
Abstract
The optimized geometry and vibrational frequencies of picolinium maleate (PM), an organic non-linear optical material, was obtained by ab initio DFT/B3LYP level with complete relaxation in the potential energy surface using 6-311G(d,p) basis sets. The Fourier-transform infrared (FT-IR) spectrum of PM was recorded in the region 4,000–400 cm−1. The harmonic vibrational frequencies were calculated and scaled values had been compared with experimental FT-IR spectrum. The optical non-linearity arose from intermolecular hydrogen bonding between centro-symmetric picolinic acid and maleic acid. The DFT study was aimed at identifying intermolecular hydrogen bonding nature and comparing the calculated values with the XRD data. Analysis of experimental 13C shielding parameters was supported by DFT theoretical calculations carried out within the gauge-including atomic orbital and the spectra estimations were performed using ChemPro 8.0. It was found that chemical shifts obtained with ChemPro 8.0 were due to the substitution effects. The charge density distribution and site of chemical reactivity of molecules were obtained by mapping electron density isosurface with electrostatic potential surfaces. The scaled frequencies are comparable with the experimental frequencies. Energy span as calculated by ZINDO method, between the highest occupied molecular orbital and the lowest unoccupied molecular orbital of PM is −8.9697 and −1.5477 eV.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Second-order nonlinear optical (NLO) materials are much attracted in recent research work because of their potential applications in the field of optoelectronics. In recent years, organic molecular NLO materials have been intensely investigated due to their high nonlinearities, rapid response to electro-optic effect as compared to inorganic NLO materials [1–5]. Organic crystals have been extensively studied due to their nonlinear optical (NLO) coefficient being often larger than those of inorganic. Combinations of amino acids with organic salts are promising materials for optical second harmonic generation. New nonlinear optical frequency conversion materials can have a significant impact on laser technology, optical communication and optical data storage technology [6]. The search for new frequency conversion materials over the past decade has concentrated primarily on organic compounds [7, 8] and many organic NLO materials with high nonlinear susceptibilities have been discovered. Amino acids are organic molecules that contain a carboxyl group (–COOH) as well as amino group (–NH2). In solid state, amino acids contain a protonated amino group (NH3+) and deprotonated carboxylic group as well as amino group (NH2). This dipolar nature exhibits peculiar physical and chemical properties in amino acids which makes them an ideal candidate for NLO application [9–12]. High performance of electro-optical switching for telecommunication and optical information processing are based on materials with high nonlinear optical properties. The search for new frequency conversion materials in recent years has concentrated on semi-organic complexes [13]. Though inorganic materials or semiorganic materials have higher thermal stability than that of organic, their molecular hyperpolarizability plays vital role for second harmonics. In the present investigation picolinium and meleate are subjected to density functional study for their intermolecular and intramolecular hydrogen bonding and π–π* interactions. For the proper assignment of vibrational fundamentals and to understand effect of hydrogen bonding on the characteristic frequencies of the title compound normal coordinate analysis has been carried out.
2 Experimental details
The FTIR spectra of PM have been recorded in the region 4,000–400 cm−1 by using KBr windows with Bruker IFS 66 V spectrometer. A KBr beam splitter and liquid nitrogen cooled mercury-cadmium-tellurium (MCT) detector have been used to collect the mid infrared spectra. The UV–vis transmission spectrum of PM crystal is recorded in the range 190 and 1,100 nm using PerkinElmer Lamda 35 UV–VIS-NIR spectrometer, covering the entire visible and near infrared region. The spectral resolution for both FTIR and UV–vis is 2 cm−1.
3 Theoretical calculations
The density functional theory (DFT) calculations were performed for the title compound, the initial geometry taken from crystallographic measurements was taken as the starting geometry for density functional theory (DFT) optimization. The geometry was optimized first, at semi-empirical level using Austin Model (AM1) method from the Chemsoft package and then at Hartree Fock (HF) and DFT level using the Becke 3 Lee–Yang–Parr (B3LYP) functional at 6-31G basis set with the help of GAUSSIAN-03 package [14]. Isotropic 1H- and 13C-nuclear magnetic shielding constants of this compound were calculated by employing the direct implementation of the gauge including-atomic-orbital (GIAO) method at the B3LYP density functional theory using 6-31G basis set. By combining the results, using graphical interface, GAUSSVIEW program with symmetry considerations, vibrational frequency assignments were made with a high degree of accuracy.
