
Abstract
In the present study, proton transfer from nitric and maleic acids to amine group (4-chloroaniline) led to hydrogen bonded crystals of 4-chloroanilinium maleate (4CAM) and 4-chloroanilinium nitrate (4CAN) which are investigated by the experimental and theoretical approaches. The molecular structures of these two compounds were optimized with the Density Functional Theory (DFT) using B3LYP function and the Hartree-Fock (HF) level with a6-311 ++G(d,p) basis set. Geometrical parameters of the molecules were also analyzed along with their intermolecular hydrogen bond, which tailors the ions. These analyses show that present molecules are stabilized through the N–H ⋯O and O–H ⋯O hydrogen bonds. The vibrational modes were computed by quantum chemical methods. Further, these modes are investigated by FT-IR and FT-Raman spectroscopy in the range of 4000–400 cm−1. The optimized molecular geometry and computed vibrational spectra are compared with experimental results, which show significant agreement. The natural bond orbital (NBO) analysis was carried out to interpret hyperconjucative interaction and intramolecular charge transfer (ICT). This analysis gives the precise insight into the nature of H-bond interactions. The chemical hardness, electronegativity and chemical potential of the molecules were determined by HOMO–LUMO plot. The frontier molecular orbitals have small band gap value, which signify the possible biological/pharmaceutical activity of the compounds.

The proton transfer from the nitric and maleic acids to amine group (of 4-chloroaniline) lead to hydrogen bonded crystals of 4-chloroanilinium maleate (4CAM) and 4-chloroanilinium nitrate (4CAN). The molecular structures of these two compounds were optimized with the Density Functional Theory (DFT) and Hartree-Fock (HF) methods. Geometrical parameters of the molecules were analyzed along with their intermolecular hydrogen bond which tailors the ions. These analyses show that the molecular aggregations are stabilized through the N–H···O and O– H···O hydrogen bonds. The vibrational modes were computed by quantum chemical methods and further investigated by FT-IR and FT- Raman spectroscopy in the range of 4000–400 cm-1.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
One of the most important reactions in chemistry and biochemistry is proton transfer between donor and acceptor.[1] These interactions are playing an important role in various chemical and biological processes such as stabilizing the biomolecular structures,[2] controlling the speed of the enzymatic reactions[3] and constructing supramolecular structures.[4] In most of the cases, the applications are ensued due to the intra- and intermolecular interactions particularly with the hydrogen bonds, which have more importance in the areas of molecular recognition, crystal engineering research and supramolecular chemistry. Understanding of these non-covalent interactions has proved to be the most valuable because of its strength and directional properties.[5] The strength and directionality of hydrogen bonding interactions are responsible for solid-state formation and other physical properties of the system.[6] Also, we continuously seek to identify hydrogen bond–enriched assemblies by means of efficient organic hydrogen bonding synthons.
Substituted anilines are known to be candidates for supramolecular synthons as they have both donor and acceptor sites, viz., nitrogen and chlorine atoms in the present case. The present work was attempted with nitrate and maleate salts of 4-chloroanilinium crystal. Though the single-crystal XRD clearly predicts the molecular structure and packing tendency of the molecule in the solid crystalline state, the spectral measurement gives an account of the strength of the intermolecular interactions by means of shifting of wavenumbers and the intensity of the peaks. Further, the molecular geometry obtained from the crystallographic data is a well-suited input for the quantum chemical calculations. By which it is possible to optimize the molecule in different environments and the corresponding vibrational modes can be calculated. The hyperconjugative interactions and band gap values can be inferred from natural bond orbitals (NBOs) and frontier molecular orbital (FMO) studies. Hence, a combined crystallographic, spectroscopic and quantum chemical calculations were attempted on 4-chloroanilinium maleate (4CAM) and 4-chloroanilinium nitrate (4CAN), and the results are summarized.
2 Experimental
2.1 Crystal Growth
Crystals of 4CAM and 4CAN were synthesized from aqueous mixtures containing 4-chloroaniline with maleic and nitric acid in the 1:1 stoichiometric ratio at room temperature by slow evaporation method, respectively. After a week, light yellow colour crystals of 4CAM and 4CAN were obtained. The qualities of the crystals were improved by recrystallization.
