
Abstract
The Qinghai-Tibet Plateau is sensitive to climate change, with ecosystems that are important with respect to scientific research. Here high-throughput DNA pyrosequencing was used to assess bacterial diversity within different alpine grassland ecosystems of the Qinghai-Tibet Plateau, China. In total, 34,759 sequences were obtained for the three ecosystems––alpine cold swamp meadow (ASM), alpine cold meadow (AM), alpine sandy grassland (ASG), and 31 phyla and a small number of unclassified bacteria were detected. The bacterial community structures were different for each alpine grassland ecosystem. The Proteobacteria and Acidobacteria were the predominant phyla in all three ecosystems. Besides this, Actinobacteria and Chloroflexi were abundant in ASM, Bacteroidetes, Gemmatimonadetes and Verrucomicrobia were abundant in AM, and Actinobacteria were abundant in ASG. In addition, the functional bacterial genera also differed with each alpine grassland ecosystem. The ASM contained more nitrifying bacteria, methane-oxidizing bacteria and sulfur- and sulfate-reducing bacteria, whereas the ASG ecosystem contained more nitrogen-fixing bacteria. Pyrosequencing provided a greater insight into bacterial diversity within different alpine grassland ecosystems than previously possible, and gave key data for the involvement of bacteria in the protection of alpine grassland ecosystems of the Qinghai-Tibet Plateau, China.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Qinghai-Tibet Plateau is an early warning area and also considered as a sensitive key region with respect to global warming (Beniston 2003; Zhang et al. 2006). This plateau is less disturbed by human activities than other regions of China or the rest of the world, and is important for scientific research (Zhang et al. 2006). In the Qinghai-Tibet Plateau, about 60 % of the area is covered by grasslands, which include alpine meadows, alpine steppes, alpine meadow steppes, and alpine sandy grasslands (Sun and Zheng 1996). The alpine grassland ecosystems are very fragile and sensitive to climate change (Klein et al. 2004), primarily because the Plateau is located in marginal land areas where the growth and distribution of plants depend heavily on local climatic conditions (Zhang et al. 1996). Recently, global warming has impacted on the process of vegetation degradation and land desertification in the Qinghai-Tibet Plateau ecosystem (Klein et al. 2007), thus a comprehensive understanding of alpine grassland ecosystems is a priority research area.
Soil microbial communities are the most species-rich components of terrestrial ecosystems, and could change in response to many different ecological factors (Torsvik and Øvreas 2002; Schweitzer et al. 2008). In these communities, bacteria are the most abundant and diverse group of organisms (Gans et al. 2005). Microbial communities are vital in terms of soil nutrient cycles and energy transformations in grassland ecosystems, controlling the development and succession of grasslands at the microcosmic level (Clark and Pawl 1970; Bardgett et al. 1997). Because of the important role microorganisms play in grassland ecosystem, the study of microorganisms in alpine grassland ecosystems of the Qinghai-Tibet Plateau is very important, but to date there are few published studies. Previous research indicated the distribution of denitrifying and nitrogen-fixing bacteria in the alpine meadow soil of the Qinghai-Tibet Plateau (Zhang et al. 2005, 2006). For any complex system, the number and relative abundance of its components are fundamental to a quantitative description of the system (Gans et al. 2005). Consequently, a more comprehensive study of microbial diversity of the different alpine grassland ecosystems of the Qinghai-Tibet Plateau is highly relevant.
