
Abstract
The ubiquitin (Ub)-proteasome system (UPS) is considered as a central protein degradation system in all eukaryotes. The UPS comprises of several factors such as Ub and Ub-like molecules, Ub hydrolases, E3 Ub ligases, and the proteasome itself. Numerous studies have demonstrated that the dysfunction of UPS plays an essential role in the pathogenesis and progression of Alzheimer’s disease (AD). Furthermore, current evidence has suggested that the UPS components can be connected with the initial stage of AD that is characterized by synaptic dysfunction, and to the late phases of AD, marked by neurodegeneration. In AD patients, the accumulations of insoluble protein in the brain can be caused by overload or dysfunction of the UPS, or by conformational alterations in the protein substrates that prevent their degradation and recognition by the UPS. Synaptic dysfunction is also caused by defective proteolysis that has found in the initial stage in AD as the UPS is widely recognized to play a pivotal role in the regular activities of synapses. Conversely, its precise cause and pathogenesis are unclear. Presently accepted medicines for AD give symptomatic relief, though they are unable to stop the progression of the disease. Besides, the components of the cellular quality control system demonstrate a significant emphasis on the advancement of targeted and effective treatments for AD. In this review, we focus on the role of UPS in the pathogenesis of AD and highlight how the UPS-linked treatments influence in the management of AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder of the central nervous system (CNS), which is featured by gradual loss of cognitive functions such as memory, attention, judgment, comprehension, language, and reasoning, ultimately resulting in severe dementia (Uddin et al. 2019a, 2019b; Kabir et al. 2019a, 2019b). Intracellular neurofibrillary tangles (NFTs) and extracellular beta-amyloid (Aβ) plaques are the main pathological hallmarks of AD (Price and Sisodia 1998; Uddin et al. 2019c, 2019d; Hossain et al. 2019; Mathew et al. 2019; Mamun et al. 2020). The Aβ plaques result from precise proteolytic processing of amyloid precursor protein (APP) that is positively controlled by the presenilins 1 (PS1) and presenilins 2 (PS2) (De Strooper et al. 1998; Al Mamun and Uddin 2020). Currently, PS1 and PS2 have been revealed to be substrates of the ubiquitin (Ub)-proteasome system (UPS) (Kim et al. 1997; Marambaud et al. 1998; Steiner et al. 1998; Johnston et al. 1998). Additionally, UPS is accountable for expressing various fundamental cellular functions.
Numerous neurodegenerative diseases such as AD, transmissible spongiform encephalopathies, Parkinson’s disease, Huntington’s disease, neurodegeneration following spinal cord injury, and amyotrophic lateral sclerosis are connected with the UPS dysfunction (LEIGH et al. 1991; Neumann et al. 2006; Uddin et al. 2018a, 2020b). In these neurodegenerative disorders, the influence of the UPS may be linked to deficits in the removal of misfolded proteins leading to the intracellular accumulation of protein, neuronal cell death, and cytotoxicity (Demuro et al. 2005; Schwartz and Ciechanover 2009; Sahab Uddin and Ashraf 2020).
The UPS also plays a central role in neuronal signaling pathways that control the release of neurotransmitter, synaptic plasticity, and synaptic membrane receptor turnover (Zhao et al. 2003; Patrick 2006). In this article, we discuss the role of UPS in the pathogenesis of AD and emphasize how the UPS-associated treatments to combat AD pathogenesis.
Molecular Biology of Ubiquitin-Proteasome System
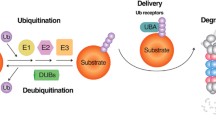
The UPS is situated in the cytosol and the nucleus. Ub is a protein of 76-amino acid residues that are vastly evolutionarily conserved in all eukaryotes (Hershko and Ciechanover 1998; Nandi et al. 2006). Moreover, the ubiquitination process is involved in 3 successive stages concerning 3 enzymes known as activating (E1), conjugating (E2), and ligating (E3) enzymes (Fig. 1). Furthermore, the 26S proteasome is a large multi-subunit complex that plays a pivotal role in the degradation of the Ub-conjugated proteins. The Ub chain is prepared by E1 and E2 enzymes, which are subsequently attached to target proteins with the help of the E3 enzyme. The ubiquitination process starts when the Ub-activating enzyme E1 triggers an Ub molecule through an adenosine triphosphate (ATP)–dependent mode. Ub is then bound with an internal E1 Cys moiety through an intermediate thiolester bond producing E1-S~Ub. Subsequently, Ub is shifted to one of the various E2 forms. Afterward, the attachment of Ub to the protein substrate is catalyzed by several E3s—a large and different set of proteins with discrete motifs (Ciechanover 1994).

General overview of the ubiquitin (Ub)-proteasome system. The ubiquitination process starts when the Ub-activating enzyme E1 triggers a Ub molecule through adenosine triphosphate (ATP)-dependent mode. Ub is then bound with an internal E1 Cys moiety through an intermediate thiolester producing E1-S~Ub. Subsequently, Ub is shifted to one of the various E2 forms. Then, the E2 interacts with a substrate-bound E3 Ub ligase, which catalyzes the transfer of Ub to a lysine residue in the substrate to generate a stable isopeptide bond. Multiple cycles of ubiquitylation can take place to form a polyubiquitin chain on the substrate. Polyubiquinated proteins are recognized and degraded by the 26S proteasome. The polyubiquitin chain is disassembled by DUBs. The free ubiquitin monomers can be reused to tag other substrates. Abbreviations used are Ub, ubiquitin; DUBs, deubiquitinating enzymes
The variety of diverse Ub protein ligase E3s could distinguish a precise substrate due to its high specificity as well as the selectivity to the UPS (Ciechanover 1998). Furthermore, the most significant recognition pattern is the destabilizing N-terminus amino acids including lysine and arginine. This distinctive N-terminus destabilizing residues could assess the half-life of an intracellular protein which is known as the N-end rule. Subsequently, multiple cycles of ubiquitylation take place to form a polyubiquitin chain on the substrate. Then, polyubiquitinated proteins are recognized and degraded by the 26S proteasome. Then, the polyubiquitin chain is disassembled by deubiquitinating enzymes (DUBs), and the free ubiquitin monomers can be reused to tag other substrates.
