
Abstract
The brain is one of organs vulnerable to aluminum insult. Aluminum toxicity is involved in neurobehavioral deficit, neuronal cell dysfunction, and death. The aim of this study are as follows: (1) to evaluate the repairing efficiency of Necrostatin-1 (Nec-1), a cell death inhibitor, and Z-VAD-FMK, a pan-caspase inhibitor, on Al-induced neurobehavioral deficit and neuronal cell death, in order to evidence the cell death inducing ability of aluminum, and (2) to primarily explore the possibility of treating neuronal cell loss–related disease, such as Alzheimer’s disease, with Nec-1 and Z-VAD in Al-induced dementia animal model. We found Nec-1 and Z-VAD-FMK alone or in combination could reduce aluminum-induced learning and memory impairment in mice. Pathohistological results indicated that Nec-1 and Z-VAD-FMK can decrease Al-induced neuronal death cell. In addition, some cell death–associated proteins in cell death signal pathway were inhibited by Nec-1 and Z-VAD-FMK in Al-exposed mice. In conclusions, Nec-1 and Z-VAD-FMK can repair the injury of learning and memory induced by aluminum in mice. Furthermore, Nec-1 was more obvious to repair the injury of learning and memory function compared with Z-VAD-FMK. Nec-1 and Z-VAD-FMK can repair the Al-induced morphological injury of cell and reduce the amounts of dead cell, and repairing effects were more significant at higher doses. The effect of Nec-1 was stronger than Z-VAD-FMK, though their mechanism was different. The combination of them had the strongest effect. Our study evidenced Al-induced neuronal necroptosis and apoptosis existing in animal model and suggested potential therapeutic effects of Nec-1 and Z-VAD-FMK on neuronal cell death in neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aluminum products are closely related to our life. It is used in industry, water treatment, food additives, and medicines such as antacids and vaccine adjuvant. Aluminum exists in the form of aluminum ions, which form insoluble compounds when absorb in living organism (Exley 2013, Walton 2014). Provided that it accumulates too much in the body, it will produce chronic toxicity and damage the body’s kidney, bone, brain, and other organs (Esparza et al. 2018). More importantly, the intake of aluminum in the brain is more than the elimination, resulting in an increase in the net content of aluminum, and this process is related to the increase of age (Walton 2014). At present, aluminum is believed to have obvious neurotoxicity, and it is suspected to have a relationship with varieties of neurodegenerative diseases, for example Alzheimer disease (AD) (Neha Singla 2013), amyotrophic lateral sclerosis, and Parkinson’s dementia of Guam (Bondy 2014, Esparza et al. 2018). AD is a progressive neurodegenerative disease with clinical manifestations of deteriorating cognitive and memory functions, and various neuropsychiatric and behavioral disorders (Liu-Seifert et al. 2015). Neuropathologic features of Alzheimer’s disease caused by aluminum accumulation in neurons included neurofibrillary tangles (NFTs) (Walton 2010), Aβ plaque formation (Yumoto et al. 2009), and reduced connections between nerve cells (Yang et al. 2019) . Neuronal cells loss is another neuropathological feature in AD (Serrano-Pozo et al. 2011) and was observed in the brain tissue of aluminum-treated mice (Qinli et al. 2013); however, the mechanism of this aluminum-induced destructive cell fate has not been clearly clarified.
Traditionally, there are two main ways of cell death, apoptosis and necrosis. Our studies have shown that the chronic toxicity of aluminum can induce neuronal cell death, causing serious neuronal loss, and thus play an important role in the development of Al-induced animal neurodegenerative model (Niu et al. 2005, Niu et al. 2007). Generally, necrosis was considered to be a passive process caused by various attacks and insults, different from apoptosis which is triggered by a series of signal molecules and is called programmed form of cell death. In recent years, authors also discovered a type of necrosis caused through precise cellular signaling pathways and named it “necroptosis” (Leist and Jaattela 2001, Zilkova et al. 2006), a non-apoptotic programmed cell death described herein. A small molecule, Necrostatin-1 (Nec-1), specifically blocks the way of necroptosis to protect the neurocyte injured by insults (Yang et al. 2018), including aluminum (Zhang et al. 2008a, b, c). The key protein in the signal pathway of programmed cell necrosis is the receptor interaction protein (RIP) (Degterev et al. 2014). RIP is an enzyme with seven kinases family members, RIP1 being the first one of the family (Humphries et al. 2015). It is complicated that the cascade of events were triggered by activated RIP1, which not only activates the transcription factor NF-κB, triggering proliferative, but also induces cells apoptosis and necroptosis; therefore, RIP1 is a key regulator of cell proliferation and death (Humphries et al. 2015). We discovered there are two cellular death pathways including apoptosis and necroptosis in dementia mouse model induced by aluminum. Moreover, Nec-1 plays a significant role in amelioration of learning and memory impairment in aluminum-induced dementia; therefore, it lays the foundation for our future research on aluminum-induced neuronal loss. In our present study, we exposed mice to aluminum solution and treated them with Nec-1, Z-VAD-FMK (a pan-apoptosis inhibitor), or both of them, to observe which type of cell death aluminum will induce, and explore possible mechanism.