4 Results and discussion
4.1 Geometry analysis
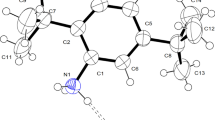
The optimized structure parameters of PM by ab initio HF and DFT (B3LYP) levels with the 6-311G basis set in accordance with the atom numbering scheme given in Fig. 1. The molecules possessing C1 point group symmetry with 72 fundamental modes of vibrations and are active in IR region. The X-ray experimental data on the geometrical parameters of PM are available in literature [15] and computed results are compared with the experimental data.

Molecular structure (with atoms numbering) of PM at B3LYP/6-311G(d,p) basis set
4.2 Conformational analysis
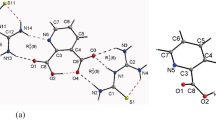
Molecular structure of PM is shown in Fig. 1. The bond length and bond angles of the structure of PM have been calculated by using B3LYP method at 6-311G(d,p) basis set. Picolinic acid forms well defined crystalline charge transfer (CT) with anion carboxylic acid through N···H···O and C···H···O hydrogen bonds. The present investigation aims to identify the direction of specific N···H···O bond between –NH2 group and O− group in the anion. The bond length between N–H···O is 1.638 Å and the observed value is at 2.28 Å. It is very close agreement with experimental data [16]. The average C–H bond length in the aromatic ring calculated by B3LYP/6-311G(d,p) is 1.067 Å. It is less than C–H bond length present outside the ring. Moreover, the calculated values of C–H bond lengths of methyl part according to B3LYP and HF methods are also in good agreement.
4.3 Molecular electrostatic potential analysis
Electrostatic potential maps illustrate the charge distributions of molecules three dimensionally. The molecular electrostatic potential (MEP) at point r in space around a molecule (in atomic units) can be expressed as:
where Z A is the charge on nucleus A, located at R A and is the electronic density function for the molecule. The MESP mapped surface of the compounds and electrostatic potential contour map for highest unoccupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) potentials are shown in Fig. 2. The scheme-coded values are then projected onto the 0.02 a.u. isodensity surface to produce a three-dimensional electrostatic potential model [17, 18]. Local negative electrostatic potentials (dotted line) signal nitrogen and oxygen atoms with lone pairs whereas local positive electrostatic potentials (continuous line) signal polar hydrogen in C–H bonds.

Electrostatic potential contour map of PM (a) HOMO and (b) LUMO potentials using 6-311G(d,p) basis set
5 Vibrational analysis
Vibrational spectral assignments have been performed on the recorded FT-IR (powder) spectrum based on the theoretically predicted wavenumbers by ab initio B3LYP. The calculated and scaled vibrational wavenumbers are collected in Table 1 with the scaling factor of 0.96 [19] for B3LYP level. All 72 fundamental modes of vibrations are active. Figure 3 shows the observed spectrum of PM crystal and the simulated spectra of PM molecule at HF/DFT level using 6-311G(d,p) basis sets.

Simulated spectrum at (a) DFT/6-311G(d,p), (b) HF/6-311G(d,p) basis sets and (c) FTIR spectrum of Picolinium maleate molecule
5.1 Carbon–carbon vibrations
In pyridine derivatives, the carbon–carbon stretching modes of vibration appear in the region 1,650–1,200 cm−1 and are determined by nature of substituent around the ring [20, 21]. Pyridines and pyrimidines absorb majorly in the region 1,600–1,450 cm−1 due to C–C vibrations. This is evidence of presence of bands around 1,500 cm−1 in spectrum of the compound [22]. The band observed at 1,472 cm−1 in the FTIR spectrum is assigned to CC stretching vibrations. The CC aromatic stretch, known as semicircle stretching predicted at 1,474 cm−1 in HF and 1,482 cm−1 in DFT are in excellent agreement with experimental observations in FTIR spectrum. The ring deformation at 730 cm−1 in HF coincides satisfactorily with the medium band at 650 cm−1 and sharp band at 890 cm−1 both in FTIR spectrum.
5.2 Hydrocarbon vibrations
C–H stretching vibrations of the pyridine ring are assigned to bands observed at 3,252 cm-1 and 3,170 cm-1, also a sharp peak at 3,099 cm−1 in IR spectrum of PM. It is noticed that C–H stretching mode do not couple with each other. The bands in the range 3,100–3,000 cm−1 are due to C–H stretching vibrations in aromatic compounds. The fundamental modes observed at 2,555 cm−1 in IR spectra are assigned to C–H stretch with combination and overtones, C–H in plane and out of plane bending vibrations are observed in the region 1,350–950 cm−1. In substituted pyridine derivatives, C–H in plane bending vibration appears in the frequency region 1,550–1,000 cm−1 [23, 24]. Normally, the bands are very weak. In our study the bands observed at 1,028, 1,330 cm−1 in FTIR spectrum are assigned to C–H in plane bending vibrations and these values are in good agreement with the calculated values. Krishnakumar et al. [25] have been assigned C–H out of plane bending vibrations in the region 986–825 cm−1 in FTIR spectrum. The bands observed at 960 in FTIR are assigned to C–H out of plane bending vibrations.