The recent development of the amplicon pyrosequencing revealed the microbial diversity in complex environments, and facilitated characterizing the diversity and distribution of soil bacterial communities (Uroz et al. 2010). In this study, pyrosequencing was used to analyze the composition of bacterial communities of different alpine grassland ecosystems of the Qinghai-Tibet Plateau, China. These different alpine grassland ecosystems represented the degree of degradation of the alpine grassland in the study area. The alpine grassland has degraded from alpine cold swamp meadow (ASM) through alpine cold meadow (AM) to alpine cold desert grassland (ASG) (Guo et al. 2007). During this degradation process, wet plant species in communities have decreased gradually and drought plants such as Poa annua expanded rapidly, and the height and coverage of plant communities have also decreased. With further degradation of alpine grassland, soil moisture decreases and the soil environment suffers extreme drought which in turn causes mesophytes in the alpine meadow to disappear, and drought-enduring plants to expand as ASM gradually changes into ASG (Guo et al. 2007; Yang et al. 2010). High-throughput tag-encoded FLX amplicon pyrosequencing was used and it revealed contrasting bacterial diversity in soils from these three alpine grassland ecosystems. The results provide key data for the involvement of bacteria in the protection of alpine grassland ecosystems of the Qinghai-Tibet Plateau, China.
Materials and methods
Sampling
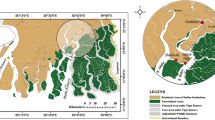
Samples were collected near Beiluhe Experimental Station of the Cold and Arid Regions Environmental and Engineering Research Institute of the Chinese Academy of Sciences on the Qinghai-Tibet Plateau (Fig. 1). The elevation is 4,628 m. The area has a typical continental highland climate. Natural alpine meadow ecosystems were divided into ASM, AM and ASG along the Beiluhe River. The dominant species investigated in three alpine meadows are listed in Table 1.

Map of sampling site
For each alpine grassland ecosystems, five subsamples were collected and mixed from the 5 cm depth layer. For each subsample, there was 100 m distance between them. The samples were put into aseptic aluminum tins, which were sealed and kept at −20 °C until further analysis. The samples were sieved through 2 mm mesh to thoroughly homogenize and remove roots and plant detritus from the samples before use.
Soil property characteristics
The total carbon and nitrogen contents of the soils were analyzed by dry combustion using a C/N analyzer (GmbH VarioEL, Elementar Analysensystem, Germany). The pH values were analyzed for 1:1 soil water mixtures using a pH meter (PT-10, Sartorius).
DNA extraction, PCR and bar-coded pyrosequencing
Soil samples from a given collection site were pooled. Genomic DNA was isolated from at least 1 g of mixed soil using the PowerSoil DNA Isolation Kit (MoBio) according to the manufacturer’s instructions. Eluted DNAs were stored at −20 °C.
To amplify a 16S rRNA gene fragment of the appropriate size and sequence variability for 454 pyrosequencing, primers B27F (5′-AGAGTTTGATCCTGGCTCAG-3′, Escherichia coli position 9–27) and B515R (5′-TTACCGCGGCTGCTGGCAC-3′, E. coli position 533–515) were chosen (Watanabe et al. 2009), and the reverse primer B515R contained the 454 Life Sciences primer A sequence. With these primers, the V1–V3 region of bacterial 16S rRNA genes was amplified. Thermal cycling consisted of an initial denaturation at 94 °C for 3 min, followed by 25 cycles of denaturation at 94 °C for 30 s, annealing at 50 °C for 30 s, and extension at 72 °C for 30 s, with a final extension of 10 min at 72 °C. PCR products were cleaned and sequenced on a GS FLX Titanium System (454 Life Sciences, Roche Applied Science).
Phylogenetic assignment, alignment and clustering of 16S rRNA gene fragments
To pass, a sequence read had to (1) include a perfect match to the sequence tag (bar-code) and the 16S rRNA gene primer; (2) be at least 200 bp in length; (3) have no undetermined bases. Phylotypes were identified using Megablast, and a representative sequence from each phylotype was aligned using SILVA (Pruesse et al. 2007). Various thresholds of sequence similarity among 16S rRNA gene sequences were commonly used as a proxy for different taxonomic levels in studies on microbial diversity with 95 and 97 % similarity to differences between genera and species (Horner-Devine et al. 2004; Roesch et al. 2007; Oakley et al. 2008). The results followed the criteria previously described to assess the quality of sequence reads (Sogin et al. 2006; McKenna et al. 2008).