Materials and Methods
Animals
Male KM mice (supplied by the Laboratory Animal Center, Shanxi Medical University, Taiyuan, China), weighing 25–30 g, aged 6 months, were kept in a regulated temperature and humidity conditions, fed with standard diet and drinking water ad libitum, with 12 h light/dark cycles (light from 8:00 am to 8:00 pm). This study was approved by the Ethics Committee for Animal Studies of Shanxi Medical University.
Animals and Treatments
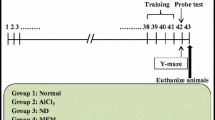
One hundred twenty healthy male KM mice were divided into three batches randomly, 40 in each batch. The first batch (Al plus Nec-1) included the following: control group (0.5% AlCl3 solution 3 μl and 10 %DMSO solution 2 μl), 0.5% AlCl3 solution 3 μl with Nec-1 2 μl in concentrations of 2 mM, 4 mM, and 8 mM (Qinli et al. 2013) treatment respectively. The second batch (Al plus Z-VAD-FMK) was composed of the control group (0.5% AlCl3 solution 3 μl and 10 %DMSO solution 2 μl) and 0.5% AlCl3 solution 3 μl with Z-VAD-FMK 2 μl in concentrations of 75 ng, 150 ng, and 300 ng treatment respectively. The third batch (Al plus Nec-1 plus Z-VAD-FMK) consisted of the control group (0.5% AlCl3 solution 3 μl and 10 %DMSO solution 2 μl), 0.5% AlCl3 solution 3 μl and 4 mM Nec-1 solution 2 μl; 0.5% AlCl3 solution 3 μl and 300 ng Z-VAD-FMK solution 2 μl; and 0.5% AlCl3 solution 3 μl and 4 mM Nec-1 + 300 ng Z-VAD-FMK solution 2 μl. The mice received 0.5% AlCl3 and Nec-1 or Z-VAD-FMK via intracerebroventricular (i.c.v.) injection once daily for 5 successive days.
Spatial Learning and Memory Functions
The Morris water maze (MWM) task was executed in 14 days after intracerebroventricular injection. Mice from the different groups underwent the test for 4 consecutive days. Each mouse was released into water toward the pool wall at one of the four starting positions of east, west, south, and north at random. The time of the mice from entering the water to finding and climbing onto the platform (all limbs climbing onto the platform) was recorded as the escape latency (LT). If the time mice spend to find the platform exceeds 60 s, the animal is guided to the platform by the experimenter and allowed to stay on platform for 10 s. Each animal was trained 4 times a day, with 15- to 20-min interval between training sessions. On the day after the last acquisition training, the platform was removed and 60-s probe test was performed. Each mouse was released into water again from the opposite side of the original platform quadrant. The time that the animal spent in the target quadrant (the quadrant where the platform was originally placed) and the number of times it entered the quadrant were recorded as indicators of spatial memory. All tests were recorded by a camera located 2 m above the water; swimming speed, time required to the hidden platform, time spend in the target quadrant, and the number of crossings were recorded.
Sample Preparation
After the learning and memory evaluation, mice were killed by decapitation. The hippocampus and cerebral cortex were removed and preserved in refrigerators at − 80 °C for further analysis. The processes of dissection were all performed on ice.
Thionin Staining
In order to observe the neuronal death induced by AlCl3 and effects of Nec-1 and (or) Z-VAD treatment, thionin staining was used as follows (Haelewyn et al. 2008): dewaxed slices were briefly immersed in water for washing 1–2 min, stained with 0.2% thionin, and placed in a 60 °C incubator for 20 min, followed by distilled water to wash at 1–2 min. After decoloration with 95% ethanol, the solution was dehydrated with serial anhydrous alcohol, cleared with xylene for 8-10 min respectively, and covered with neutral gum.
Immunohistochemical Assay
The brains were removed from the head of mice and immediately fixed with 4% neutral formaldehyde, followed by paraffin embedding for standby. The paraffin-embedded samples were serially sectioned; each slice was about 4-μm thick. Sections were detected by using SP immunohistochemical commercial assay kit (Beijing Zhongshan Jinqiao Biotechnology Co., Ltd. Beijing, China). The staining was carried out in accordance with experimental protocol. Briefly, after deparaffinization, the slides were soaked in 0.01 M/L citric acid buffer and heated by microwave for 13 min to expose the antigen, and then, 3% H2O2 inactivated endogenous protease 10 min. Primary RIP1 antibody (1:100 dilution), secondary antibody (1:200 dilution), and streptomycin avidin-peroxidase solution were added successively and incubated at 37 °C for 2 h in each step, with PBST for 10 min × 3 times in each step. Finally, for DAB staining, sections were observed under optical microscope. PBS was used as negative control instead of primary antibody.
Western Blot Analysis
Extraction of mouse hippocampal tissue protein was conducted with a Tissue Protein Extraction Kit (CoWin Biotech Co., Beijing, China). The hippocampus was homogenized in ice-cold Tissue Protein Extraction Reagent and a mixture of protease inhibitors. The supernatant was obtained after centrifugation at 10,000g for 15 min at 4 °C. The protein concentrations were quantified by using a BCA (bicinchoninic acid) assay (CoWin Biotech Co., Beijing, China).
