosono T, Yamada C, Manga M, Wang C-Y, Tanimizu M (2020) Stable isotopes show that earthquakes enhance permeability and release water from mountains. Nat Commun 11:2776
Hossain S, Hosono T, Ide K, Matsunaga M, Shimada J (2016a) Redox processes and occurrence of arsenic in a volcanic aquifer system of Kumamoto Area, Japan. Environ Earth Sci 75(9):740
Hossain S, Hosono T, Yang H, Shimada J (2016b) Geochemical processes controlling fluoride enrichment in groundwater at the western part of Kumamoto area, Japan. Water Air Soil Pollut 227(10):385
Hunter AG (1998) Intracrustal controls on the coexistence of tholeiitic and calc-alkaline magma series at Aso Volcano, SW Japan. J Petrol 39:1255–1284
Ide K, Hosono T, Hossain S, Shimada J (2018) Estimating silicate weathering timescales from geochemical modeling and spring water residence time in the Kirishima volcanic area, southern Japan. Chem Geol 488:44–55
Join J-L, Coudray J, Longworth K (1997) Using principal components analysis and Na/Cl ratios to trace groundwater circulation in a volcanic island: the example of Reunion. J Hydrol 190:1–18
Kagabu M, Matsunaga M, Ide K, Momoshima N, Shimada J (2017) Groundwater age determination using 85Kr and multiple age tracers (SF6, CFCs, and 3H) to elucidate regional groundwater flow systems. J Hydrol Reg Stud 12:165–180
Kamata H (1997) Geology of the Miyanoharu district. With Geological Sheet Map at 1:50000, Geological Survey of Japan, p 127 (in Japanese with English abstract)
Kaneko K, Kamata H, Koyaguchi T, Yoshikawa M, Furukawa K (2007) Repeated large-scale eruptions from a single compositionally stratified magma chamber: an example from Aso volcano, Southwest Japan. J Volcanol Geotherm Res 167:160–180
Kawagoshi Y, Suenaga Y, Chi NL, Hama T, Ito H, Duc LV (2019) Understanding nitrate contamination based on the relationship between changes in groundwater levels and changes in water quality with precipitation fluctuations. Sci Total Environ 657:146–153
Kenoyer GJ, Bowser CJ (1992) Groundwater chemical evolution in a sandy silicate aquifer in northern Wisconsin: 1. Patterns and rates of change. Water Resour Res 28:579–589
Koh D-C, Chae G-T, Ryu J-S, Lee S-G, Ko K-S (2016) Occurrence and mobility of major and trace elements in groundwater from pristine volcanic aquifers in Jeju Island, Korea. Appl Geochem 65:87–102
Koh D-C, Genereux DP, Koh G-W, Ko K-S (2017) Relationship of groundwater geochemistry and flow to volcanic stratigraphy in basaltic aquifers affected by magmatic CO2, Jeju Island, Korea. Chem Geol 467:143–158
Kudo K, Nagamatsu T, Shimada J, Kabeya N, Tanaka N (2013) Subsurface storage in different land use catchments evaluated by deuterium excess. In: Proceedings of H01, IAHS-IAPSO-IASPEI Assembly, Gothenburg, Sweden, IAHS Publ. 359, pp 371–376
Kumamoto City Waterworks and Sewerage Bureau (2008) Annual report of water quality test 2008, Kumamoto City, p 246 (in Japanese)
Kumamoto Prefecture and Kumamoto City (1995) Integrated Groundwater Survey Report of the Kumamoto Area, Kumamoto Prefecture and Kumamoto City, p 122 (in Japanese)
Kumamoto Prefecture and Kumamoto City (2005) The investigation report for the groundwater preservation and management in the Kumamoto area. Kumamoto Prefecture and Kumamoto City, p 86 (in Japanese)
Kumamoto Prefecture Geological Map Compilation Committee Report (2008) Kumamoto Prefecture geological map (1:100000) and the explanations, Kumamoto Prefecture Industry Association, Geological Survey of Japan, p 118 p
Lipman PW (1967) Mineral and chemical variations within an ash flow sheet from Aso caldera, south-western Japan. Contr Mineral Petrol 16:300–327
Matsumoto A, Uto K, Ono K, Watanabe K (1991) K-Ar age determinations for Aso volcanic rocks-concordance with volcano stratigraphy and application to pyroclastic flows. Progr Abstr Volcanol Soc Jpn 2:73
McKenzie J, Siegel D, Patterson W, McKenzie J (2001) A geochemical survey of spring water from the main Ethiopian Rift Valley, southern Ethiopia: implications for well head protection. Hydrogeol J 9:265–272
Meybeck M (1987) Global chemical weathering from surficial rocks estimated from river dissolved loads. Am J Sci 287:401–428
Miyoshi M, Furukawa K, Shinmura T, Shimono M, Hasenaka T (2009) Petrography and whole-rock geochemistry of pre-Aso lavas from the caldera wall of Aso volcano, central Kyushu. J Geol Soc Jpn 115:672–687 (in Japanese with English abst.)