Ubiquitin-Proteasome System in the Pathogenesis of Alzheimer’s Disease
Degradation of protein is mainly carried out by proteasomes in the cytosol and the nucleus of all cells (Lecker et al. 2006; Uddin et al. 2018b). In the nervous system, these processes are regulated by the Ub-proteasome pathway (UPP). The damage of the UPP-dependent protein degradation system leads to the development of several neurodegenerative diseases such as AD. In recent times, many researchers have observed that UPS has an impact on the AD pathogenesis, and ubiquitinated proteins are greatly present in AD patients (Gentier and van Leeuwen 2015; Tramutola et al. 2016). UPS controls not only the metabolism of Aβ but also the degradation of tau through the 26S proteasome. Conversely, the malfunction of these proteins in the neurons can cause both the aggregation of ubiquitinated proteins and the modifications in the combination of proteasome subunits, which reduces the function of proteasome and α-secretase, triggering the generation of Aβ. Nonetheless, the precise fundamental mechanism of this progression remains unclear. The activity of proteasome is reduced in the diverse parts of the AD brain including, the inferior parietal lobe, the superior and middle temporal gyri, and the parahippocampal gyrus, which specifies the functional failure of UPP throughout the AD pathogenesis (Necchi et al. 2011).
Furthermore, the relationship between the impairment of synaptic plasticity and the UPP is more widely investigated in AD (Vriend et al. 2015; Cheng et al. 2016). There are some cellular events such as the oxidation of DUBs, the aggregation of mutated Ub, the changes of proteasome subunits, and the downregulation of E1 and E2 enzymes are found both in transgenic mice with Aβ and in AD patients (Choi et al. 2015). The accumulations of Aβ and the hyperphosphorylation of tau, as well as neurodegeneration in AD, are closely connected with the dysfunction of UPS (Fig. 2). The impairment of UPS leads to the generation of Aβ by inducing the activity of α-secretase in AD neurons (Gentier and van Leeuwen 2015). Normally toxic Aβ is generated by inducing β-secretase targeted APP cleavage and produces a C-terminus portion (Uddin and Kabir 2019). Then the γ-secretase plays an essential role in the cleavage of this portion and generates toxic Aβ40 and Aβ42 portions (Uddin et al. 2020a). The inhibitors of the proteasome can reduce the activity of β-secretase by the upregulation of APP-C99 (Renziehausen et al. 2015). The primary sign for the pathological connection between UPS and tau is resulting from the recurrent colocalization and the aggregation of Ub in paired helical filaments (PHFs) and NFTs. Research detected that polyubiquitinated tau within PHF is primarily in the Lys48-related poly-Ub form, which is the most recognized degradation signal. Moreover, this powerfully advocates the role of UPS-targeted tau removal in defense against the pathogenesis of AD (Cripps et al. 2006).

The ubiquitin-proteasome system in the pathogenesis of Alzheimer’s disease. Intracellular neurofibrillary tangles (NFTs) and extracellular beta-amyloid (Aβ) plaques are the main pathological hallmarks of AD. The figure shows only increased generation of Aβ42 (a splice variant of Aβ) due to the mutations of APP and PS gene, whereas the formation of tau takes place owing to the mutations of MPAT and GRN gene. The roles of the UPS in the steps leading to AD pathogenesis are shown in blue boxes. Furthermore, the ubiquitin mutant UBB + 1 is also connected with AD, although it is vague that how the mutation contributes to discrete pathologies in many patients remains mysterious. Abbreviations used are: Aβ, amyloid beta; AD, Alzheimer’s disease; APP, amyloid precursor protein; GRN, progranulin, MAPT, microtubule-associated protein tau; PHFs, paired helical filaments; PS, presenilin; Ub, ubiquitin; UPS, ubiquitin-proteasome system
However, the direct connection between the UPS and the AD pathogenesis was acknowledged with the detection of a frameshift mutation in the Ub transcript that leads to the elongation of the molecule with 20 amino acids moiety UBB + 1 (Fig. 2), which had been selectively found in AD patients who were affecting with late-onset AD (van Leeuwen et al. 1998). UBB + 1 is an effective acceptor for polyubiquitination, although it could not be activated by E1 (because of the absence of vital G76 moiety) and be shifted to a substrate or to another Ub portion. The resultant chain of poly-Ub is difficult to disassemble by DUBs, especially isopeptidase T (Lam et al. 2000) which needs for its function in a manifested G76 moiety at the proximal Ub portion. Moreover, the aggregated poly-Ub chains block the degradation of the proteasome (Lindsten et al. 2002) that leads to the apoptosis of neurons (Bardag-Gorce et al. 2002). Hence, UBB + 1 expression rises noticeably with aging in the brain, which can possibly result in dominant suppression of the UPS, leading to the aggregation of toxic proteins with the neuropathologic outcome including AD.