Each lane loaded with 50 μg of protein was separated by 8% SDS-PAGE gel, then the proteins were transferred electrophoretically onto polyvinylidene difluoride membranes, which were soaked in 5% fat-free milk in PBST solution at 37 °C for 3 h to block non-specific binding and incubated overnight at 4 °C with following antibodies: caspase-3, LC3-II, RIP, NF-κB 1:500 (caspase-3, LC3-II, RIP, NF-κB; Santa Cruz, CA, USA), or mouse β-actin 1:600 (1:600, CoWin Biotech Co., Beijing, China). After rinsing 3 times for 10 min each in PBST, the blots were incubated for 2 h with a secondary antibody: anti-rabbit IgG (1:2000, CoWin Biotech Co., Beijing, China) or anti-mouse IgG (1:2000, CoWin Biotech Co., Beijing, China). The signal were detected using the ECL Western blot detection kit (CoWin Biotech Co., Beijing, China) after washing three times for 10 min in PBST. The blots were quantified using densitometry analysis.
Statistical Analysis
Data are expressed as the means ± SEM. Statistical differences were assessed by one-way analysis of variance (ANOVA). The post hoc Duncan test was carried out where appropriate. The level for a statistically significant difference was set at P < 0.05.
Result
Effects of Necrostatin-1 and (or) Z-VAD-FMK on Learning and Memory in Al-Induced Dementia Mouse Model
The first batch, as we describe previously, 0.5% Al solution 3 μl, was treated to produce models of dementia, and 2 mM, 4 mM, and 8 mM Nec-1 were administrated respectively to different groups for 5 days. After 2 weeks of treatment, all the groups were tested by Morris water maze for evaluating learning and memory capacity. In Al plus 4-mM and Al plus 8-mM Nec-1–administrated groups, there was a marked decrease in escape latency, compared with the only Al-exposed group. Treatment with Nec-1 in concentrations of 2 mM, 4 mM, and 8 mM significantly improved the spatial probe time of Al-exposed mice in comparison with only Al-exposed group. (See in Table 1 and Fig. 1.)

The results of Morris water maze test treated with different concentrations of Nec-1 in mice exposed to aluminum (10 mice in each group). In the 4-mM and 8-mM Nec-1 administration groups, there was a marked decrease in escape latency during the 4-day place navigation test, compared with the 0.5% Al-exposed group (P < 0.01). Treatment with Nec-1 in concentrations 2 mM, 4 mM, and 8 mM significantly improved the spatial probe time in comparison with 0.5%Al-exposured group. *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group
The second batch were as follows: control group (0.5% AlCl3) and 0.5% AlCl3 with Z-VAD-FMK in concentrations of 75 ng, 150 ng, and 300 ng treatment respectively. We can see the results from Table 2 and Fig. 2. The effect of the Al plus 300-ng Z-VAD-FMK–administrated mice demonstrated obviously shorter escape latency time than the other three groups, though other groups did not show significant ones. Meanwhile, administration with Z-VAD-FMK in concentrations of 75 ng, 150 ng, and 300 ng significantly improved the spatial probe time of Al-exposed mice, as compared with only Al-exposed group.

The results of Morris water maze test treated with different concentrations of Z-VAD-FMK in mice exposed to aluminum (10 mice in each group). It demonstrated obviously that the escape latency time of the 300-ng Z-VAD-FMK group is shorter than the other three groups (P < 0.01). Meanwhile, administration with Z-VAD-FMK in concentrations of 75 ng, 150 ng, and 300 ng significantly improved the spatial probe time, as compared with 0.5% Al-exposed group (P < 0.01). *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group
Based on the results of Figs. 1 and 2, in the third batch, we choose 4 mM Nec-1 and 300 ng Z-VAD-FMK, which showed obvious inhibition effects, to observe the combined effect of them. This batch included the following: control group (0.5% AlCl3), 0.5% AlCl3 with 4-mM Nec-1 group, 300-ng Z-VAD-FMK group, and Nec-1 + Z-VAD-FMK group respectively. Nec-1 group and Nec-1 + Z-VAD-FMK group indicated distinctly increase in escape latency time, compared with the Al-exposed group. Moreover, the Nec-1 executed more obvious inhibition on Al-induced behavior deficit than Z-VAD-FMK did. In spatial probe test, time in target quadrant, combination of Nec-1 plus Z-VAD-FMK treatment on Al-exposed group revealed dramatic difference, as compared with the Al-treated group, which was showed in Table 3 and Fig. 3.