5.3 Nitro-carbon vibrations
Primary aromatic amine with nitrogen directly in the ring absorbs strongly at 1,330–1,260 cm−1 due to stretching of the phenyl carbon–nitrogen bond [26]. Silverstein et al. [27] assigned C–N stretching absorption in the region 1,342–1,220 cm−1. The spectra of benzene and pyridyl substituted compounds show bands in the region of 1,260–1,210 cm−1. In analogy with previous work, the band appears at 1,226 cm−1 and band at 1,284 cm−1 in FTIR spectrum of PM is assigned to C–N symmetric and asymmetric stretching mode of vibrations, respectively. The FTIR spectrum shows complicated pattern of absorption in the region of 3,400–2,700 cm−1 originated from N–H stretching mode involved in N–H···O bonds between picolinic acid and mealic acid. The band observed at 1,310 cm−1 in infrared is assigned to C–N stretching mode vibrations coupled with the in-plane pyridine ring deformation. The band at 1,425 cm−1 is assigned to C–N stretching with some contribution from N–H···O stretch. Medium to weak intensity bands in the frequency range 600–400 cm−1 are due to C–N–C deformation vibrations. The peaks at 570, 540, 480, and 420 cm−1 are assigned for C–N–C deformation [28]. The assignments of C–N in-plane and out-of-plane bending vibrations made in this study are also supported by literature [29].
5.4 Carbonyl vibrations
The interaction of carbonyl group with the hydrogen donor group does not produce such drastic change in the frequency C=O stretch, as done by interaction of NH stretch. The carbonyl group of maleic acid may interact with other atoms in the molecule and gives rise to a complex pattern of spectrum. As the intermolecular hydrogen bond in maleic acid is very strong, it forms crystalline maleate of various organic molecules through hydrogen bonding and π–π interactions, but acts as a acceptor [30]. The band due to C=O stretching vibration is observed in the region 1,850–1,550 cm−1 due to tautomerism, pyrimidines substituted with hydroxyl groups are generally in the keto form and therefore have a strong band due to carbonyl group [31]. In the present work, a sharp band at 1,743 in FTIR spectrum is assigned to C=O stretching vibrations. In general, the characteristic C=O stretching vibrations of cyclic ketones are found over a relatively wide range depending on the ring size.
5.5 Bending vibrations
The strong band in FTIR at 1,446 cm−1 is assigned to N–HO stretching while the bands below 400 cm−1 in the calculated spectra are assigned to N–HO in and out of plane bending vibrations. Normally, the deformation modes of C–N–H occur in the region 1,450–1,320 cm−1. Hence, in the present work, the bands appear at 1,330 and 1,420 cm−1 in FTIR spectrum are assigned to C–N–H bending modes of vibrations. The vibrational spectra of substituted phenols and its derivatives N–C–H bending modes are observed in the region 1,500–1,425 cm−1. The band appears at 1,450 cm−1 in the FTIR spectrum of PM is assigned to N–C–H bending mode of vibration. C–H deformation frequencies in benzene and its derivatives are found to occur in the region 1,200–1,050 cm−1. In the present study the bands observed at 1,090 and 1,210 cm−1 in FTIR of PM are assigned to C–C–H deformation. The C–C–C bending mode always occurs below 600–480 cm−1 in isopropyl benzenes. In the present work, the band observed at 540 cm−1 and the band at 575 cm−1 are assigned to C–C–C bending modes of vibration.
5.6 Scale factor
Comparison of frequencies calculated at HF/6-311G(d,p) and B3LYP/6-311G(d,p) basis sets with experimental values reveals that theoretical values give reasonable deviations from the experimental values (see Table 1). To determine the scale factors, the procedure used previously [32] has been followed, that minimizes residual separating experimental and theoretically predicted vibrational frequencies. The optimum scale factors for vibrational frequencies have been determined by minimizing the residual
where \( \lambda_{wi}^{T} \), ith theoretical harmonic frequency and \( \nu_{i}^{E} \), ith experimental fundamental frequency (in cm−1), respectively, and N is the number of frequencies included in the optimization which leads to
The scale factors used in this study minimized the deviations between the computed and experimental frequencies.