Prospective Targets of Ubiquitin-Proteasome System for Alzheimer’s Disease
Numerous ubiquitination enzymes are prospective targets for AD therapies that control not only the Aβ metabolism but also the UPS in AD brains. E2-25K is one of the distinctive Ub-conjugating enzymes that is upregulated and leads to the toxicity of Aβ (Song et al. 2008). Aβ raises the E2-25K/Hip-2 expression that subsequently stabilizes the caspase-12 and apoptotic protein by suppressing the activity of proteasome (Song et al. 2003, 2008). The expression of E2-25K/Hip-2 knockdown inhibits neuronal cell death in AD mice model and in cultured neurons. In a study, Lonskaya et al. revealed that the intracellular aggregation of Aβ and damaged proteasome activity could be restored by the Ub E3 ligase parkin (Lonskaya et al. 2012, 2014). Furthermore, parkin has the capability to defend neurons against diverse insults, which leads to the prevention of AD (Fig. 3). The expression of parkin can decrease the level of Aβ, and it also reverses damaged long-term potentiation and behavioral aberrations of the AD model mouse by reversing the deleterious effects of Aβ on the proteasome.

Potential therapeutic targets of ubiquitin-proteasome system for AD. Ubiquitin proteasomal dysfunction causes the accumulation of Aβ and tau, which leads to neuronal cell death and ultimately resulting in Alzheimer’s disease. The polyubiquitinated tail is the best potential therapeutic targets for parkin, UCHL1, CHIP, HRD1 because it predominantly regulates substrate specificity and selectivity and plays an important in the management of AD. Abbreviations used are AD, Alzheimer’s disease; Aβ, amyloid beta; UCHL1, ubiquitin carboxy-terminal hydrolase L1; CHIP, C-terminus of HSC70-interacting protein; HRD1, The HMG-CoA reductase degradation protein 1
Moreover, parkin increases beclin-dependent autophagy by which it helps the removal of Aβ (Khandelwal et al. 2011). Conversely, Ub carboxyl-terminal hydrolase 1 (UCHL1) has been found in Ub-enriched inclusion bodies in AD brains. According to the study by Zhang et al., the overexpression of UCHL1 enhances contextual memory and recovers synaptic activities in APP/PS1 model mouse (Zhang et al. 2015) by decreasing the β-secretase enzyme 1 (BACE1) levels and subsequently reduces the cleavage products of BACE1 (APPC-end portion C99 and Aβ) (Zhang et al. 2012; Guglielmotto et al. 2012). BACE1 is a novel substrate of E3 ligase C-terminus of Hsc70-interacting protein (CHIP) that advocates the destabilization of BACE1 by attaching through the U-box domain of CHIP (Singh and Pati 2015). Additionally, CHIP decreases the level of BACE1 by enhancing its ubiquitination and proteasomal degradation that subsequently reduces the processing of APP and Aβ generation in neurons. Moreover, the level of HMG-CoA reductase degradation protein 1 (HRD1) is considerably reduced in the cerebral cortex of AD human model, which is inversely connected with the generation of Aβ (Saito et al. 2010; Gerakis et al. 2016). The increased expression of HRD1 also enhances the ubiquitination and degradation of APP that results in reduced Aβ generation (Kaneko et al. 2010).
Crosstalk of Alzheimer’s Targets and Ligands—Ubiquitin-Proteasome System Components
Advancement of drugs and treatment strategies that target UPS components would need a well comprehending of the role of proteolysis in the progression of AD. The proteasome, Ub-conjugating enzymes, and DUBs are potential drug targets in the UPS. Furthermore, E3s are excellent prospective therapeutic targets amid other enzymes since they predominantly regulate substrate specificity (Fig. 3). As the substrate-binding area possesses specificity to E3s and allosteric modification of this area using small molecules can cause specificity to E3s either increase or decrease affinity to specific substrates, which is one of the ways of regulating aggregation of the ubiquitylated substrate (Upadhya and Hegde 2005). Moreover, selective engineering of the UPS components allows modification of their transfer to a precise affected area and consequent degradation of particular aggregated ubiquitylated proteins or protein accumulates could offer a substitute strategy to small allosteric molecules (Upadhya and Hegde 2005). Conversely, no robust E3 agent has been applied in AD until now.
In addition, DUBs can be prospective drug targets when a specific role for these enzymes in AD is recognized. The regulation of specific DUBs by small molecules improves the deubiquitylation of poly-Ub chains of mutant UBB + 1 that can be an additional probability since these chains suppress proteasomes in the AD brains (Lam et al. 2000). Activation of the proteasome is another uninvestigated arena for novel drug discovery. Even though numerous proteasome inhibitors are available, however, there are no potent drugs that can improve the proteasome activity. As the aberrant accumulation of protein and the inhibition of proteasome are the usual hallmarks of AD and other neurodegenerative disorders, improvement of proteasome function with the aid of small molecules can be an effective method to eliminate the accumulations that aggregate in the AD brain (Upadhya and Hegde 2005). Resveratrol is a naturally occurring polyphenol found abundantly in grapes, grape juice, and red wine, which is suspected to possess antioxidant and neuroprotective activities (Savaskan et al. 2003; Jang and Surh 2003; Han et al. 2004). Numerous researches demonstrated that resveratrol has a strong anti-amyloidogenic activity by decreasing the levels of Aβ-produced and delays Aβ-induced toxicity in different experimental models (Savaskan et al. 2003; Jang and Surh 2003; Han et al. 2004; Marambaud et al. 2005). It has also been reported that resveratrol works by enhancing the intracellular degradation of Aβ using a mechanism that connects the proteasome (Marambaud et al. 2005). Ultimately, these investigations recommend a potential application for this compound in the management of AD.