Results of Morris water maze test in Al-exposed mice treated with combination of Nec-1 and Z-VAD-FMK (10 mice in each group). There are four groups, 0.5% AlCl3, 0.5% AlCl3 with 4-mM Nec-1 group, 300-ng Z-VAD-FMK group, and Nec-1 + Z-VAD-FMK group respectively. Nec-1 group and Nec-1 + Z-VAD-FMK group indicate distinctly increase in escape latency time, compared with the Al-exposed group (P < 0.05). Moreover, improvement of Nec-1 on behavior deficit of Al-exposed mice was more obvious than Z-VAD-FMK did. In spatial probe test, time in target quadrant in combination treatment group revealed dramatic difference, as compared with the AlCl3-treated group. *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group
Pathomorphology and Thionin staining
In the cortical area of the aluminum-exposed group, the perinuclear and cytoplasmic vacuoles were degenerated, and the cell outline was blurred. The number of cells in the hippocampus was diminished, and morphology of cells changed greatly including messy cell distribution, cell junction loosened, nucleus condensed (characteristics of apoptosis), and ring around the cell. Some cells showed necrosis characteristics, like nucleus aggregation, concentration, and dissolution. However, the abovementioned cell structure impairment was alleviated after the Z-VAD or Nec-1 alone or in combination administration, and the effect of the combination is more obvious (Fig. 4).

Morphological and pathological changes of hippocampal CA3 area in Z-VAD or Nec-1 alone or in combination administration in Al-exposed mice (H&E × 400). After 0.5% AlCl3 (a) exposure, the number of cells in the hippocampus was diminished; in the meantime, neuronal morphological damage includes messy cell distribution, cell junction loosened, condensed body nucleus apoptosis, and necrosis characteristics, like nucleus concentration and dissolution. Cell structure impairment was alleviated after the 300 ng Z-VAD (C) or 4 mM Nec-1 alone (b) or in combination (d) administration, and the effect of the combination is more obvious
Nissl, one of the characteristic structures of neurons, can be clearly identified by thionin staining. Thionin staining of CA3 area of mice hippocampus were presented in Fig. 5. In aluminum-exposed group, the cells loosely distributed with blurred cell membrane boundaries, and the number of nissl body in the hippocampus decreased. With Nec-1 and the Z-VAD treatment, aluminum-exposed cells distributed orderly with intact cell membrane, and the number of nissl body was increased in a dose-dependent manner, especially in the combination of 4-mM Nec-1 and 300-ng Z-VAD group (Fig. 5).

Thionin staining of CA3 area of mice hippocampus in Z-VAD or Nec-1 alone or in combination administration (× 400). a The cell structure was destroyed by aluminum. The cells loosely distributed with blurred cell membrane boundaries, and the number of nissl body in the hippocampus decreased. With the 4-mM Nec-1 (b) and the 300-ng Z-VAD (c) treatment, aluminum-exposed cells distributed orderly with intact cell membrane, and the number of nissl body was increased, especially in the combination of Nec-1 and Z-VAD group (d)
The previous research of our team demonstrated that aluminum could cause neuronal apoptosis in morphology (Zhang et al. 2008a, b, c) and proposed possible mechanisms (Zhang et al. 2014). In this experiment, neuronal cells, originated from Al-exposed mice with Nec-1 treatment, were observed under electron microscope. The nucleus of the aluminum-exposed group was pyknotic and the nuclear membrane was released. With the increase of Nec-1 dose, the aforementioned morphological impairment was gradually reduced and the nuclear membrane recovered gradually and became intact (Fig. 6).

Neuronal cells, originated from Al-exposed mice (A) with 2 mM Nec-1 (b), 4 mM Nec-1 (c), and 8 mM Nec-1 (d) treatment, were observed under electron microscope. The nucleus of the aluminum-exposed group was pyknotic and the nuclear membrane was released. The abovementioned morphological impairment was gradually reduced and the nuclear membrane became gradually intact in a dose-dependent manner
Effects of Nec-1 and (or) Z-VAD on the Expression Levels of Cell Death–Related Proteins in the Hippocampus
To evaluate the effect of the Nec-1 treatment on Al-induced neuronal cell death, Western blot was used to analyze the expression of cell death–related proteins in the cerebral cortex of Al-exposed mice. The RIP protein, which is a key protein in signal pathway of necroptosis, was obviously declined in 4-mM Nec-1–treated Al-exposed group than other three groups. Cleaved caspase-3, as the executer of apoptosis, manifested a downward trend, especially in 4-mM and 8-mM Nec-1 administration groups. The LC3-II, an autophagy-specific marker, demonstrated a downward expression in a dose-dependent manner in mice exposed with 0.5% Al and treated with 2 mM, 4 mM, and 8 mM Nec-1. By contrast, for expression of NF-κB, there was no distinct difference in all four groups (Table 4 and Fig. 7).

Relative expression of cell death–associated proteins in the cerebral cortex of Al-exposed mice treated with different concentrations of Nec-1 (5 mice in each group). The RIP protein was obviously declined in 4-mM Nec-1 plus Al-treated groups than other three groups (P < 0.05). Cleaved caspase-3 manifested a downward trend, especially in 4-mM and 8-mM Nec-1 administration groups (P < 0.01). The LC3-II demonstrated a downward expression in a dose-dependent manner in mice administrated with 0.5% Al plus 2 mM, 4 mM, and 8 mM Nec-1 (P < 0.01). There is no distinct difference in all four groups for expression of NF-κB. *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group
Z-VAD is the pan-inhibitor of the apoptosis. The 300 ng Z-VAD could result in cleaved caspase-3 decrease in Al-exposed mice cortex treated with 300 ng Z-VAD, while NF-κB revealed a downward expression in a dose-dependent manner in mice exposed with 0.5% Al and treated with 75 ng, 150 ng, and 300 ng Z-VAD. The expression of LC3-II and RIP protein increased significantly in Al-exposed mice treated with 150 ng and 300 ng Z-VAD respectively (Table 5 and Fig. 8).