2.2 Density Measurement
The density of the grown crystals were measured by sink and swim method (flotation technique) using a liquid mixture of carbon tetrachloride and xylene. Initially, carbon tetrachloride (5 mL) was taken in a test tube and a good-quality three-dimensional crystal was placed on it. Due to high density of the liquid, the crystal began to float. Then, drops of xylene were added drop by drop with continuous agitation to get uniform density over the liquid. When the density of the crystal matches that of liquid mixture, the crystal levitates to the middle of the test tube. Then, the density of the liquid was found with specific gravity bottle from the concept of relative density. Thus, the densities of the crystals are found to be 1.495 mg.m−3 for 4CAM and 1.465 mg.m−3 for 4CAN.
2.3 Single-Crystal XRD Studies
The entire crystallographic calculations of 4CAM were performed done using single-crystal X-ray diffraction by Bruker SMART APEX CCD area detector diffractometer (graphite- monochromated, MoK α = 0.71073 Å). A good-quality single-crystal was selected from the grown crystals and used for the single crystal X-ray diffraction studies. Consequently, the full data collection of the crystal was carried out. The cell refinement and data reduction were done with XCAD4 software.[7] The structure was solved and refined with the SHELXL97 program.[8] All the H atoms were discernible in the difference electron density maps. Nevertheless, the aryl H atoms were constrained and refined in the riding atom approximation: C −H = 0.93,Å and Uiso(H) = 1.2Ueq(C). The other H atoms, which are involved in the N −H⋯O and O–H …O hydrogen bonds, were located from electronic density map and refined isotropically. Graphical molecular illustrations were done with ORTEP3 for Windows[9] and Mercury[10] packages.
Though good-quality crystals of 4CAN were obtained, the attempts of single crystal X-ray diffraction was not fruitful due to the hygroscopic nature of the crystal.
2.4 Vibrational Spectroscopic Measurements
A Jasco spectrometer FT-IR, model 410 under a resolution of 4 cm−1 and with a scanning speed of 2 mm/sec was used for IR spectral measurements. The samples were prepared using pellet technique and the spectrum was recorded over the range 4000–400 cm−1. The FT-Raman spectrum was recorded in the frequency range of 4000–50 cm−1 using a BRUKER RFS 27 FT-Raman Microscope module with the resolution of 2 cm−1. The Nd: YAG Laser source was operated at 1064 nm for the excitation.
2.5 Computational details
The geometrical optimization for 4CAM and 4CAN molecules was carried out theoretically by 6-311 ++ G(d,p) level on an Intel Core i5/3.20 GHz computer using Gaussian 09W[11]program package without any constraint.[12] Initial geometry was taken from the single-crystal X-ray studies and it was minimized (optimized) by Hartree −Fock (HF) method using the 6-311 ++G (d,p) basis set. The molecular geometries were also optimized by the Density Functional Theory (DFT) using the Becke’s three-parameters exchange functional (B3)[13] in combination with the Lee–Yang–Parr correlation functional (LYP).[14] It is accepted as a cost-effective approach for the computation of molecular structure, vibrational frequencies and energies of optimized structures. The optimized structural parameters were used in the vibrational frequency calculations at the same level to characterize all stationary points as minima. Then vibrationally averaged nuclear positions of the structure were used for harmonic vibrational frequency calculations resulting in IR and Raman frequencies. Finally, the calculated normal mode vibrational frequencies provide thermodynamic properties through the principle of statistical mechanics. By combining the analysis preformed using the GAUSSVIEW program[15] with symmetry considerations, vibrational frequency assignments were made with a high degree of accuracy. There is always some ambiguity in defining internal coordination. However, the defined coordinate from the complete set matches quite well with the motions observed using the GAUSSVIEW program. Also, the NBO calculations were carried out using HF and DFT methods with the 6-311 ++G (d,p) basis set to get more detailed information about the chemical bonds and hydrogen bonding interaction within the ions. The FMOs have been computed and analyzed by HF and DFT method with the 6-311 ++G (d,p) basis set.