Statistical analysis
Rarefaction, Chao1 and Ace were used as richness estimators for 3, 5 and 10 % dissimilarity levels using DOTUR (Roesch et al. 2007).
Results
Table 2 shows the properties of the different soils. The water content was higher in the ASM, up to 31.84 %, and less in ASG, which reached 6.95 %. The ASM also contained more C and N, which reached 3.34 and 1.75 %, respectively. The C and N contents were least in ASG, which reached 1.81 and 0.68 ‰, respectively. The pH values also differed, with ASM exhibiting lowest values while the AM had the highest value.
The richness estimators to predict the number of species and genera are shown in Table 3. In total, 34,759 valid sequences were obtained by pyrosequencing experiments. According to each estimator, all the results revealed that the richness order of each samples was as follows: ASM > ASG > AM (Table 3).
The relative abundance of phyla in each soil is indicated in Fig. 2. In total, 31 phyla and a low number of unclassified bacteria at the phylum level were detected with the various study samples. In these, the Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Gemmatimonadetes, Planctomycetes and Verrucomicrobia were abundant at more than 5 %. The study also detected a low proportion of some phyla and candidate divisions, whose abundance was lower than 1 %. The predominant bacteria were different with each ecological landscape. In the ASM ecosystem, the predominant soil bacteria were the Proteobacteria, which were subdivided into α-, β-, γ-, δ-, and ε-Proteobacteria classes, with the abundance of the α-Proteobacteria reaching 17.2 %. Then followed the Acidobacteria, Actinobacteria and Chloroflexi, whose abundance exceeded 10 %. In the AM sample, the predominant bacteria were Proteobacteria, Acidobacteria, Bacteroidetes, Gemmatimonadetes and Verrucomicrobia. The Actinobacteria took predominant status in the ASG sample, whose abundance was up to 34.2 %. Meanwhile, the Proteobacteria and Acidobacteria were also abundant in ASG. The bacterial community was different with different alpine grassland ecosystems. For example, the ε-Proteobacteria, Chlamydiae, Deferribacteres and Lentisphaerae were only detected in ASM. Simultaneously, candidate division SR1 was only detected in AM, and Tenericutes was only detected in ASG.

Relative abundance of phyla and proteobacterial classes for each soil library
Figure 3 shows the relative abundance of each genus for each soil library. As the number of genera was too large to show in one figure, the genera with more than 1 % relative abundance were picked out. As the figure indicates, a large proportion of unclassified genera were detected in each soil library. In the ASM soil, this accounted for 49.1 %. Besides these, the predominant genus was different in each sample. In the ASM soil, the Nordella, Paenisporosarcina and Prosthecomicrobium took the predominant status, and their relative abundance was over 2 %. The Chloracidobacterium took the predominant status in AM, accounting for 12.4 %. The abundance of Arthrobacter reached 16.4 % in the ASG sample, which made it the most abundant genus in this soil. Meanwhile, the Aquiflexum, Chthoniobacter, Echinicola, Flavisolibacter, Gillisia, Micrococcus, Mycobacterium, Pirellula, Rhodobium, Rhodoplanes, Segetibacter and Terrimonas were also abundant in the study samples. The Aquiflexum, Echinicola and Gillisia were only found in the AM sample.

Relative abundance of genera for each soil library. The genera with >1 % relative abundance are showed in this figure
Some known functional bacterial genera are shown in Table 4. The ASM contained more nitrifying bacteria, such as Nitrobacter, Nitrospira, and some unclassified genera belonging to the families Nitrospiraceae and Nitrosomonadaceae. It also contained more methane-oxidizing bacteria and sulfur- and sulfate-reducing bacteria. The ASG ecosystem contained more nitrogen-fixing bacteria. Meanwhile, AM contained the least nitrifying, nitrogen-fixing and methane-oxidizing bacteria.