Relative expression of cell death–associated proteins in the cerebral cortex of Al-exposed mice treated with different concentrations of Z-VAD (5 mice in each group). The cleaved caspase-3 and NF-κB revealed a downward expression. Especially, the NF-κB decline in a dose-dependent manner in mice administrated with 0.5% Al plus 75 ng, 150 ng, and 300 ng Z-VAD (P < 0.01). The expression of LC3-II and the RIP protein began to increase significantly in Al plus 150 ng and 300 ng Z-VAD, respectively (P < 0.01). *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group
When both Z-VAD and Nec-1 in combination administrated the Al-exposed mice, cleaved caspase-3 in the brain declined distinctly, compared with the Z-VAD and Nec-1 treatment alone (Fig. 9). However, the expression of RIP protein did not show change with both Z-VAD and Nec-1, as compared with the Al-exposed group, although the separate effects of Z-VAD and Nec-1 could reduce protein expression of RIP. Results of immunohistochemical assay to detect RIP protein expression was shown in Fig. 10. Single treatment of Z-VAD and Nec-1 could upregulate the expression of NF-κB protein, but the combination of the two drugs reduced NF-κB protein expression. LC3-II protein was not statistically significant in either Z-VAD or Nec-1 alone or in both combination group in comparison with Al-exposed mice’ brain (Table 6 and Fig. 9).

Relative expression of cell death–associated proteins in the cerebral cortex of Al-exposed mice treated with combined Z-VAD and Nec-1 (5 mice in each group). When both Z-VAD and Nec-1 in combination administrated the Al-treated mice, cleaved caspase-3 declined distinctly, compared with the treatment of Z-VAD and Nec-1 alone (P < 0.01). The expression of RIP protein had no change with both Z-VAD and Nec-1 (P > 0.05), although the separate effects of Z-VAD and Nec-1 could reduce protein expression of RIP (P < 0.01). Single treatment of Z-VAD and Nec-1 could upregulate the expression of NF-κB protein (P < 0.01), but the combination of the two drugs reduces NF-κB protein expression (P < 0.05). Compared with the Al-exposed mice’ brain, there was no statistically significant in LC3-II protein in either Z-VAD or Nec-1 alone or in both combination group (P > 0.05). *P < 0.05 compared with 0.5% Al group; **P < 0.01 compared with 0.5% Al group

Immunohistochemical changes of RIP1 in hippocampal CA3 area of Z-VAD, or Nec-1 alone, or in combined administration of them in Al-exposed mice (IHC × 400) (5 mice in each group). The positive immune response of RIP1 protein in the hippocampal CA3 region of mice in each group was localized in cytoplasm, which presented a brown-yellow or yellow staining. The staining of Nec-1 + Al-treated tissue (b) was weaker than that of the Al-treated one (a), and the staining of Z-VAD-FMK + Al-treated tissue (c) showed more brown-yellow color compared with the Al-treated tissue (a). There was no significant change in the Nec-1 + Z-VAD-FMK + Al combinedly treated tissue (d) compared with the Al-treated one (a)
Discussion
Animal models have been the primary method of studying various physiological disorders and pathological manifestations. It is the inevitable research tools for the study of drug treatment and toxic reactions caused by metals and other chemicals, especially for assessing nervous system function. AD is a progressive neurodegenerative disease with deteriorating learning and memory functions. A lot of studies confirmed that aluminum can cause learning and memory impairment in animals and human (Walton 2012, Qinli et al. 2013, Silva et al. 2013, Singla and Dhawan 2017). In this experiment, aluminum maltolate was injected into mice by intracerebroventricular injection. After 2 weeks, the mice showed difficulty in learning and memory, which was similar to the report of Neha Singla (Singla and Dhawan 2017). The loss of neurons is one of several possible causes of memory impairment (Serrano-Pozo et al. 2011). Another important cause for memory damage is free radicals produced in Al-induced dementia (Singla and Dhawan 2013). The free radicals of oxygen, also known as reactive oxygen species (ROS), account for about 95% of free radicals. Our previous research had confirmed that a large number of ROS were produced after mitochondrial destruction in Al exposure neural cells, thus accelerating cell death (Zhang et al. 2005).
Apoptosis is a typically programmed form of cell death, but in recent years, emerging evidence has indicated a necrosis caused by precise cellular signaling pathways, which was named “necroptosis” (Cho 2018), which can be inhibited by a small chemical molecule—necrostatin-1 (Nec-1). In vitro, Nec-1 can reduce the rate of cell necrosis in SHSY-5Y cell induced by aluminum (Zhang et al. 2008a, b, c). In vivo, Nec-1 also can alleviate learning and memory impairment caused by Al in mice (Qinli et al. 2013). In this study, treatment of different concentrations of Nec-1 or Z-VAD-FMK in aluminum-exposed mice showed that Nec-1 and Z-VAD-FMK could ameliorate the impairment of spatial learning and memory function induced by aluminum in a dose-dependent manner, more importantly, the effect of Nec-1 was more obvious than Z-VAD-FMK. At the same time, the effect of Nec-1 combined with Z-VAD-FMK was better than that of the Nec-1 alone.