3 Results and Discussions
3.1 Molecular Geometry
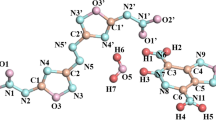
The solid-state molecular and crystal structures of 4CAM are illustrated from the single-crystal XRD studies. The asymmetric part of the 4CAM contains a 4-chloroanilinium cation and a maleate anion shown in figure 1. As 4CAN is prepared with 4-chloroaniline and nitric acid, the crystals are expected to possess 4-chloroanilinium cation and a nitrate anion in the asymmetric part. The optimized structures of both the molecular assemblies are shown in figure 2. In both the compounds, the transfer of proton from the inorganic and organic acid leads to protonation on the NH2 sites of the 4-chloroaniline and it forms the 4-chloroanilinium cation. This protonation on the N site of the cation is confirmed from the elongated C −N bond distance [1.456 (3) Å] in 4CAM.[16] The protonated bond distance is compared with the theoretically computed bond distances. The optimized C–N bond distances are 1.418 and 1.426Å in HF and DFT levels, respectively, for 4CAN and 1.415 and 1.425 Å by the HF and DFT levels, respectively, for 4CAM (table 1). In theoretical calculations the C–N bond distances of the molecules are observed to be smaller than the single crystal XRD study which is due to the effect of N–H ⋯O intermolecular hydrogen bonds. Even though the hydrogen atom seems to have deviated from the N atom of the cation, there must be some attraction between cation and anion which is confirmed by the Mulliken charge distribution analysis. The bond geometry between N, H and O atoms which connect the ion pairs, are shown scheme 1. From the Mulliken charge distribution analysis of the two complexes, N atom is more electronegative than the O atom. These N atoms of the cations are attracting the proton from acids and it forms the N–H ⋯O intermolecular hydrogen bond. The N–H ⋯O angle is 180° and 179° at the HF and DFT/B3LYP methods, respectively, for 4CAN and 172° in both methods for 4CAM, whereas the experimental counterpart is only 158 (2)°. Hence, this intermolecular hydrogen bonding effect is perceived to be dominant in the DFT/B3LYP and HF methods than in the experimental methods.

The molecular structure of the 4CAM with the numbering scheme for the atoms and 50% probability displacement ellipsoids.

Optimized molecular geometry and atomic numbering scheme by HF (a) DFT; (b) methods for 4CAM and HF; (c) DFT; (d) for 4CAN.

Atomic charges and molecular geometry parameters calculated from experimental and theoretical calculations of the N–H· · ·O hydrogen bond.
In 4CAM, the planes of the cation and maleate anion are inclined at an angle 15.6 (1)° in the single crystal XRD, these planar angles are calculated at 73.6° and 73.9° in HF and DFT methods respectively. These deviations occur due to the reorientation of N–H ⋯O intermolecular hydrogen bond. The C–O bond distances of the carboxylate group from the maleate anion are 1.182–1.302 Å at the HF level and 1.209–1.330 Å by the B3LYP method, whereas in experimental methods it is 1.234 (2)–1.267 (2) Å (table 2). The planarity of the maleate anion is confirmed by the torsion angles.
In 4CAN, the nitrate group exists in a planar orientation with the O–N–O angles varying from 115.5 to 127.6° in HF level and from 115.1 to 128° in the DFT/ B3LYP method. The average bond lengths varied at 1.189–1.318 and 1.222–1.379 Å in the HF and DFT/ B3LYP methods respectively (table 3). One of the N–O bond distances (1.318 Å in HF and 1.379 Å in B3LYP) in the nitrate anion increases more than the literature value (∼1.240 Å). This elongation is due to the N–H ⋯O hydrogen bond between the cation and anion. It shows the possible charge transfer between donor N and acceptor O atoms. This hydrogen bonding effect is less in the HF level, which is revealed in the computed spectra. The –NH2 stretching mode of NH\(_{\mathrm {3}}^{\mathrm {+}}\) group is observed to be low-intense peak in the HF level, whereas it is the strongest among the computed vibrational peaks in the DFT/B3LYP level.
The optimized C–C bond lengths of phenyl ring are in the falling range 1.381 −1.392 Å, according in the theoretical calculations, whereas the single-crystal XRD reveals the C–C bond lengths in the range of 1.368–1.376 Å. On comparison, it is observed that most of the optimized bond lengths are slightly deviated from the experimental values. Because theoretical calculations were carried out for the molecules in the free gaseous phase and the experimental results corresponding to the molecules are in the solid crystalline state.