Targeting the Ubiquitin-Mediated Protein Degradation in Alzheimer’s Disease
The aggregation of toxic Aβ is controlled by the quality control systems of the cell (autophagy, molecular chaperones, and the components of UPS) (Morawe et al. 2012). Ub-mediated protein degradation takes place through two chief catabolic systems, for instance, the autophagy (endosomal/lysosomal system) and the ATP-dependent, non-lysosomal proteolysis system termed UPS.
Autophagy
Lysosomes damage normal and accumulated proteins through autophagy, which are generally observed under injury or stress conditions. Autonomous changes in the endocytic pathway trigger the lysosomal system and raise the quantity and density of lysosomes as well as the expression of the gene (Cai and Yan 2013). Furthermore, the latter effect plays a central role in the synthesis of all types of lysosomal hydrolases, such as cathepsin (Nixon et al. 2001). Lysosomal cathepsin B is upregulated both by the modulator 2-Phe-Ala-diazomethyl ketone (PADK) and by the accumulation of protein. Moreover, systemic administration of PADK enhances the activity of cathepsin B that raises the removal of intracellular Aβ and reduces its extracellular aggregation. Therefore, regulators of lysosomal activity exhibit great potential for treating neurodegenerative disorders including AD.
Autophagy decreases the aggregation and expedites the removal of regular/mutant Aβ (Cai and Yan 2013). The Aβ, which is produced in endosomes and autophagic vacuoles, is transferred to lysosomes wherein it is removed through lysosomal proteolysis under standard conditions (Yang et al. 2011). In a study by Yang et al., reported that in TgCRND8 transgenic mice (Yang et al. 2011), increasing lysosomal proteolysis enhanced the removal of autophagy substrates that decreased extracellular and intracellular levels of Aβ and recovered many cognitive dysfunctions. Furthermore, Cecarini et al. (Cecarini et al. 2012) exposed in human SH-SY5Y neuroblastoma cells stably transfected either 717 valine-to-glycine APP-mutated gene or with wild-type APP gene (Cecarini et al. 2012), and increased expression of the APP mutant isoform associated with a rise in oxidative stress as well as a remodeled pattern of protein degradation, with both significant suppression of proteasome activities as well as impairment in the autophagic flux.
In addition, rapamycin suppresses motor activity and increases autophagy; thereby it could be beneficial in stopping or recovering AD pathology. Rapamycin also suppresses the formation of NFT and the phosphorylation of tau, as well as decreases cognitive dysfunctions (Cai and Yan 2013). Therefore, agents that trigger autophagy can decrease or remove protein accumulations (Lane et al. 2012). The two proteolytic systems including UPS and autophagy-lysosomal pathway (ALP) are primarily accountable for the quality control of cellular protein in neurons and their significant roles in the pathogenesis of AD. Both the UPS and ALP pathways control proteostasis, forming a single network to maintain the homeostasis of protein (Balch et al. 2008). Even though the UPS and ALP are deliberated for a long time as independent mechanisms, numerous studies show close crosstalk as well as coordination between both pathways (Korolchuk et al. 2010). Likewise, Aβ and C-terminal membrane fragment β are the two main detrimental proteins for neuronal function, and these two proteins are removed by the UPS and ALP pathways (Bustamante et al. 2013; Xiao et al. 2015; Wang et al. 2017; González et al. 2017; Yang et al. 2017). Moreover, the growing number of evidence stated that some specific enzymes of the ubiquitylation machinery play a pivotal role in both degradation pathways. Any interferences in normal molecular features of the UPS and ALP pathways are predominantly related to pathophysiological conditions that instigate the aggregation of abnormal proteins, for example in various neurodegenerative disorders, like AD.
Molecular Chaperones—Heat Shock Proteins
Molecular chaperones and the UPS are considered as the first and the second lines of defense against misfolded protein and accumulation. Chaperones control the folding of freshly synthesized proteins as well as the refolding or transport of misfolded proteins to protein degradation systems (Morawe et al. 2012). Higher molecular weight heat shock proteins (Hsps) (> 43 kD) is ATP-dependent, while lower molecular weight Hsps (12–43 kD) is ATP-independent. Numerous investigations (Wilhelmus et al. 2007; Salminen et al. 2011; Takalo et al. 2013; Ou et al. 2014; Blair et al. 2014) have revealed that the chaperone system can be targeted to advance treatment approaches for controlling AD (Jinwal et al. 2010). Generally, chaperones attach to tau and Aβ toxic protein and control their degradation. In addition, not only Hsp90 but also Hsp70 takes part in the metabolism of APP (Gao and Hu 2008).
Hsp70 is ATP-dependent and the main target for treating AD. Elevated levels of Hsp70 suppress its ATPase activity and maybe a successful treatment approach for AD (Jinwal et al. 2010). Bobkova et al. showed that the recombinant human Hsp70 decreased the formation of Aβ plaque in 5XFAD transgenic mice (Bobkova et al. 2013). Furthermore, methylene blue suppresses Hsp70 and raises the removal of tau at very high concentrations (O’Leary et al. 2010). Many studies demonstrated that curcumin was able to enhance the activity of Hsp70 and Hsp90 that could suppress or linger the formation of amyloid and decrease neuronal cell death (Mishra and Palanivelu 2008; Maiti et al. 2014). Moreover, Bcl2-linked athanogene proteins are a family of co-chaperones that forms a complex with tau and Hsp70 and can suppress the degradation of tau in cell cultures, raising the levels of both APP and tau (Elliott et al. 2007, 2009).