Mori K, Tawara Y, Tada K, Hosono T, Shimada J, Matsunaga M, Tosaka H (2016) Numerical modeling for simulating fate and reactive transport processes of nitrogen in watershed and discussion on applicability to actual fields. J Groundw Hydrol 58(1):63–86 (in Japanese with English abstract)
Mukherjee A, Bhattacharya P, Shi F, Fryer EA, Mukherjee BA, Xie MZ, Jacks G, Bundschuh J (2009) Chemical evolution in the high arsenic groundwater of the Huhhot basin (Inner Mongolia, PR China) and its difference from the western Bengal basin (India). Appl Geochem 24:1835–1851
Nesbitt HW, Wilson RE (1992) Recent chemical weathering of basalts. Am J Sci 292:740–777
Nordstrom DK, Ball JW, Donahoe RJ, Whittemore D (1989) Groundwater chemistry and water-rock interactions at Stripa. Geochim Cosmochim Acta 53:1727–1740
Okumura A, Hosono T, Boateng D, Shimada J (2018) Evaluations of the downward velocity of soil water movement in the unsaturated zone in a groundwater recharge area using δ18O tracer: the Kumamoto region, southern Japan. Geologia Croatia 71(2):65–82
Ono K, Watanabe K (1985) Geological Map of Aso Volcano (1:50,000). In: Geological Map of Volcanoes 4. Geological Survey of Japan (in Japanese with English abstract)
Ono K, Matsumoto Y, Miyahisa M, Teraoka Y, Kambe N (1977) Geology of the Taketa district. With Geological Sheet Map at 1:50000, Geological Survey of Japan, p 157 (in Japanese with English)
Ono M, Shimada J, Ichikawa T, Tokunaga T (2011) Evaluation of groundwater discharge in Lake Ezu, Kumamoto, based on radon in water. Jap J Limnol 72:193–210
Ono M, Tokunaga T, Shimada J, Ichiyanagi K (2013) Application of continuous 222Rn monitor with dual loop system in a small lake. Groundwater 51:706–713
Parkhurst DL, Appelo CAJ (1999) User's guide to PHREEQC (version 2)—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations: US Geological Survey Water-Resources Investigations Report 99-4259, p 312
Piper AM (1944) A graphic procedure in the geochemical interpretation of water-analyses. Trans Am Geophys Union 25:914–923
Plummer LN, Busby JF, Lee RW, Hanshaw BB (1990) Geochemical modeling of Madison aquifer in parts of Montana, Wyoming, and South Dakota. Water Res Res 26(9):1981–2014
Schulte P, van Geldern R, Freitag H, Karim A, Négrel P, Petelet-Giraud E, Probst A, Probst JL, Telmer K, Veizer J, Barth JAC (2011) Applications of stable water and carbon isotopes in watershed research: weathering, carbon cycling, and water balances. Earth Sci Rev 109:20–31
Stallard RF, Edmond JM (1987) Geochemistry of the Amazon: 3. Weathering chemistry and limits to dissolved inputs. J Geophys Res Oceans 92(C8):8293–8302
Tanaka K, Funakoshi Y, Hokamura T, Yamada F (2010) The role of paddy rice in recharging urban groundwater in the Shira River Basin. Paddy Water Environ 8:217–226
Taniguchi M, Shimada J, Uemura T (2003) Transient effects of surface temperature and groundwater flow on subsurface temperature in Kumamoto Plain, Japan. Phy Chem Earth 28:477–486
Taniguchi M, Burnett K, Shimada J, Hosono T, Wada CA, Ide K (2019) Recovery of lost nexus synergy via payment for environmental services in Kumamoto, Japan. Front Environ Sci 7:28
Tardy Y (1971) Characterization of the principal weathering types by the geochemistry of waters from some European and African crystalline massifs. Chem Geol 7:253–271