3.2 Mulliken Charge Analysis
In general, Mulliken atomic charge calculation has an important application of quantum chemical calculations to molecular systems. It plays a vital role in the packing of crystals in the solid state by means of intermolecular interaction and it has significant influence on dipole moment, polarizability, electronic structure and vibrational modes.[17] The Mulliken charge analyses of molecules 4CAM and 4CAN are calculated at the HF and DFT/ B3LYP levels for the molecule under study which are given in table 4 (a), 4 (b) and 4 (c) and the corresponding population analysis graph is shown in figure 3. The charge distribution of 4-chloroanilinium cation shows that the five carbon atoms attached to hydrogen atoms (C1 C3, C4, C5 and C6) are negative. Out of five carbon atoms in the 4-chloroanilinium cation, the carbon C5 atom has more negativity than the others, because the carbon (C5) atom is attached to NH\(_{\mathrm {3}}^{\mathrm {+}}\) group. The elongation of C −N bond distance due to the protonation is reiterated with the electronegative repulsion between these two atoms (C5 and N16). Interestingly, another carbon (C2) atom from the cation is attached in the chlorine (Cl11) atom and both of them have electropositive charge. The characteristic feature of Cl atom is electropositive and it does not participate in any hydrogen bond. In the present case, both the methods predict that the Cl atom of the cation (Cl11) is positive (0.277 e in HF and 0.340 e in DFT) for 4CAM and (0.275 e in HF and 0.335 e in DFT) for 4CAN. This may be accorded with the formation of hydrophobic region in the crystal with the presence of the Cl atoms and the absence of N −H⋯Cl hydrogen bond.
In the 4CAM anion, all the oxygen atoms have electronegative charges. Out of four oxygen atoms, O19 (–0.435 e in HF and 0.332 e in DFT) and O25 (–0.387 e in HF and –0.274 e in DFT) have more electronegativity than the others. These two oxygen atoms behave as a donor and acceptor in the O–H ⋯O intramolecular hydrogen bond and these intramolecular hydrogen bonds are also observed in single-crystal XRD.
The hydrogen atom in the 4CAN anion has more electropositive charges (0.491e in HF and 0.469 e in DFT) because it is surrounded by two electronegative atoms, viz., nitrogen (N16) and oxygen (O14). This hydrogen atom plays a key role in the solid state through the N–H ⋯O intermolecular hydrogen bond.

The atomic charges plots by HF (a) and DFT; (b) methods for 4CAM and HF; (c) DFT; (d) methods for 4CAN.
3.3 Vibrational Analysis
Vibrational spectroscopy can give valuable information about the hydrogen bonding forces and strength of the intermolecular bonds in addition to symmetry of the individual species. The effect of hydrogen bond is very important in crystal environment. The strong, normal and weak hydrogen bonds cause red shifting of stretching mode vibrations and blue shifting of deformation mode vibrations with different wave numbers depending on their strength. Normally, the vibrational shifts of the stretching modes are greater than that of the deformation modes. This indicates that the linear distortion is much greater than the angular distortion.
The molecular structures of 4CAM and 4CAN have various functional groups such as –NH\(_{\mathrm {3}}^{\mathrm {+}}\), -CH, -C-N, -C-C-, -OH, -C =O, disubstituted benzene ring, etc. The vibrational bands of these groups are expected to change in their intensity and position due to their environments.[18] Complete vibrational analyses of the 72 fundamental modes of 4CAM and 51 fundamental modes of 4CAN are attempted and predicted by the quantum chemical calculations and compared with their experimental vibrational spectra. The optimized structural parameters are used in the calculation of vibrational frequencies to characterize all stationary points as minimum. The calculated vibrational wave numbers, measured by FT-IR and FT-Raman band positions with their corresponding assignments for the molecules 4CAM and 4CAN, are tabulated in tables 5 and 6, respectively. The FT-IR and FT-Raman spectra of the compounds are shown in figures 4 and 5, respectively.

Comparative representation of FT-IR spectra of 4CAM and 4CAN.

Comparative representation of FT-Raman spectra of 4CAM and 4CAN.