In addition, Hsp90 controls the stabilization and the misfolding of neurotoxic proteins and expedites tau pathology in AD (Sarah Kishinevsky et al. 2013). Suppressing Hsp90 decreases the levels of Ser/Thr-mutated tau, hyperphosphorylated tau, and the kinases, which are involved in continuous hyperphosphorylation (Jinwal et al. 2011). Furthermore, the inhibition of Hsp90 triggers small Hsps, Hsp70, and heat shock factors. This suppression also expedites the binding of Hsp70 with aberrant proteins to produce a complex that is ubiquitinated by CHIP and damaged via proteolysis. Therefore, the suppression of Hsp90 raises the degradation of tau and maybe a prospective therapeutic approach for tau-based neurodegeneration in AD (Sarah Kishinevsky et al. 2013).
Conclusions
The components of UPS are the key factors for the treatment of AD. The UPS also influences in the AD pathology through several mechanisms. Comprehensive knowledge of the mechanisms of proteasomal function is crucial for the advancement of novel therapeutic as well as diagnostic approaches for the management of AD. Additionally, for the advancement of effective medicines also needs in-depth knowledge of the role of proteasome inhibition and how neuronal cell death takes place in AD brains. Although, Aβ inhibits proteasomes in vitro, however, it remains unclear whether AD patients exhibit the same pattern. Moreover, future treatments of AD may decrease the accumulation of protein (both Aβ and tau) by targeting specific UPS components such as the Ub, polyubiquitinated tail, DUBs, and 26S proteasome. Finally, for the development of novel treatments of AD, we need to entirely comprehend in what way the UPS components interact in the degradation of the protein.
References
Al Mamun A, Uddin MS (2020) KDS2010: a potent highly selective and reversible MAO-B inhibitor to abate Alzheimer’s disease. Comb Chem High Throughput Screen 23. https://doi.org/10.2174/1386207323666200117103144
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319(80):916–919. https://doi.org/10.1126/science.1141448
Bardag-Gorce F, van Leeuwen FW, Nguyen V, French BA, Li J, Riley N, McPhaul L, Lue YH, French SW (2002) The role of the ubiquitin–proteasome pathway in the formation of Mallory bodies. Exp Mol Pathol 73:75–83. https://doi.org/10.1006/EXMP.2002.2451
Blair LJ, Sabbagh JJ, Dickey CA (2014) Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin Ther Targets 18:1219–1232. https://doi.org/10.1517/14728222.2014.943185
Bobkova NV, Garbuz DG, Nesterova I et al (2013) Therapeutic effect of exogenous Hsp70 in mouse models of Alzheimer’s disease. J Alzheimers Dis 38:425–435. https://doi.org/10.3233/JAD-130779
Bustamante HA, Rivera-Dictter A, Cavieres VA et al (2013) Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS One 8. https://doi.org/10.1371/journal.pone.0083096
Cai Z, Yan L-J (2013) Rapamycin, autophagy, and Alzheimer’s disease. J Biochem Pharmacol Res 1:84–90
Cecarini V, Bonfili L, Cuccioloni M et al (2012) Crosstalk between the ubiquitin–proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim Biophys Acta 1822:1741–1751. https://doi.org/10.1016/j.bbadis.2012.07.015
Cheng B, Anand P, Kuang A et al (2016) N-Acetylcysteine in combination with IGF-1 enhances neuroprotection against proteasome dysfunction-induced neurotoxicity in SH-SY5Y cells. Parkinsons Dis 2016:1–12. https://doi.org/10.1155/2016/6564212
Choi J, Gao J, Kim J et al (2015) The E3 ubiquitin ligase idol controls brain LDL receptor expression, ApoE clearance, and Aβ amyloidosis. Sci Transl Med 7:314ra184. https://doi.org/10.1126/scitranslmed.aad1904
Ciechanover A (1994) The ubiquitin-proteasome proteolytic pathway. Cell 79:13–21. https://doi.org/10.1016/0092-8674(94)90396-4
Ciechanover A (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 17:7151–7160. https://doi.org/10.1093/emboj/17.24.7151
Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ (2006) Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem 281:10825–10838. https://doi.org/10.1074/jbc.M512786200
De Strooper B, Saftig P, Craessaerts K et al (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391:387–390. https://doi.org/10.1038/34910
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280:17294–17300. https://doi.org/10.1074/jbc.M500997200