Thomas JM, Welch AH, Dettinger MD (1996) Geochemistry and isotope hydrology of representative aquifers in the great basin region of Nevada, Utah, and Adjacent States. US Geological Survey Professional Report. 1409-C
Tomiie K, Iwasa Y, Maeda K, Otsuzuki M, Yunoue T, Kakimoto R, Kawagoshi Y (2009) Present status and future of groundwater contamination by nitrate-nitrogen in Kumamoto City. J Water Environ Technol 7:19–28
Watanabe K (1978) Studies on the Aso pyroclastic flow deposits in the region to the west of Aso caldera, southwest Japan, I: geology of the Aso-4 pyroclastic flow deposits. Mem. Fac. Educ., Kumamoto Univ., Nat. Sci., vol 27, pp 97–120
Watanabe K (1979) Studies on the Aso pyroclastic flow deposits in the region to the west of Aso caldera, southwest Japan, II: petrology of the Aso-4 pyroclastic flow deposits. Mem. Fac. Educ., Kumamoto Univ., Nat. Sci., vol 28, pp 75–112
White AF, Claassen HC, Benson LV (1980) The effect of dissolution of volcanic glass on the water chemistry in a tuffaceous aquifer, Rainer Mesa, Nevada. US Geol. Surv. Water-Supply Paper 1535-Q
White AF, Blum AE, Schulz MS, Vivit DS, Stonestrom DA, Larsen M, Murphy SF, Eberl D (1998) Chemical weathering in a tropical watershed, Luquillo Mountains, Puerto Rico: I. Long-term versus short term weathering fluxes. Geochim Cosmochim Acta 62:209–226
Wood WW (1981) Guidelines for collection and field analysis of ground-water samples for selected unstable constituents. U.S. Geol. Surv. Techniques Water-Resour. Invest. Book 1 (Chapter D2)
Wood WW, Low WH (1986) Aqueous geochemistry and diagenesis in the eastern Snake River plain aquifer system, Idaho. Geol Soc Am Bull 97:1456–1466
Yamada M, Ohsawa S, Kazahaya K, Yasuhara M, Takahashi H, Amita K, Mawatari H, Yoshikawa S (2011) Mixing of magmatic CO2 into volcano groundwater flow at Aso volcano assessed combining carbon and water stable isotopes. J Geochem Explor 108(1):81–87
Zeng X, Hosono T, Ohta H, Niidome T, Shimada J, Morimura S (2016) Comparison of microbial communities inside and outside of a denitrification hotspot in confined groundwater. Int Biodeterior Biodegrad 114:104–109
Acknowledgements
We thank Dr. Kimpei Ichiyanagi in Hydrology laboratory, Kumamoto University for his valuable comments and suggestions during seminar discussions. We are humbly grateful to Dr. Makoto Kagabu in Nagasaki University for extending his help in illustrating the maps and figures for this manuscript. The authors acknowledge Prof. Masaki Hayashi in the University of Calgary, Canada for his informal reviews of previous draft and suggestions to improve the manuscript. We are also thankful to Dr. Heejun Yang and Ms. Midori Matsunaga for their cordial help during field survey and for logistic support. This study was funded by a CREST project (JST: Japan Science and Technology Agency) with additional supports from the JSPS Grant-in-Aid for Scientific Research B (17H01861).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hosono, T., Hossain, S. & Shimada, J. Hydrobiogeochemical evolution along the regional groundwater flow systems in volcanic aquifers in Kumamoto, Japan. Environ Earth Sci 79, 410 (2020). https://doi.org/10.1007/s12665-020-09155-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12665-020-09155-4