Necroptosis, different from necrosis and apoptosis, is characterized by autophagic activation and necrotic cell morphology (Wu et al. 2015), which is regulated by a series of signaling pathways. Degterev et al. (2005) show that necrostatin-1, as the first type of necroptosis inhibitors, blocks the formation and occurrence of necroptosis by restraining the dimerization of receptor interacting protein-1 (RIP1) kinase domain, which can activate the RIP1; therefore, RIP1 kinase is the key enzyme of regulating necroptosis (Degterev et al. 2008).
RIP1 is a Ser/Thr kinase that is required for the integration of intracellular and extracellular stress signals in various membranes (Degterev et al. 2008). The RIP1 protein consists of three major domains which have different functions (Degterev et al. 2008). The C-terminal death domain (RIP1-DD) is important for the affinity response of death receptors, and the central region is associated with activation of NF-κB (Christofferson et al. 2014). The N-terminal Ser/Thr kinase domain is necessarily required for necroptosis, not necessarily for apoptosis and activation of NF-κB (Christofferson et al. 2014). Necrostatin-l can lock the active fragment of RIP1 in an inactive state, thereby inhibiting the functional activity of RIP1 and the formation of necroptosis (Degterev et al. 2008, Xie et al. 2013). Previous studies have found that necrostatin-1 (Nec-1) can effectively inhibit cell necrosis induced by the death receptor pathway, rather than apoptosis induced by TNF-α. Nec-1 mainly inhibits the activity of RIP1 by competing with ATP to bind to RIP1, but Nec-1 does not affect the activated NF-κB activity stimulated by TNF-α (Degterev et al. 2008, Xie et al. 2013). Nec-1 could decrease the expression of RIP1 protein in Al-induced neuronal cell death, but Z-VAD-FMK increased its expression in the same model. When both Z-VAD and Nec-1 in combination administrated the Al-induced neuronal cell death, its expression was also decreased. It might be suggested that Nec-1 enabled the active fragment of RIP l to be locked in an inactive state (Degterev et al. 2008), while Z-VAD-FMK did not have this function.
Certain members of the cysteinyl aspartate–specific protease (caspase) family are involved in the molecular signaling of apoptosis, of which caspase-3 is the last executor of apoptosis (Green and Llambi 2015). Our previous study proved Al administration resulted in inhibition of hippocampal long-term potentiation (LTP) and the expression of caspase-3 protein increase (Zhang et al. 2016). In addition to caspase-3, the expression of caspase-9, caspase-8, caspase-6, and caspase-7 proteins were increased in the aluminum-induced dementia model (Singla and Dhawan 2015). Z-VAD-FMK is a small peptide inhibitor designed according to the active structure of caspase, and its mechanism of action is to inhibit its activity by alkylating the cysteine residue of the caspase-3 active center. Nec-1 and Z-VAD-FMK can significantly reduce the expression of apoptosis-related protein caspase-3, probably because Nec-1 blocks the apoptotic pathway by inhibiting RIP1, while Z-VAD-FMK can specifically block the apoptotic pathway. It indicates that RIP1 activates caspase-8 and then performs apoptosis (Grootjans et al. 2017. When caspase-8 function is inhibited, RIP1 and RIP3 form necrotic complex to stimulate programmed necrosis, and apoptosis is inhibited (Grootjans et al. 2017).
Autophagy is a metabolic mechanism for the degradation of large amounts of protein in cells (Kim et al. 2017), and it is also a caspase-independent cell death (Green and Llambi 2015). During the necroptosis, autophagy is also formed, but it is a follow-up result of necroptosis, not the cause of necroptosis (Degterev et al. 2005), because necrostatin-1 inhibits the occurrence of autophagy by inhibiting the upstream of necroptosis signaling pathway, not directly inhibits the occurrence of autophagy itself (Degterev et al. 2005, Degterev et al. 2008). LC3 has two types, I and II. When autophagy does not occur, LC3, which is expressed conventionally in the cell, is processed into cytosolic soluble type I LC3. When autophagy occurs, type I LC3 undergoes ubiquitin-like processing modification process and binds to phosphatidylethanolamine (PE) on the surface of the autophagic membrane to form type II LC3. LC3-II is always located on the membrane of intracellular autophagosomes, and its content is proportional to the number of autophagic vacuoles (Yukiko Kabeya et al. 2000). Therefore, the amount of type II LC3 is proportional to the number of autophagic vesicles. Nec-1 also significantly decreased the expression of autophagocytic protein LC3-II, but Z-VAD increased its expression. It may be that Nec-1 can block necroptosis of cells, reducing autophagy expression, while Z-VAD inhibited cell apoptosis which had no effect on the inflammatory response induced by “necroptosis.”