3.3.1 N–H Vibrations
In general, the N–H stretching vibrations from the primary amines occur in the region 3600–3300 cm−1.[19,20]In the present case, 4-choloanilinium cation has more intense peak calculated at 3588 cm−1 and 3504 cm−1 in B3LYP level to assign the NH2 asymmetric and symmetric stretching modes, respectively, for the molecules 4CAN. Similarly, the asymmetric and symmetric –NH2 stretching occurred as the less-intense peak at 3586 and 3499 cm−1 for the molecule 4CAM. In 4CAN, the medium band observed at 3078 cm−1 in Raman spectrum is assigned to the N–H stretching. This stretching vibration was calculated as a very strong band at 3324 and 2907 cm−1 in HF and B3LYP level. In 4CAM, the same stretching vibration is observed as a weak band at 3041 cm−1 in IR spectrum and strong band at 3043 cm−1 in Raman spectrum. This corresponding vibration is calculated at 3324 and 2971 cm−1 in HF and B3LYP methods, respectively. These calculated stretching vibrations are occurred as a very strong peak in theoretical methods due to the N–H ⋯O hydrogen bond. But, in the case of experimental IR spectra, the N–H ⋯O hydrogen bond trace is observed as a very broad spectrum around 3200–2200 cm−1 for both the molecules. The occurrences of sharp and broad peaks of the spectrum in the theoretical and experimental methods are due to the fact that the theoretical calculations are carried out in isolated gas state whereas the crystalline state is dominated with the excessive hydrogen bonding interactions. As the hydrogen atom is placed between two heavier electronegative atoms (donor and acceptor) the resulted vibration is observed at a very strong intensity peak for the N–H ⋯O band in theoretical computation. It confirms the relocation of proton from the acids to amino group.
The scissoring vibration of the amine group appears at 1630–1610 cm−1. As one of the H atoms of the amine group is strongly involved in the N–H ⋯O intermolecular interactions between ion pairs, the NH\(_{\mathrm {3}}^{\mathrm {+}}\) group vibration is reduced to –NH\(_{\mathrm {2}}^{\mathrm {}}\)molecular vibration. In molecule 4CAN, NH2of NH\(_{\mathrm {3}}^{\mathrm {+}}\) scissoring vibration was calculated at 1661 and 1641 cm−1 in DFT/B3LYP method. The vibration coincides with 4CAM at 1663 cm−1. In both the molecules, the HF level is more deviated from the DFT level. The vibrational mode mentioned above is not observed in the experimental spectra. In 4CAN, the medium band at 1601 cm−1 in Raman spectrum is assigned to the NH2 rocking vibration and the corresponding vibration is predicted at 1772 and 1628 cm−1 in HF and B3LYP method, respectively. In lower wavenumber regions, most of the bands are observed in the scissoring, rocking and wagging vibrations. It is in good agreement with the experimental results.
3.3.2 C–H, C–C and C–N vibrations
The aromatic C–H stretching vibrations are observed in the region 3100–3000 cm−1.[21]This is the characteristic region for identification of the C–H stretching vibrations. Also, the bands are not affected appreciably by the nature of the substituents in this region. The asymmetric C–H stretching is calculated at 3595 and 3204 cm−1 in HF and DFT/B3LYP methods, respectively for 4CAN. In 4CAM, the same vibration is observed at 3725 and 3204 cm−1 in HF and DFT/B3LYP methods respectively. The C–H symmetric stretching vibration is predicted at 3364 cm−1 in HF level and 3203 cm−1 in DFT/B3LYP method for 4CAN and 3363 cm−1 in HF and 3203 cm−1 in DFT/B3LYP methods for 4CAM. These asymmetric stretching vibrations are not observed in the experimental results. In 4CAM, the strong peak observed at 3082 cm−1 in Raman spectrum is assigned to the C–H stretching vibration; this stretching vibration is computed at 3332 cm−1 in the HF level and at 3167 cm−1 in the B3LYP level.
The substituted benzene ring has six C–C stretching vibrations. These vibrations mainly involve ‘quadrant stretching’ of the phenyl C–C bonds. But there is a small interaction with C–H in-plane bending vibration. There are two quadrant-stretching components in substituted benzenes are expected to appear in the regions 1620–1585 and 1590–1565 cm−1, respectively. The 1600 cm−1 doublet region is not frequency sensitive to the nature of substitution at ortho, meta and para positions. For these types of substitutions, the quadrant stretching vibrations are infrared inactive because all atoms from the ring are moving to the opposite directions. The C–C stretching vibrations mixed with NH2 deformation modes and observed in the modes of 41 and 42 for 4CAN molecule and 57 for 4CAM molecule. In 4CAM, the weak band at 1242 cm−1 in Raman spectrum and IR inactive is assigned to the C −N stretching mode. These modes are mixed with C–H in-plane deformation modes. The same vibration is computed at 1326 and 1262 cm−1 in the HF and B3LYP methods respectively. It is matched well with the 4CAN molecule.