Discussion
Here, bacterial diversity is analyzed in ASM, AM and ASG soils, characterized by a gradient of moisture contents, obtained from alpine grassland ecosystems from the Qinghai-Tibet Plateau. During degradation of alpine grassland, the soil water content and total C and N contents decreased from ASM through AM to ASG. However, the respective pH values did not show a similar trend. Pyrosequencing results revealed differences in bacterial diversity in these different ecosystems. All the richness estimators used in this study, including rarefaction, ACE and Chao, showed that the soil bacterial community diversity was highest in ASM, and least in the AM ecosystem. At 5 % dissimilarity, the rarefaction data could accurately predict the number of genera (Roesch et al. 2007), with a value of between 2,000 and 5,000 in a gram of soil (Schloss and Handelsman 2006). In this study, rarefaction revealed values of 4,405, 3,040 and 3,624 corresponding to ASM, AM and ASG with 5 % dissimilarity, respectively. In a previous study, Fierer and Jackson (2006) found that bacterial diversity was unrelated to site temperature, latitude, and other variables that typically predict plant and animal diversity. The diversity and richness of soil bacterial communities differed by ecosystem type, and these differences could largely be explained by soil pH, which was highest in neutral soils and lower in acidic soils (Fierer and Jackson 2006). In this study, the soil bacterial communities also differed by ecosystem type, and its diversity was highest in neutral soils and lower in alkaline soils.
Bacterial communities are recognized as one of the major components of soil functions, playing a key role in its sustainability (Uroz et al. 2010). In this study, 29, 19 and 24 bacterial phyla were found, with a small proportion of unclassified bacteria at the phylum level, corresponding to ASM, AM and ASG ecosystems, respectively. These results gave a deeper insight of bacterial diversity in the alpine meadow ecosystems of the Qinghai-Tibet Plateau, China. Pyrosequencing detected nine candidate divisions, such as BRC1, OP3 and OP11. Phylum- or division-level bacterial lineages with no pure culture isolates are referred to as ‘candidate divisions’, a measure proposed to classify environmental bacterial 16S rRNA gene sequences that cannot be affiliated with any of the recognized bacterial divisions (Hugenholtz et al. 1998). The number of recognized candidate divisions may greatly increase when the use of 454 pyrosequencing technology with extended read lengths becomes routine in phylogenetic surveys of bacterial diversity (Cole et al. 2009; Glöckner et al. 2010). For any complex system, the number and relative abundance of its components are fundamental to a quantitative description of the system (Gans et al. 2005). The large number of candidate divisions discovered in this study suggests that the bacterial diversity of the Qinghai-Tibet Plateau exceeds our current knowledge of the extent of soil bacterial diversity. Based on this, these data provide further understanding of alpine grassland ecosystems of the Qinghai-Tibet Plateau.
Pyrosequencing results also provided insight into the differences between bacterial community structures in different alpine grassland ecosystems. The Proteobacteria and Acidobacteria took predominant status in all three ecosystems. Besides this, the Actinobacteria and Chloroflexi were also abundant in the ASM ecosystem. In the study of Colorado Rocky Mountains, Costello and Schmidt (2006) found 14 bacterial phyla in alpine tundra wet meadow soil, with the Proteobacteria, Acidobacteria and Chloroflexi taking the predominant status, but also with a large proportion of Actinobacteria. This is comparable with this study, which indicates that a related cold and water-saturated soil environment has a similar bacterial community structure, and this community may exhibit a similar ecosystem function. In addition, the bacterial community structure was different for the three tested ecosystems. The ε-Proteobacteria, Chlamydiae, Deferribacteres and Lentisphaerae were only detected in ASM. The predominant bacterial communities for AM were Bacteroidetes, Gemmatimonadetes and Verrucomicrobia, and the candidate division SR1 was only detected in this ecosystem. Tenericutes were only detected in the ASG ecosystem, where Actinobacteria were also significantly abundant, reaching 34.2 % of the total. Spore formation is common among members of the phylum Actinobacteria, and this allows Actinobacteria to tolerate drought (Gao and Gupta 2005), consistent with there being more of these bacteria in the ASG ecosystem. It was recognized that the composition of microbial communities can have important effects on ecological processes (Klironomos 2002; Lipson and Schmidt 2004), and the differences in microbial communities consequently reflect the different alpine grassland ecosystems of the Qinghai-Tibet Plateau, China. These differences are not only exhibited in the phylum level, but also in the genus level. In the ASM soil, the Nordella, Paenisporosarcina and Prosthecomicrobium assume the predominant status, whereas the Chloracidobacterium took the predominant status in AM, and the Arthrobacter were predominant in the ASG soil. These differences may reflect different vegetation types, carbon availability, nutrient availability, and soil moisture, which could influence microbial community composition at local scales (Fierer and Jackson 2006).