6 Molecular polarizability
It is proposed that investigation should be done to see the effect of basis set on molecular polarizability of PM. The first hyperpolarizability is a third-rank tensor that can be described by 10 components in a 3 × 3 × 3 matrix due to Lleinmen symmetry [33–36]. The complete equations for calculating the magnitude of total static dipole moment μ, mean polarizability α0, anisotropy of polarizability Δα and mean first hyperpolarizibility β0 using the x, y, z components are given by the following equation
where E 0 is energy of unperturbed molecules, F α is field at origin and μα, μαβ and βαβγ are components of dipole moment polarizability and first hyperpolarizability, respectively.
The total static dipole moment μ, mean polarizability α0, anisotropy of polarizability Δα and mean first hyperpolarizability β0, using the x, y, z components are defined as
the DFT calculated first hyperpolarizability of PM is 237.082 esu and dipole moment is −8.590 in Debye are shown in Table 2. The calculated first hyperpolarizability of PM is much higher than that of urea, and the material is best suited for NLO applications.
7 UV–vis analysis
Results of ZINDO calculations of electronic transition energies of PM molecule along with band assignments are presented in Table 3 and they have been compared with experimental data of PM in solvent as shown in Fig. 4. The wavelength belonging to HOMO–LUMO transition and thus maximum wavelength is at 296 nm. The present experiment shows one intense band at 297 nm (sh), and a weak band is observed at 405 nm, respectively. In ZINDO, we have found that it has a sharp band at 346 nm with the configuration of 0.041 (H − 2 → L + 1) and 0.016 (H → L + 1) excited state respectively [37].

(a) Simulated spectrum using ZINDO basis set and (b) UV spectrum of Picolinium maleate crystal
7.1 HOMO, LUMO energy gap
Both HOMO and LUMO are the main orbitals which take part in chemical stability
-
HOMO energy = −8.9697 eV
-
LUMO energy = −1.5477 eV
-
HOMO–LUMO energy gap = −10.5174 eV
The HOMO and LUMO energy gap explains the eventual charge transfer interactions taking place within the molecule.
7.2 Ionization potential
The ionization potential and chemical hardness of the molecule have been calculated using Koopman’s theorem [38] and are given by
where IP ≈ −E(HOMO), EA ≈ −E(LUMO); IP = ionization potential (eV), EA = electron affinity (eV).
The ionization potential has been calculated for PM (5.258 eV) and the effect of substitution of maleic acid in picolinic acid decreases the ionization potential of picolinic acid. One can also relate the stability of the molecule to hardness, which means that the molecule with least HOMO–LUMO gap is more reactive.
8 NMR analysis
The gauge including atomic orbital (GIAO) method is used in calculations of NMR shielding constants. The shielding constants are frequently converted to NMR chemical shifts by calculating a reference compound, which is picolinic acid. The gross atomic charges of PM are given in Table 4. This data would be useful in identifying the preferred position of the molecule for nucleophilic attack due to charge transfer between molecules [39]. The carbon attached to N and O has lesser electron density than other carbons and hence these are more unshielded. From calculated values, atoms C7 and C17 have nucleophilic centers that share charge transfer during crystallization by hydrogen bonding. Further, the bond distance between C–O···H is 1.428 A confirming nucleophilic attraction between maleic acid and picolinic acid. Hence, we compare the gross atomic charges of PM with calculated 1H and 13C NMR chemical shifts.
9 Conclusions
Attempts have been made in the present work to obtain the proper frequency assignments for the crystal PM from FT-IR and UV–vis spectra. The equilibrium geometries, harmonic frequencies and IR spectra of PM have been determined and analyzed both at HF and DFT levels of theory utilizing 6-311G(d,p) basis set, giving allowance to the lone pairs through diffuse functions. Comparison between calculated vibrational frequencies and experimental values indicates that both the methods of B3LYP/6-311G(d,p) and HF/6-311G(d,p) can predict IR and UV spectra of the title compound well. Any discrepancy noted between observed and calculated frequencies may be due to the fact that calculations have been actually done on a single molecule in gaseous state contrary to experimental values recorded in the presence of intermolecular interactions.