3.3.3 O–H Vibrations of Maleate Anion
In general, free O–H stretching vibrations appeared around 3600 cm−1.[22]In the present case, the intense band is computed at 3748 cm−1 and 3290 cm−1 in the HF and DFT/B3LYP methods respectively (table 5). These results show that the DFT/B3LYP method gets downshifted to the literature value which is due to the strong O–H ⋯O intramolecular interaction in the maleate anion. It confirms the migration of the proton transfer between the two O atoms of the anion. The O–H...O stretching vibration in the intramolecular hydrogen bonds is crucial in the crystal packing. The medium band observed at 1685 cm−1 in Raman spectrum is assigned to O–H in-plane bending vibration. The theoretically computed frequency for O–H in-plane bending vibration by DFT/B3LYP method matches well with the experimental results. The medium band at 1460 cm−1 in IR spectrum is assigned to the O–H in-plane bending vibration mixed with C–H in-plane bending vibration. The O–H outof-plane bending vibration is observed at the weak band at 1089 cm−1 in the IR spectrum and the medium band at 1094 cm−1 in the Raman spectrum; this vibration is mixed with NH2 rocking vibration. In lower wavenumber regions most of the bands assigned to the O–H in-plane bending vibrations are mixed with the C–H in-plane bending vibration.
3.3.4 Vibrations of the Nitrate Anion
In the free state the nitrate group has D3h symmetry and its normal modes of vibrations are \(A_{1}^{\prime }\) (ν 1), \(A_{2}^{\prime \prime }(\nu _{\mathrm {2}})\) and \(E^{\prime }\)(ν 3 and ν 4). In which, ν 1 is Raman active, ν 2is IR active, ν 3 and ν 4are both IR and Raman active. In the free state of the NO\(_{\mathrm {3}}^{\mathrm {-}}\) anion, the symmetric stretching vibration occurs at 1049 cm−1 the symmetric bending vibration appears at 530 cm−1 and ν 3 and ν 4 (asymmetric stretching and bending modes) occur at 1355 and 690 cm−1, respectively.[23–25]
The medium band at 1752 cm−1 in IR spectrum is assigned to the N =O stretching vibration. The same vibration is computed at 1928 and 1449 cm−1 in HF and 1734 and 1328 cm−1 in DFT/B3LYP methods respectively (table 6). The weak band at 1042 cm−1 in IR spectrum and medium band at 1047 cm−1 in Raman spectrum is assigned to the N–O in-plane bending vibration. The corresponding vibration is computed at 1155 and 1032 cm−1 in the HF and DFT/B3LYP methods respectively. The out-of-plane deformation is observed at 859 (HF) and 787 cm−1 (DFT/B3LYP) method. These deformation modes are mixed with the NH2 wagging vibration. These results show migration of proton transfer between the cation and nitrate group of the anion. This out-of-plane deformation is not present in the experimental results.
3.4 NBO Analysis
In the NBO analysis, the electronic structures are explained in terms of a set of occupied Lewis and a set of non-Lewis localized orbitals.[26] Also, delocalization effects can be identified by the presence of off-diagonal elements of the Fock matrix in the NBO basis. The NBO method gives information about interactions in both filled and virtual orbital spaces that could enhance the analysis of intra- and intermolecular interactions.