Differences in functional bacterial genera present in the different alpine grassland ecosystems of the Qinghai-Tibet Plateau were also investigated in this study. The ASM ecosystem contained more nitrifying bacteria, methane-oxidizing bacteria and sulfur- and sulfate-reducing bacteria. Sulfur- and sulfate-reducing bacteria are of great significance in the carbon and sulfur cycles of aquatic environments (Vladár et al. 2008), and sulfate reduction may contribute to more than 20 % of total anaerobic mineralization (Vladár et al. 2008). This is likely reflected in the functional genera in ASM soil. Moreover, the role of sulfur- and sulfate-reducing bacteria in freshwater sediments was considered to be less important than that of methanogenic bacteria (Kuivila et al. 1989), that could oxidize more than 90 % of the methane produced before it reaches the atmosphere (Roslev and King 1996; Popp et al. 2000). The net methane emissions are the results of two opposite processes: methane production by methanogenic archaea and methane oxidation by methanotrophic bacteria (Philippot et al. 2009). The most important sink for methane in swamps is aerobic methane-oxidizing bacteria, which use methane as their sole energy and carbon source (Knoblauch et al. 2008), so this may be the reason for their wide distribution in the ASM ecosystem. Global warming has impacted on the process of vegetation degradation and land desertification, and caused alpine grassland to degrade from ASM through alpine cold meadow AM to alpine cold desert grassland ASG in the Qinghai-Tibet Plateau ecosystem (Guo et al. 2007; Klein et al. 2007). The high content of methane-oxidizing bacteria in ASM indicates that more methane will be released to the atmosphere during this degradation of alpine grassland. It is a well-accepted fact that methane is a powerful greenhouse gas, and causes about 25 times more global warming than a molecule of CO2 (IPCC 2007). Thus, these increased methane emissions during this degradation process are likely to further aggravate global warming. The nitrifying bacteria can cause nitrogen loss (Van Der Heijden et al. 2008), and the higher N content in ASM that also has a lower proportion of nitrogen-fixing bacteria may reflect the high vegetation coverage. Moreover, the ASG ecosystem contained more nitrogen-fixing bacteria, but has a low nitrogen content, and this may be due to its lower vegetation coverage. AM soil contained the least nitrifying, nitrogen-fixing and methane-oxidizing bacteria. The results provide an initial insight into bacterial function in the different alpine grassland ecosystems of the Qinghai-Tibet Plateau.
This study reports the first use of high-throughput DNA pyrosequencing to assess bacterial diversity in different alpine grassland ecosystems of the Qinghai-Tibet Plateau, China. The results indicate high bacterial diversity in this environment, and that bacterial communities also differ within the different ecosystems. The outcome is a greater insight into bacterial diversity within different alpine grassland ecosystems than previously possible, and provides key data for the involvement of bacteria in the protection and ecological restoration of alpine grassland ecosystems of the Qinghai-Tibet Plateau, China.