Elliott E, Tsvetkov P, Ginzburg I (2007) BAG-1 associates with Hsc70·tau complex and regulates the proteasomal degradation of tau protein. J Biol Chem 282:37276–37284. https://doi.org/10.1074/jbc.M706379200
Elliott E, Laufer O, Ginzburg I (2009) BAG-1M is up-regulated in hippocampus of Alzheimer’s disease patients and associates with tau and APP proteins. J Neurochem 109:1168–1178. https://doi.org/10.1111/j.1471-4159.2009.06047.x
Gao X, Hu H (2008) Quality control of the proteins associated with neurodegenerative diseases. Acta Biochim Biophys Sin Shanghai 40:612–618. https://doi.org/10.1111/j.1745-7270.2008.00441.x
Gentier RJ, van Leeuwen FW (2015) Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer’s disease. Front Mol Neurosci 8:47. https://doi.org/10.3389/fnmol.2015.00047
Gerakis Y, Dunys J, Bauer C, Checler F (2016) Aβ42 oligomers modulate β-secretase through an XBP-1s-dependent pathway involving HRD1. Sci Rep 6:37436. https://doi.org/10.1038/srep37436
González AE, Muñoz VC, Cavieres VA, Bustamante HA, Cornejo VH, Januário YC, González I, Hetz C, daSilva L, Rojas-Fernández A, Hay RT, Mardones GA, Burgos PV (2017) Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. FASEB J 31:2446–2459. https://doi.org/10.1096/fj.201600713R
Guglielmotto M, Monteleone D, Boido M, Piras A, Giliberto L, Borghi R, Vercelli A, Fornaro M, Tabaton M, Tamagno E (2012) Aβ1-42-mediated down-regulation of Uch-L1 is dependent on NF-κB activation and impaired BACE1 lysosomal degradation. Aging Cell 11:834–844. https://doi.org/10.1111/j.1474-9726.2012.00854.x
Han YS, Zheng WH, Bastianetto S, Chabot JG, Quirion R (2004) Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons: involvement of protein kinase C. Br J Pharmacol 141:997–1005. https://doi.org/10.1038/sj.bjp.0705688
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479. https://doi.org/10.1146/annurev.biochem.67.1.425
Hossain MF, Uddin MS, Uddin GMS, Sumsuzzman DM, Islam MS, Barreto GE, Mathew B, Ashraf GM (2019) Melatonin in Alzheimer’s disease: a latent endogenous regulator of neurogenesis to mitigate Alzheimer’s neuropathology. Mol Neurobiol 56:1–22. https://doi.org/10.1007/s12035-019-01660-3
Jang J-H, Surh Y-J (2003) Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic Biol Med 34:1100–1110. https://doi.org/10.1016/s0891-5849(03)00062-5
Jinwal UK, Koren J, O’Leary JC et al (2010) Hsp70 ATPase modulators as therapeutics for Alzheimer’s and other neurodegenerative diseases. Mol Cell Pharmacol 2:43–46
Jinwal UK, Trotter JH, Abisambra JF, Koren J 3rd, Lawson LY, Vestal GD, O'Leary JC 3rd, Johnson AG, Jin Y, Jones JR, Li Q, Weeber EJ, Dickey CA (2011) The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J Biol Chem 286:16976–16983. https://doi.org/10.1074/jbc.M110.182493
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898. https://doi.org/10.1083/jcb.143.7.1883
Kabir MT, Abu Sufian M, Uddin MS et al (2019a) NMDA receptor antagonists: repositioning of memantine as multitargeting agent for Alzheimer’s therapy. Curr Pharm Des 25:3506–3518. https://doi.org/10.2174/1381612825666191011102444
Kabir MT, Uddin MS, Begum MM et al (2019b) Cholinesterase inhibitors for Alzheimer’s disease: multitargeting strategy based on anti-Alzheimer’s drugs repositioning. Curr Pharm Des 25:3519–3535. https://doi.org/10.2174/1381612825666191008103141
Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y (2010) Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid- generation. J Neurosci 30:3924–3932. https://doi.org/10.1523/JNEUROSCI.2422-09.2010
Khandelwal PJ, Herman AM, Hoe H-S, Rebeck GW, Moussa CE (2011) Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated A in AD models. Hum Mol Genet 20:2091–2102. https://doi.org/10.1093/hmg/ddr091
Kim T-W, Pettingell WH, Hallmark OG, Moir RD, Wasco W, Tanzi RE (1997) Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J Biol Chem 272:11006–11010. https://doi.org/10.1074/jbc.272.17.11006
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584:1393–1398
Lam YA, Pickart CM, Alban A et al (2000) Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc Natl Acad Sci 97:9902–9906. https://doi.org/10.1073/pnas.170173897
Lane RF, Shineman DW, Steele JW et al (2012) Beyond amyloid: the future of therapeutics for Alzheimer’s disease. Adv Pharmacol 64:213-71.
Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J Am Soc Nephrol 17:1807–1819. https://doi.org/10.1681/ASN.2006010083
Leigh PN, Whitwell H, Garofalo O et al (1991) Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Brain 114:775–788. https://doi.org/10.1093/brain/114.2.775
Lindsten K, de Vrij FMS, Verhoef LGGC, Fischer DF, van Leeuwen F, Hol EM, Masucci MG, Dantuma NP (2002) Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol 157:417–427. https://doi.org/10.1083/jcb.200111034
Lonskaya I, Shekoyan AR, Hebron ML et al (2012) Diminished parkin solubility and co-localization with intraneuronal amyloid-β are associated with autophagic defects in Alzheimer’s disease. J Alzheimers Dis 33:231–247. https://doi.org/10.3233/JAD-2012-121141
Lonskaya I, Hebron ML, Desforges NM et al (2014) Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med 92:373–386. https://doi.org/10.1007/s00109-013-1112-3
Maiti P, Manna J, Veleri S, Frautschy S (2014) Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed Res Int 2014:1–14. https://doi.org/10.1155/2014/495091
Mamun AA, Uddin MS, Mathew B, Ashraf GM (2020) Toxic tau: structural origins of tau aggregation in Alzheimer’s disease. Neural Regen Res 15:1417–1420. https://doi.org/10.4103/1673-5374.274329
Marambaud P, Ancolio K, Lopez-Perez E, Checler F (1998) Proteasome inhibitors prevent the degradation of familial Alzheimer’s disease-linked presenilin 1 and potentiate A beta 42 recovery from human cells. Mol Med 4:147–157
Marambaud P, Zhao H, Davies P (2005) Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J Biol Chem 280:37377–37382. https://doi.org/10.1074/jbc.M508246200
Mathew B, Parambi DGT, Mathew GE et al (2019) Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch Pharm (Weinheim) 352:1–13. https://doi.org/10.1002/ardp.201900177
Mishra S, Palanivelu K (2008) The effect of curcumin (turmeric) on Alzheimer’s disease: an overview. Ann Indian Acad Neurol 11:13–19. https://doi.org/10.4103/0972-2327.40220
Morawe T, Hiebel C, Kern A, Behl C (2012) Protein homeostasis, aging and Alzheimer’s disease. Mol Neurobiol 46:41–54. https://doi.org/10.1007/s12035-012-8246-0
Nandi D, Tahiliani P, Kumar A, Chandu D (2006) The ubiquitin-proteasome system. J Biosci 31:137–155. https://doi.org/10.1007/bf02705243
Necchi D, Lomoio S, Scherini E (2011) Dysfunction of the ubiquitin–proteasome system in the cerebellum of aging Ts65Dn mice. Exp Neurol 232:114–118. https://doi.org/10.1016/j.expneurol.2011.08.009
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(80):130–133. https://doi.org/10.1126/science.1134108
Nixon RA, Mathews PM, Cataldo AM (2001) The neuronal endosomal-lysosomal system in Alzheimer’s disease. J Alzheimers Dis 3:97–107
O’Leary JC, Li Q, Marinec P et al (2010) Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener 5:45. https://doi.org/10.1186/1750-1326-5-45
Ou J-R, Tan M-S, Xie A-M et al (2014) Heat shock protein 90 in Alzheimer’s disease. Biomed Res Int 2014:1–7. https://doi.org/10.1155/2014/796869
Patrick GN (2006) Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol 16:90–94. https://doi.org/10.1016/j.conb.2006.01.007
Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 21:479–505. https://doi.org/10.1146/annurev.neuro.21.1.479
Renziehausen J, Hiebel C, Nagel H, Kundu A, Kins S, Kögel D, Behl C, Hajieva P (2015) The cleavage product of amyloid-β protein precursor sAβPPα modulates BAG3-dependent aggresome formation and enhances cellular proteasomal activity. J Alzheimers Dis 44:879–896. https://doi.org/10.3233/JAD-140600
Sahab Uddin M, Ashraf GM (2020) Quality control of cellular protein in neurodegenerative disorders. IGI Global, Hershey
Saito R, Kaneko M, Okuma Y, Nomura Y (2010) Correlation between decrease in protein levels of ubiquitin ligase HRD1 and amyloid-beta production. J Pharmacol Sci 113:285–288. https://doi.org/10.1254/jphs.10118sc
Salminen A, Ojala J, Kaarniranta K, Hiltunen M, Soininen H (2011) Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer’s disease. Prog Neurobiol 93:99–110. https://doi.org/10.1016/j.pneurobio.2010.10.006
Sarah Kishinevsky AC, Wenjie Lou JKI, Koren J et al (2013) Chaperone-dependent neurodegeneration: a molecular perspective on therapeutic intervention. J Alzheimer’s Dis Park s10. https://doi.org/10.4172/2161-0460.S10-007
Savaskan E, Olivieri G, Meier F, Seifritz E, Wirz-Justice A, Müller-Spahn F (2003) Red wine ingredient resveratrol protects from β-amyloid neurotoxicity. Gerontology 49:380–383. https://doi.org/10.1159/000073766
Schwartz AL, Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96. https://doi.org/10.1146/annurev.pharmtox.051208.165340
Singh AK, Pati U (2015) CHIP stabilizes amyloid precursor protein via proteasomal degradation and p53-mediated trans-repression of β-secretase. Aging Cell 14:595–604. https://doi.org/10.1111/acel.12335
Song S, Kim S-Y, Hong Y-M, Jo DG, Lee JY, Shim SM, Chung CW, Seo SJ, Yoo YJ, Koh JY, Lee MC, Yates AJ, Ichijo H, Jung YK (2003) Essential role of E2-25K/Hip-2 in mediating amyloid-β neurotoxicity. Mol Cell 12:553–563. https://doi.org/10.1016/j.molcel.2003.08.005
Song S, Lee H, Kam T-I, Tai ML, Lee JY, Noh JY, Shim SM, Seo SJ, Kong YY, Nakagawa T, Chung CW, Choi DY, Oubrahim H, Jung YK (2008) E2-25K/Hip-2 regulates caspase-12 in ER stress–mediated Aβ neurotoxicity. J Cell Biol 182:675–684. https://doi.org/10.1083/jcb.200711066