The delocalization effects of the present molecules can be identified by the diagonal elements of the Fock matrix in the NBO basis. The strengths of these delocalization interactions, E (2), are estimated by second-order perturbation theory. The second-order Fock matrix was calculated to evaluate the donor–acceptor interactions.[27] These interactions result in loss of occupancy from the localized NBO of the idealized Lewis structure into an empty non-Lewis orbital. For each donor (i) and acceptor (j), the stabilization energy E (2) associated with the delocalization i→j is estimated as
where q i is the donor orbital occupancy, ε i and ε j are the orbital energies and F(i, j) is an off- diagonal NBO Fock matrix element. NBO analysis provides an efficient method for studying intra- and intermolecular bonding and interaction among bonds and also provides a convenient basis for investigating charge transfer or conjugative interaction in molecular systems. Some electron donor orbital, acceptor orbital and the interacting stabilization energy resulting from the second-order micro-disturbance theory are reported.[28,29]
NBO analyses of both the molecules have been carried out by the B3LYP/HF method with 6-311 ++ G(d,p) basis set, in order to explain the intramolecular, re-hybridization and delocalization of electron density within the molecules. In the NBO analysis of the hydrogen bonded systems, the most significant element is the charge transfer between the lone pairs of the proton acceptor and the anti-bonds of the proton donor. In case of the present molecular structures, intramolecular charge transfer leads to C–C bond in the aromatic ring. As a result of this intramolecular charge transfer, the aromatic ring of the molecules is stabilized, which is due to the N–H ⋯O intermolecular hydrogen bonds. The interaction between bonding and anti-bonding of C–C bond has large stabilization energy due to the hyperconjugative interactions of π (C–C) →π* (C–C). The intermolecular hyperconjugative interactions π (C1–C2) →π* (C3–C4) lead to stabilization of 39.47 kJ/mol in HF and 19.45 kJ/mol in DFT method for 4CAN and 38.46 kJ/mol in HF and 18.52 kJ/mol in DFT method for 4CAM. This interaction enhances further conjugation with anti-bonding orbital of π*(C5–C6), which leads to strong delocalization energies for both the molecules. The same kind of interaction was identified as π (C3–C4), which is presented in tables TS1 and TS2 (supplementary material) for both the molecules. The lone pair chlorine atom in both molecules has the highest occupancies and the stabilization energies also increase. But in the present case, the chlorine atom does not participate in any intermolecular hydrogen bonds because the chlorine atom has an electropositive charge. Much of the delocalization takes place in the aromatic ring, which is due to the proton transfer between the cation and anion. This larger delocalization of the 4-chloroaninline cation stimulates the bioactivity nature.
3.5 HOMO and LUMO analysis
In general, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are called as frontier molecular orbitals. These orbitals give information about the chemical hardness, electro-negativity and chemical potential of the molecule.[30] The HOMO and LUMO energies of the molecules 4CAM and 4CAN were calculated by the B3LYP/HF methods with basis set of 6-311 ++ G(d,p) as listed in table TS3 (supplementary material). The corresponding energy level diagrams are shown in figures 6 and 7, respectively. From these figures, it is observed that the energy levels 320 and 429 in 4CAM and 4CAN are in the energy ranges of -104.843 a.u. to 219.354 a.u. and -101.563 a.u. to 215.760 a.u. in HF and DFT method, respectively, for both molecules. The energy difference between HOMO and LUMO analyses is called as energy gap, which is an important feature for stability of the structures. The calculated energy values of LUMO and HOMO are 0.027 a.u. and -0.328 a.u. for 4CAM at the HF level and -0.109 a.u. and -0.250 a.u. by the DFT method, -0.028 a.u. and - 0.330 a.u. at the HF level, -0.079 a.u. and -0.250 a.u. in DFT level for 4CAN. The frontier molecular orbital energy gap value of 4CAM molecule is 0.355 a.u. at the HF and 0.141 a.u. at the DFT level and the same energy gap of 4CAN is 0.358 a.u./ 0.171 a.u. in the HF/B3LYP method. The results show that the molecules are having a small frontier energy gap. This small energy gap is associated with a high chemical reactivity, because a huge amount of charge transfer occurs within the molecule. The narrow energy gap between HOMO and LUMO supports the possible bioactivity of the molecule.

Energy level diagram of molecular orbits of 4CAM by (a) HF and; (b) B3LYP levels.

Energy level diagram of molecular orbits of 4CAN by (a) HF and; (b) B3LYP levels.
4 Conclusions
The experimental and theoretical investigations reveal the proton transfer between the 4-chloroanilinium cation and maleate and nitrate anions. This proton transfer provides the support for the molecular assemblies dominated by the strong inter and intramolecular N–H ⋯O and O–H ⋯O hydrogen bonds. The optimized molecular structure, vibrational frequencies and NBO analysis of 4-chloroanilinium salts were found out by quantum chemical calculations at the HF and DFT/B3LYP methods invoking 6-311 ++G(d,p) basis sets. The shifting of vibrational bands due to the intermolecular hydrogen bonds was analyzed with experimental and theoretically computed vibrational spectra. Natural bond orbital analysis indicates the strong intermolecular interactions and in an agreement with the experimental intermolecular hydrogen bonding results. Natural bond orbital analysis of the molecule confirms that the charge transfer caused by π electron cloud movement from donor to acceptor must be responsible for the bioactivity. The value of the energy gap between the HOMO and LUMO gived the information about chemical softness, chemical hardness, electronegativity and chemical potential of the molecules.