Conclusions
The pyrosequencing used in this study assessed the bacterial diversity within different alpine grassland ecosystems of the Qinghai-Tibet Plateau. The results revealed that the bacterial community structures were different for each alpine grassland ecosystem. The Proteobacteria and Acidobacteria were the predominant phyla in all three ecosystems. Besides this, Actinobacteria and Chloroflexi were abundant in ASM, Bacteroidetes, Gemmatimonadetes and Verrucomicrobia were abundant in AM, and Actinobacteria were abundant in ASG. In addition, the functional bacterial genera also differed with each alpine grassland ecosystem. The ASM contained more nitrifying bacteria, methane-oxidizing bacteria and sulfur- and sulfate-reducing bacteria, whereas the ASG ecosystem contained more nitrogen-fixing bacteria. Pyrosequencing revealed a greater insight into bacterial diversity within different alpine grassland ecosystems than previously possible, and provided key data for the involvement of bacteria in the protection of alpine grassland ecosystems of the Qinghai-Tibet Plateau, China.
References
Bardgett RD, Leemans DK, Cook R, Hobbs PJ (1997) Seasonality of soil biota of grazed and ungrazed hill grasslands. Soil Biol Biochem 29(8):1285–1294
Beniston M (2003) Climatic change in mountain regions: a review of possible impacts. Climatic Change 59(1–2):5–31
Clark FE, Pawl EA (1970) The microflora of grassland. Adv Agron 22:375–435
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucl Acid Res 37:141–145
Costello EK, Schmidt SK (2006) Microbial diversity in alpine tundra wet meadow soil: novel Chloroflexi from a cold, water-saturated environment. Environ Microbiol 8(8):1471–1486
Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proc Nat Acad Sci USA 103(3):626–631
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390
Gao B, Gupta RS (2005) Conserved indels in protein sequences that are characteristic of the phylum Actinobacteria. Int J Syst Evol Microbiol 55(6):2401–2412
Glöckner J, Kube M, Shrestha PM, Weber M, Glöckner FO, Reinhardt R, Liesack W (2010) Phylogenetic diversity and metagenomics of candidate division OP3. Environ Microbiol 12(5):1218–1229
Guo Z, Niu F, Zhan H, Wu Q (2007) Changes of grassland ecosystem due to degradation of permafrost frozen soil in the Qinghai Tibet Plateau. Acta Ecol Sin 27:3294–3301
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM (2004) A taxa-area relationship for bacteria. Nature 432:750–753
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR (1998) Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180(2):366–376
IPCC (2007) Climate change 2007: mitigation of climate change. Contribution of working group III to the fourth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge
Klein JA, Harte J, Zhao X (2004) Experimental warming causes large and rapid species loss, dampened by simulated grazing, on the Tibetan Plateau. Ecol Lett 7(12):1170–1179
Klein JA, Harte J, Zhao X (2007) Experimental warming, not grazing, decreases rangeland quality on the Tibetan plateau. Ecol Appl 17(2):541–557
Klironomos JN (2002) Feedback with soil biota contributes to plant rarity and invasiveness in communities. Nature 417:67–70
Knoblauch C, Zimmermann U, Blumenberg M, Michaelis W, Pfeiffer E (2008) Methane turnover and temperature response of methane-oxidizing bacteria in permafrost-affected soils of northeast Siberia. Soil Biol Biochem 40(12):3004–3013
Kuivila KM, Murray JW, Devol AH (1989) Methane production, sulfate reduction and competition for substrates in the sediments of Lake Washington. Geochim Cosmochim Acta 53(2):409–416
Lipson DA, Schmidt SK (2004) Seasonal changes in an alpine soil bacterial community in the Colorado Rocky mountains. Appl Environ Microbiol 70(5):2867–2879
McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu ZZ, Lozupone CA, Hamady M, Knight R, Bushman FD (2008) The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog 4:22–28