References
X Q Wang et al. J. Crystal Growth 234 469 (2002)
X L Duan et al. Cryst. Res. Technol. 37 1066 (2002)
M H Jiang and Q Fang Adv Mater 11 1147 (1999)
J B Gaudry et al. Chem. Phys. Lett. 324 321 (2000)
T Rasheed and S Ahmad Indian J. Phys. 85 239 (2011)
D S Chemla and J Zyss Nonlinear Optical Properties of Organic Materials and Crystals (New York: Academic Press) (1987)
S R Marder, J W Perry and W P Schaefer Science 245 626 (1989)
P M Anbarasan, P S Kumar, K Vasudevan, R Govindan and V Aroulmoji Indian J. Phys. 85 1477 (2011)
D Eimerl, S Velsko, L Davis, F Wang, G Loiacona and G Kennedy IEEE Quantum Electron. 25 179 (1989)
G R Kumar, S G Raj, R Mohan and R Jayavel J. Crystal Growth 275 1947 (2005)
C H S Lin, N Gabas, J P Canselier and G Pope J. Crystal Growth 191 791 (1998)
A Deepthy and H L Bhat J. Crystal Growth 226 287 (2001)
S Gunasekaran, G Anand, R A Balaji, J Dhanalakshmi and S Kumaresan Pramana – J. Phys. 75 683 (2010)
M J Frisch et al. Gaussian Inc (Wallingford: CTPA) (2001)
P Pandi et al. Spectrochim. Acta A 98 7 (2012)
A U Rani, N Sundaraganesan, M Kurt, M Cinar and M Karabacak Spectrochim. Acta A 75 1523 (2010)
R H Petrucci, W S Harwood, F G Herring and J D Madura General Chemistry: Principles & Modern Applications, 9th edn. (New Jersey: Pearson Education Inc) (2007)
I G Csizmadia Theory Practice of MO Calculations on Organic Molecules (Amsterdam: Elsevier) (1976)
J A Pople et al. Int J. Quantum Chem. Quantum Chem. Symp. 15 269 (1981)
L J Bellamy The Infrared Spectra of Complex Molecules, 3rd edn. (New York: Wiley) (1975)
G Varsanyi Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives (London: Adam Hilger) (1974)
S Gunasekaran, S R Varadhan and K Manoharan Asian J. Phys. 4 12 (1993)
G Socrates Infrared and Raman Characteristic Group Frequencies, 3rd edn. (Chichester: John Wiley & Sons Ltd) (2001)
A P Upadhya and K N Upadhyay Indian J. Pure Appl. Phys. 20 672 (1982)
V Krishnakumar, N Surumbarkuzhali and S Muthunatesan Spectrochim. Acta A 71 1810 (2009)
R Saxena, L D Kauedpal and G N Mathur J. Polym. Sci. A: Polym Chem 40 3559 (2002)
R M Silverstein, G Clayton Basslor and T C Morill Spectrometric Identification of Organic Compounds (New York: John Wiley) (1981)
G Socrates Infrared characteristics group frequencies, Ed. (Meerut: Pragati Prakashan) (1992)
S Mohan, N Sundaraganesan and J Mink Spectrochim. Acta A 47 1111 (1991)
P Pandi, G Peramaiyan, M Krishna Kumar, R Mohan Kumar and R Jayavel Spectrochim. Acta Part A 88 77 (2012)
S Kumar, A K Rai, S B Rai and D K Rai Indian J. Phys. 84 563 (2010)
V Arujunan, I Saravanan, Mariusz K Marchewka and S Mohan Spectrochim. Acta Part A 92 305 (2012)
M Tommasini, C Castiglioni, M Del Zoppo and G Zerbi J. Mol. Structure 480 179 (1999)
C R Zhang, H S Chem and G H Wang Chem. Res. Chin. Univ. 20 640 (2004)
P S Kumar, K Vasudevan, A Prakasam, M Geetha and P M Anbarasan Spectrochim. Acta A 77 45 (2010)
D A Kleinman Phys. Rev. 126 1977 (1962)
S Gunasekaran, S Kumaresan, R Arunbalaji, G Anand, S Seshadri and S Muthu J. Raman Spectroscopy 40 1675 (2009)
T A Koopmans Physica 1 104 (1933)
S Gunasekaran, R A Balaji, S Kumaresan, G Anand and S Srinivasan Canadian J. Anal. Sci.Spectr. 53 149 (2008)
Acknowledgments
The authors are thankful to Prof. Dr. R. Mohan Kumar, Department of Physics, Presidency College, Chennai, Tamil Nadu, India-600005, for supplying the XRD data and FTIR, UV details used in this study. The authors are also thankful to Prof. E. M. Subramanian, Department of Chemistry, Pachaiyappas College for Men, Kanchipuram, Tamil Nadu, India-631503, for valuable discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gunasekaran, S., Kannan, K.S., Loganathan, S. et al. Structural studies on picolinium maleate crystal by density functional methods. Indian J Phys 87, 1189–1197 (2013). https://doi.org/10.1007/s12648-013-0363-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12648-013-0363-8