The NF-κB/Rel family exists in the cytoplasm in the form of a homodimer or heterodimer in combination with its inhibitory protein IκB, which has a dual effect inducing apoptosis and expression of anti-apoptotic proteins on nerve cells. In injured neurons, some two way–regulated signal transduction pathways involved in survival and repair mechanisms are activated, and protein kinase cascades are concentrated into transcription factors, which directly regulate gene expression. Nuclear factor kappa B has been found to play an important role in neurodegenerative diseases (Barbara Kaltschmidt et al. 1999). Kaltschmidt et al. (Barbara Kaltschmidt et al. 1999) believe that low concentrations of amyloid β protein components can activate nuclear factor kappa B to protect neuron, while high concentrations of amyloid β protein have neurotoxic effects, suggesting that nuclear factor kappa B may be involved in both toxic and neuroprotective effects in Alzheimer’s disease. Neha Singla found that NF-κB protein and mRNA were obviously elevated after Al exposure (Neha Singla 2013). Based of Al exposure, Nec-1 can increase the expression of NF-κB protein in a certain range, but Z-VAD-FMK can decrease its expression. When Nec-1 and Z-VAD-FMK are combined, NF-κB expression is significantly decreased, which may be related to the regulation of NF-κB itself, because of the duality of its effect on nerve cells, which was involved in toxic and neuroprotective effect in nerve cells.
As mentioned above, it can be seen that “necroptosis” and apoptosis are two independent pathways in the death receptor pathway, and they may have a crosstalk through some regulatory factors such as NF-κB. Besides, it can be inferred that Nec-1 may improve the Al-induced learning and memory impairment in mice, which may block the “necroptosis” of neuronal cells by blocking RIP1, alleviated the damaged cell structure, and improved the learning and memory impairment in Al-exposed mice. These results evidenced necroptosis existing in Al-induced neurotoxicity. Meanwhile, Z-VAD-FMK may improve the Al-induced learning and memory impairment by inhibiting caspases, hence reduce neuronal apoptosis, and evidenced apoptosis existing in in Al-induced neurotoxicity too. Both of them reduce neuronal cell death, though the effect of necrostatin-1 was stronger than Z-VAD-FMK; the combined effect of them was the best. At the same time, NF-κB may be involved in the process, but the specific mechanism remains to be further studied.
In conclusion, aluminum exposure can induce neuronal cell death, including apoptosis and necroptosis in mice. Necrostatin-1 and Z-VAD-FMK may have potential therapeutic value for neurodegenerative disease in which neuronal cell death is a main pathological characteristic.
References
Barbara Kaltschmidt MU, Wellmann H, Volk B, Kaltschmidt C (1999) Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci U S A 96(16):9409–9414
Bondy SC (2014) Prolonged exposure to low levels of aluminum leads to changes associated with brain aging and neurodegeneration. Toxicology 315:1–7
Cho YS (2018) The role of necroptosis in the treatment of diseases. BMB Rep 51(5):219–224
Christofferson DE, Li Y, Yuan J (2014) Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol 76:129–150
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321
Degterev A, Zhou W, Maki JL, Yuan J (2014) Assays for necroptosis and activity of RIP kinases. Methods Enzymol 545:1–33
Esparza, J. L., M. Gómez and J. L. Domingo (2018). Role of melatonin in aluminum-related neurodegenerative disorders: a review. Biol Trace Elem Res.
Exley C (2013) Human exposure to aluminium. Environ Sci Process Impacts 15(10):1807–1816
Green DR, Llambi F (2015) Cell death signaling. Cold Spring Harb Perspect Biol 7(12)
Grootjans S, Vanden Berghe T, Vandenabeele P (2017) Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ 24(7):1184–1195
Haelewyn B, Alix P, Maubert E, Abraini JH (2008) NMDA-induced striatal brain damage and time-dependence reliability of thionin staining in rats. J Neurosci Methods 168(2):479–482
Humphries F, Yang S, Wang B, Moynagh PN (2015) RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ 22(2):225–236
Kim, M., A. Ho and J. H. Lee (2017). Autophagy and Human Neurodegenerative Diseases-A Fly’s Perspective. Int J Mol Sci 18(7).
Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2(8):589–598
Liu-Seifert H, Siemers E, Sundell K, Price K, Han B, Selzler K, Aisen P, Cummings J, Raskin J, Mohs R (2015) Cognitive and functional decline and their relationship in patients with mild Alzheimer’s dementia. J Alzheimers Dis 43(3):949–955
Neha Singla DKD (2013) Zinc, a neuroprotective agent against aluminum-induced oxidative DNA injury. Mol Neurobiol 48:1–12
Niu PY, Niu Q, Zhang QL, Wang LP, He SE, Wu TC, Conti P, Di Gioacchino M, Boscolo P (2005) Aluminum impairs rat neural cell mitochondria in vitro. Int J Immunopathol Pharmacol 18(4):683–689
Niu Q, Zhang Q, Niu P, Shi Y (2007) Study of neurocytes apoptosis in vitro culture rat induced by aluminum. Wei Sheng Yan Jiu 36(4):407–413
Qinli Z, Meiqing L, Xia J, Li X, Weili G, Xiuliang J, Junwei J, Hailan Y, Ce Z, Qiao N (2013) Necrostatin-1 inhibits the degeneration of neural cells induced by aluminum exposure. Restor Neurol Neurosci 31(5):543–555
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1(1):a006189
Silva AFJ, Aguiar MSS, Carvalho OSJ, Santana LDNS, Franco ECS, Lima RR, Siqueira NVMD, Feio RA, Faro LRF, Gomes-Leal W (2013) Hippocampal neuronal loss, decreased GFAP immunoreactivity and cognitive impairment following experimental intoxication of rats with aluminum citrate. Brain Res 1491:23–33
Singla N, Dhawan DK (2013) Zinc protection against aluminium induced altered lipid profile and membrane integrity. Food Chem Toxicol 55:18–28
Singla N, Dhawan DK (2015) Zinc down regulates Apaf-1-dependent Bax/Bcl-2 mediated caspases activation during aluminium induced neurotoxicity. Biometals 28(1):61–73
Singla N, Dhawan DK (2017) Zinc Improves cognitive and neuronal dysfunction during aluminium-induced neurodegeneration. Mol Neurobiol 54(1):406–422
Walton JR (2010) Evidence for participation of aluminum in neurofibrillary tangle formation and growth in Alzheimer’s disease. J Alzheimers Dis 22(1):65–72
Walton, J. R. (2012). CHAPTER 2: cognitive deterioration and related neuropathology in older people with Alzheimer’s disease could result from life-long exposure to aluminium compounds. Issues in Toxicology: 31-82.
Walton JR (2014) Chronic aluminum intake causes Alzheimer’s disease: applying Sir Austin Bradford Hill’s causality criteria. J Alzheimers Dis 40(4):765–838
Wu J-R, Wang J, Zhou S-K, Yang L, Yin J-l, Cao J-P, Cheng Y-B (2015) Necrostatin-1 protection of dopaminergic neurons. Neural Regen Res 10(7):1120–1124
Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J, Shi Y (2013) Structural basis of RIP1 inhibition by necrostatins. Structure 21(3):493–499
Yang C, Li T, Xue H, Wang L, Deng L, Xie Y, Bai X, Xin D, Yuan H, Qiu J, Wang Z, Li G (2018) Inhibition of necroptosis rescues SAH-induced synaptic impairments in hippocampus via CREB-BDNF Pathway. Front Neurosci 12:990
Yang MH, Chen SC, Lin YF, Lee YC, Huang MY, Chen KC, Wu HY, Lin PC, Gozes I, Tyan YC (2019) Reduction of aluminum ion neurotoxicity through a small peptide application - NAP treatment of Alzheimer’s disease. J Food Drug Anal 27(2):551–564
Yukiko Kabeya NM, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagyosome membranes after processing. EMBO 19(21):5720–5728
Yumoto S, Kakimi S, Ohsaki A, Ishikawa A (2009) Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. J Inorg Biochem 103(11):1579–1584
Zhang QL, Niu PY, Niu Q, Wang LP (2005) Effect of aluminum on neuronal mitochondria of rats. Wei Sheng Yan Jiu 34(6):674–677
Zhang QL, Niu PY, Shi YT, Zhang HM, Wang F, Zhang L, Niu Q (2006) Role of Bcl-2 and Bax protein contents and their gene expression in Al-induced neurons apoptosis. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 24(10):582–586
Zhang QL, Boscolo P, Niu PY, Wang F, Shi YT, Zhang L, Wang JWLP, Gioacchinol MDI, Conti P, Li QY, Niu Q (2008a) How do rat cortical cells cultured with aluminum die. Necrosis or apoptosis. Int J Immunopathol Pharmacol 21(I):107–115
Zhang QL, Niu Q, Ji XL, Conti P, Boscolo P (2008b) Is necroptosis a death pathway in aluminum-induced neuroblastoma cell demise? Int J Immunopathol Pharmacol 21(4):787–796
Zhang QL, Niu Q, Ji XL, Conti P, Boscolo P (2008c) Is necroptosis a death pathway in aluminum-induced neuroblastoma cell demise? International journal of immunopathology and pharmacology 21(4):787–796
Zhang Q, Li N, Jiao X, Qin X, Kaur R, Lu X, Song J, Wang L, Wang J, Niu Q (2014) Caspase-3 short hairpin RNAs a potential therapeutic agent in neurodegeneration of Al exposed animal model. Curr Alzheimer Res 11(10):961–970
Zhang H, Yang X, Qin X, Niu Q (2016) Caspase-3 is involved in aluminum-induced impairment of long-term potentiation in rats through the Akt/GSK-3beta pathway. Neurotox Res 29(4):484–494
Zilkova M, Koson P, Zilka N (2006) The hunt for dying neurons: insight into the neuronal loss in Alzheimer’s disease. Bratisl Lek Listy 107(9-10):366–373
Funding
This study was supported by the Natural Science Foundation of China (NSFC, 81430078).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the Ethics Committee for Animal Studies of Shanxi Medical University.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hao, Yx., Li, Mq., Zhang, Js. et al. Aluminum-induced “mixed” cell death in mice cerebral tissue and potential intervention. Neurotox Res 37, 835–846 (2020). https://doi.org/10.1007/s12640-019-00123-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-00123-w