Oakley BB, Fiedler TL, Marrazzo JM, Fredricks DN (2008) Diversity of human vaginal bacterial communities and their association with clinically-defined bacterial vaginosis. Appl Environ Microbiol 74(15):4898–4909
Philippot L, Hallin S, Börjesson G, Baggs EM (2009) Biochemical cycling in the rhizosphere having an impact on global change. Plant Soil 321(1–2):61–81
Popp TJ, Chanton JP, Whiting GJ, Grant N (2000) Evaluation of methane oxidation in the rhizosphere of a Carex dominated fen in north-central Alberta, Canada. Biogeochemistry 51(3):259–281
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Pelies J, Glockner FO (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl Acid Res 35(21):7188–7196
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, Daroub SH, Camargo FAO, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Roslev P, King GM (1996) Regulation of methane oxidation in a freshwater wetland by water table changes and anoxia. FEMS Microbiol Ecol 19(2):105–115
Schloss PD, Handelsman J (2006) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72(10):6773–6779
Schweitzer JA, Bailey JK, Fischer DG, LeRoy CJ, Lonsdorf EV, Whitham TG, Hart SC (2008) Plant–soil–microorganism interactions: heritable relationship between plant genotype and associated soil microorganisms. Ecology 89(3):773–781
Sogin ML, Morrison HG, Huber JA, Welch MD, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Nat Acad Sci USA 103(32):12115–12120
Sun H, Zheng D (1996) Formation and evolution of Qinghai-Xizang Plateau. Shanghai Science and Technology Press, Shanghai
Torsvik V, Øvreas L (2002) Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol 5(1):240–245
Uroz S, Buée M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ Microbiol Rep 2(2):281–288
Van Der Heijden MGA, Bardgett RD, Van Straalen NM (2008) The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11(3):296–310
Vladár P, Rusznyák A, Márialigeti K, Borsodi AK (2008) Diversity of sulfate-reducing bacteria inhabiting the rhizosphere of Phragmites australis in Lake Velencei (Hungary) revealed by a combined cultivation-based and molecular approach. Microbial Ecol 56(1):64–75
Watanabe K, Nagao N, Toda T, Kurosawa N (2009) The dominant bacteria shifted from the order ‘‘Lactobacillales’’ to Bacillales and Actinomycetales during a start-up period of large-scale, completely-mixed composting reactor using plastic bottle flakes as bulking agent. World J Microbiol Biotechnol 25(5):803–811
Yang Z, Ou YH, Xu X, Zhao L, Song M, Zhou C (2010) Effects of permafrost degradation on ecosystems. Acta Ecologica Sinica 30(1):33–39
Zhang XS, Yang DA, Zhou GS, Liu CY, Zhang J (1996) Model expectation of impacts of global climate change on biomes of the Tibetan Plateau. In: Omasa K, Kai K, Taoda H, Uchijima Z, Yoshino M (eds) Climate change and plants in East Asia. Springer-Verlag, Tokyo, pp 25–38
Zhang Y, Wang H, Li D, Xiao Q, Liu X (2005) Molecular diversity and phylogenetic analysis of nitrogen-fixing (nifH) genes in alp prairie soil of Sanjiangyuan natural reserve. Acta Microbiol Sin 45(2):166–171
Zhang Y, Li D, Wang H, Xiao Q, Liu X (2006) The diversity of denitrifying bacteria in the alpine meadow soil of Sanjiangyuan natural reserve in Tibet Plateau. Chin Sci Bull 51(10):1245
Acknowledgments
This project was supported by National Basic Research Program (973) of China (No. 2012CB026105), National Natural Science Foundation of China (No. 31170465, 31100365), China Postdoctoral Science Fund (No. 2012M512053), Foundation for Excellent Youth Scholars of CAREERI, CAS, and UK BBSRC China Partnering Grant BB/J020419/1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, W., Wu, X., Liu, G. et al. Tag-encoded pyrosequencing analysis of bacterial diversity within different alpine grassland ecosystems of the Qinghai-Tibet Plateau, China. Environ Earth Sci 72, 779–786 (2014). https://doi.org/10.1007/s12665-013-3001-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-013-3001-z