
Abstract
Current therapeutic approaches of Alzheimer’s disease (AD) are symptomatic and of modest efficacy, and there is no available effective cure or prevention of AD; hence, the need arise to search for neuroprotective agents to combat AD. The current study aimed at investigating the neuroprotective effect of nanodiamond (ND), adamantine-based nanoparticles, in aluminum-induced cognitive impairment in rats, an experimental model of AD. AD was induced by aluminum chloride (17 mg/kg, p.o. for 6 weeks) and confirmed by Morris water maze and Y-maze behavioral tests. Biochemical and histological analyses of the hippocampus were also performed. Aluminum-treated rats showed behavioral, biochemical, and histological changes similar to those associated with AD. ND improved learning and memory and reversed histological alterations. At the molecular levels, ND mitigated the increase of hippocampal beta-amyloid (Aβ42) and beta-site amyloid precursor protein cleaving enzyme-1 (BACE1) together with down-regulation of phosphorylated tau protein. It also modulated the excitatory glutamate neurotransmitter level. Furthermore, ND boosted the brain-derived neurotrophic factor (BDNF) and mitochondrial transcription factor-A (TFAM), suppressed the proinflammatory cytokine tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), and curbed oxidative stress by hampering of inducible nitric oxide synthase (iNOS). Moreover, ND augmented the hippocampal levels of phosphorylated signal transducer and activator of transcription-3 (p-STAT3) and B cell leukemia/lymphoma-2 (Bcl-2) anti-apoptotic protein while diminished nuclear factor-kappaB (NF-κB) and caspase-3 (casp-3) expression. These findings indicate the protective effect of ND against memory deficits and AD-like pathological aberrations probably via modulating NF-kB and STAT3 signaling, effects mediated likely by modulating N-methyl-D-aspartate (NMDA) receptors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disease that is characterized by progressive impairment of memory and other cognitive functions leading to complete incapacity and death [1]. The prominent morphological hallmarks of AD are extracellular deposition of amyloid-β (Aβ) protein as senile plaques, intraneuronal accumulation of neurofibrillary tangles formed as a result of abnormal hyperphosphorylation of cytoskeletal tau protein, and massive neuronal death [2]. The hippocampus is one of the earliest brain areas to be affected by these pathologies [3, 4]. Current therapeutic approaches of AD are symptomatic and of modest efficacy, and there is no available effective cure or prevention of AD [5].
Nanotechnology has shown promising applications in targeted drug delivery for AD, and several nanocarrier systems have been studied in recent years to increase the bioavailability and efficacy of different AD therapeutic agents [6]. Nanodiamond (ND) has emerged as a promising engineered nanomaterial for a wide range of biomedical applications due to its unique structural, chemical, and biological properties [7]; it could adsorb or conjugate with nucleic acids and drugs for gene/drug delivery [8]. ND is a member of the structural family of nanocarbons called diamondoids which are category of most promising molecular building blocks in nanomedicine [9].
Adamantane is the smallest cage structure of the diamondoids, consisting of 10 carbon atoms and 16 hydrogen atoms. Memantine is an adamantane-based neuroprotective drug that slows down progression of AD by preferentially blocking excessive stimulation of N-methyl-D-aspartate (NMDA) glutamate receptors without disrupting normal neuronal activity [10, 11]. Overstimulation of NMDA receptors plays an important role in glutamate-induced excitotoxic neuronal cell death, while normal activity of these receptors plays an essential role in the long-term potentiation which is the major physiological basis of learning and memory [12]. ND possesses an adamantane nucleus similar to that of memantine; this raises the possibility that it might have an ameliorative effect in AD via modulating NMDA receptors. It has been demonstrated that ND undergoes systemic distribution, passes through the BBB [13], and internalized by neurons [14]. Noteworthy, ND has been shown to promote neuronal growth and differentiation of neural stem cells [15, 16]. Besides, it exhibited a protective effect against ultraviolet radiation in both cell cultures and mouse models [17]. Aluminum salts have been used extensively as food additives as well as in the treatment of drinking water, and chronic aluminum toxicity has been implicated in the development of AD [18]. Based on the present rationale, this study investigated the neuroprotective effect of ND in aluminum-induced cognitive impairment in rats, an experimental model of AD.
Materials and Methods
Drugs, Chemicals, and Kits
Nanodiamond powder was obtained from Van Moppes, Geneva, Switzerland. Memantine hydrochloride was obtained from Marcyrl Pharmaceuticals, Cairo, Egypt. Aluminum chloride was obtained from BDH, Poole, UK. Enzyme-linked immunosorbent assay (ELISA) kits were used in the present study for estimation of amyloid beta 1–42, inducible nitric oxide synthase, and nuclear factor-kappa B (EIAab Science, Wuhan, China); beta site amyloid peptide precursor cleaving enzyme 1, mitochondrial transcription factor A, and caspase-3 (CUSABIO, Wuhan, China); phosphorylated tau protein (Elabscience, Wuhan, China); tumor necrosis factor-α (IBL-America, MN, USA); interleukin-6 (RayBiotech, GA, USA); glutamate (Abnova, Taipei, Taiwan), brain-derived neurotrophic factor (Kamyia Biomedical Co, Seattle, WA, USA); phosphorylated signal transducer and activator of transcription3 (Abcam, Cambridge, UK); and B cell leukemia/lymphoma 2 protein (USCN Life Science, Wuhan, China). All other chemicals were of the highest purity and analytical grade.