References
Deechongkit S, Nguyen H, Powers E T, Dawson P E, Gruebele M and Kelly J W 2004 Nature 430 101
Lopez N, Vos T E, Arif A M, Shum W W, Noveron J S and Miller J S 2006 Inorg. Chem. 45 4325
Senes A, Ubarretxena-Belandia I and Engelman D M 1998 Proc. Natl. Acad. Sci. USA p. 9056
Parker L L, Houk A R and Jensen J H 2006 J. Am. Chem. Soc. 128 9863
Aakeroy C B and Seddon K R 1993 Chem. Soc. Rev. 22 397
Desiraju G R 1989 In Crystal Engineering: The Design of Organic Solids (Amsterdam: Elsevier)
Harms K and Wocadlo S 1995 In XCAD4 (Germany: University of Marburg)
Sheldrick G M 2008 Acta Cryst. A 64 112
Farrugia L J 1997 J. Appl. Cryst. 30 565
Macrae C F, Edgington P R, McCabe P, Pidcock E, Shields G P, Taylor R, Towler M and Van de Streek J 2006 J. Appl. Cryst. 39 453
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam J M, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas O, Foresman J B, Ortiz J V, Cioslowski J and Fox D J 2013 Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT
Schlegel H B 1982 J. Comput. Chem. 3 214
Hohenberg P and Kohn W 1964 Phys. Rev. B 136 864
Becke A D 1993 J. Chem. Phys. 98 5648
Dennington R, Keith T and Millam J 2009 J. Semichem Inc., Shawnee Mission KS
Anitha R, Athimoolam S, Asath Bahadur S and Gunasekaran M 2012 Acta Cryst. E 68 o959
Sidir I, Sidir Y G, Kumalar M and Tasal E 2010 J. Mol. Struct. 964 134
Ploug-Sørensen G and Andersen E K 1983 Acta Cryst. C 39 112
Kurt M, Yurdakul M and Yurdakul S 2004 J. Mol. Struct. (Theochem.) 711 25
Krishnakumar V and John Xavier R 2005 Spectrochim. Acta Part A 61 253
Socrates G 1980 In Infrared Characteristics Group Frequencies (New York: John Wiley)
Lampert H, Mikenda W and Karpten A 1997 J. Phys. Chem. A 101 2254
Banwell C N and Mccash E M 1995 In Fundamentals of Molecular Spectroscopy 4th ed. (New Delhi: Tata McGraw Hill)
Dollish F R, Fateley W G and Bentley F F 1973 In Characteristic Raman Frequencies of Organic-Compounds (New York: Wiley)
Rosenthal M R 1973 J. Chem. Educ. 50 331
Reed A E, Curtis L A and Weinhold F A 1988 Chem. Rev. 88 899
Szafran M, Komasa A and Adamska E B 2007 J. Mol. Struct. (THEOCHEM) 827 101
James C, Amal Raj A, Reghunathan J. I H and Jayakumar V S 2006 J. Raman Spectrosc. 37 1381
Na L J, Rang C Z and Fang Y Z J 2005 Zhejiang Univ. Sci. B 6 584
Fleming I 1976 In Frontier Orbitals and Organic Chemical Reactions (New York: John Wiley)
Acknowledgement
The authors SA and SSK thank the Department of Science and Technology, SERB for the financial support of this work in the form of Fasttrack Research Project scheme.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information
X-ray crystallography data of the compound, 4CAM, is available in Cambridge Structural Database (CSD), Ref No: 877151.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
ANITHA, R., GUNASEKARAN, M., KUMAR, S.S. et al. Structural and vibrational spectral studies on hydrogen bonded salts: 4-chloroanilinium maleate and nitrate. J Chem Sci 127, 1435–1450 (2015). https://doi.org/10.1007/s12039-015-0914-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-015-0914-y