Steiner H, Capell A, Pesold B, Citron M, Kloetzel PM, Selkoe DJ, Romig H, Mendla K, Haass C (1998) Expression of Alzheimer’s disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J Biol Chem 273:32322–32331. https://doi.org/10.1074/jbc.273.48.32322
Takalo M, Haapasalo A, Natunen T, Viswanathan J, Kurkinen KM, Tanzi RE, Soininen H, Hiltunen M (2013) Targeting ubiquilin-1 in Alzheimer’s disease. Expert Opin Ther Targets 17:795–810. https://doi.org/10.1517/14728222.2013.791284
Tramutola A, Di Domenico F, Barone E et al (2016) It is all about (U)biquitin: role of altered ubiquitin-proteasome system and UCHL1 in Alzheimer disease. Oxidative Med Cell Longev 2016:1–12. https://doi.org/10.1155/2016/2756068
Uddin MS, Kabir MT (2019) Emerging signal regulating potential of genistein against Alzheimer’s disease: a promising molecule of interest. Front Cell Dev Biol 7:1–12. https://doi.org/10.3389/fcell.2019.00197
Uddin MS, Al Mamun A, Asaduzzaman M et al (2018a) Spectrum of disease and prescription pattern for outpatients with neurological disorders: an empirical pilot study in Bangladesh. Ann Neurosci 25:25–37. https://doi.org/10.1159/000481812
Uddin MS, Stachowiak A, Al Mamun A et al (2018b) Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci 10:1–18. https://doi.org/10.3389/fnagi.2018.00004
Uddin MS, Al Mamun A, Kabir MT et al (2019a) Nootropic and anti-Alzheimer’s actions of medicinal plants: molecular insight into therapeutic potential to alleviate Alzheimer’s neuropathology. Mol Neurobiol 56:4925–4944. https://doi.org/10.1007/s12035-018-1420-2
Uddin MS, Kabir MT, Al Mamun A et al (2019b) APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol Neurobiol 56:2450–2465. https://doi.org/10.1007/s12035-018-1237-z
Uddin MS, Mamun AA, Labu ZK et al (2019c) Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J Cell Physiol 234:8094–8112. https://doi.org/10.1002/jcp.27588
Uddin MS, Mamun AA, Takeda S et al (2019d) Analyzing the chance of developing dementia among geriatric people: a cross-sectional pilot study in Bangladesh. Psychogeriatrics 19:87–94. https://doi.org/10.1111/psyg.12368
Uddin MS, Kabir MT, Tewari D et al (2020a) Emerging signal regulating potential of small molecule biflavonoids to combat neuropathological insults of Alzheimer’s disease. Sci Total Environ 700:1–11. https://doi.org/10.1016/j.scitotenv.2019.134836
Uddin MS, Mamun AA1, Jakaria M et al (2020b) Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci Total Environ 707:1–12. https://doi.org/10.1016/j.scitotenv.2019.135624
Upadhya S, Hegde A (2005) Ubiquitin-proteasome pathway components as therapeutic targets for CNS maladies. Curr Pharm Des 11:3807–3828. https://doi.org/10.2174/138161205774580651
van Leeuwen FW, de Kleijn DPV, van den Hurk HH et al (1998) Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer’s and down patients. Science 279(80):242–247. https://doi.org/10.1126/science.279.5348.242
Vriend J, Ghavami S, Marzban H (2015) The role of the ubiquitin proteasome system in cerebellar development and medulloblastoma. Mol Brain 8:64. https://doi.org/10.1186/s13041-015-0155-5
Wang BJ, Her GM, Hu MK, Chen YW, Tung YT, Wu PY, Hsu WM, Lee H, Jin LW, Hwang SL, Chen RP, Huang CJ, Liao YF (2017) ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer’s disease. Proc Natl Acad Sci U S A 114:E3129–E3138. https://doi.org/10.1073/pnas.1618804114
Wilhelmus MMM, de Waal RMW, Verbeek MM (2007) Heat shock proteins and amateur chaperones in amyloid-Beta accumulation and clearance in Alzheimer’s disease. Mol Neurobiol 35:203–216
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL, Czerniewski L, Ballabio A, Cirrito JR, Diwan A, Lee JM (2015) Neuronal-targeted TFEB accelerates lysosomal degradation of app, reducing Aβ generation and amyloid plaque pathogenesis. J Neurosci 35:12137–12151. https://doi.org/10.1523/JNEUROSCI.0705-15.2015
Yang D-S, Stavrides P, Mohan PS et al (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 134:258–277. https://doi.org/10.1093/brain/awq341
Yang C, Cai CZ, Song JX, Tan JQ, Durairajan SSK, Iyaswamy A, Wu MY, Chen LL, Yue Z, Li M, Lu JH (2017) NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 13:2028–2040. https://doi.org/10.1080/15548627.2017.1379633
Zhang M, Deng Y, Luo Y et al (2012) Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem 120. https://doi.org/10.1111/j.1471-4159.2011.07644.x
Zhang M, Cai F, Zhang S, Zhang S, Song W (2015) Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci Rep 4:7298–7296. https://doi.org/10.1038/srep07298
Zhao Y, Hegde AN, Martin KC (2003) The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr Biol 13:887–898. https://doi.org/10.1016/s0960-9822(03)00332-4
Acknowledgements
The authors concede the support by the Pharmakon Neuroscience Research Network, Dhaka, Bangladesh.
Funding
This project was supported by the Pharmakon Neuroscience Research Network, Dhaka, Bangladesh.
Author information
Authors and Affiliations
Contributions
AAM and MSU conceived the original idea and designed the outlines of the study. AAM, MSU, and MTK prepared the draft of the manuscript. AAM prepared the figures for the manuscript. SK, MSS, BM, and MA participated in the literature review of the manuscript. AAM, MSU, AR, and GMA participated in revision and improved the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Al Mamun, A., Uddin, M.S., Kabir, M.T. et al. Exploring the Promise of Targeting Ubiquitin-Proteasome System to Combat Alzheimer’s Disease. Neurotox Res 38, 8–17 (2020). https://doi.org/10.1007/s12640-020-00185-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-020-00185-1