Preparation of ND
Nanodiamond powder was treated with concentrated acid mixture of 68 % HNO3:98 % H2SO4 (1:9 v/v) at 100 °C for 48 h. Neutralization and washing were performed through several cycles of centrifugations (10,000 rpm for 5 min) in deionized water and ultrasonic dispersion of the pellet. The ND pellet was suspended in deionized water to form stable colloidal dispersion (after 3 months, neither sedimentation nor aggregation was observed). Size distribution was measured by dynamic light scattering at a scattering angle of 165° using a Delsa Nano C Particle Size apparatus (Beckman Coulter, Brea, CA, USA). Scattering data were collected for 70 individual measurements at a constant scattering angle and averaged for each sample. The distribution number determined for the treated ND was 15 ± 5 nm.
Animals
Adult male albino Wistar rats weighing 200–250 g were used in the present study. They were allowed acclimatization period for 2 weeks prior to testing. Animals were kept under controlled environmental conditions, room temperature (24–27 °C), constant humidity (60 ± 10 %), with alternating 12-h light and dark cycles in the animal facility of the Faculty of Pharmacy, Cairo University, Cairo, Egypt. Standard pellet diet (El-Nasr Co., AboZaabal, Egypt) and water were allowed ad libitum.
Experimental Design
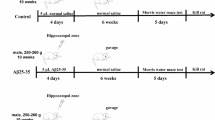
Forty-nine rats were randomly divided into seven groups (seven rats/group) and were treated for 6 weeks as follows: group 1, normal group received vehicle orally (distilled water); group 2, treated orally with AlCl3 (17 mg/kg/day) to induce AD-like pathology; group 3, injected interpretonially (i.p.) with ND (4 mg/kg/day); group 4, treated orally with memantine (5 mg/kg/day); and groups 5, 6, and 7 were treated with AlCl3 and either memantine, ND, or their combination, respectively. Five days before the end of the experiment, rats were trained on the Morris water maze for four consecutive days, and on the fifth day, they were subjected to the probe test as well as the Y-maze test (Fig. 1).

Schematic representation of the experimental design. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
Behavioral Tests
Morris Water Maze Test
Spatial learning and memory were investigated using Morris water maze paradigm [19–21] in a circular pool, 150 cm in diameter and 60 cm high, filled with water (27 ± 1 °C) to a depth of 40 cm. The pool was divided into four quadrants and a movable escape platform (8-cm diameter) located in the center of a fixed quadrant. Four points equally distributed along the perimeter of the tank were served as starting locations. The apparatus was located in a room with numerous extra-maze clues, posters, and objects that remained constant throughout the experiment. Rats were allowed training sessions, each 120 s thrice a day for four consecutive days. During each acquisition trial, animals were left free to find the platform in the designated quadrant. Once the rat located the platform, it was permitted to remain on it for 10 s; while if an animal failed to reach the platform within 120 s, it was placed on the platform for 30 s. On the fifth day, a probe test (retrieval trial) was performed where the platform was removed and each rat was allowed to explore the pool for 60 s; the time spent by the animal in the target quadrant searching for the hidden platform was regarded as the index of retrieval. The passing times through the target quadrant were also recorded.
Y-Maze Spontaneous Alternation Test
The impact of different treatments on spatial recognition was further verified by assessing the spontaneous alternation behavior in Y-maze test [22, 23]. The maze was made of painted wood and consisted of three identical arms (40 cm long, 35 cm high, and 12 cm wide) positioned at equal angles and labeled A, B, and C. Rats were placed at the end of one arm and were allowed to move freely through the maze during a 5-min session. The pattern of entrance into each arm in the maze was visually monitored for every rat. Arm entry was considered to be complete when the hind paws of the rat were completely placed in the arm. Maze arms were thoroughly cleaned between tasks to remove residual odors. Alternation was defined as successive entries into the three arms on overlapping triplet set (i.e., ABC, CBA, and BCA). Total arm entries and number of alternations were recorded and used to calculate the spontaneous alternation percentage (SAP) using the following equation: SAP (%) = [(number of alternations)/(total arm entries − 2)] × 100. Same arm returns (SARs) were also recorded.
Tissue Sampling
Six weeks from the beginning of experiments and 24 h following Morris water maze probe test and Y-maze task, the rats were euthanized and the brains were quickly dissected. The hippocampi were harvested and frozen at −80 °C. The hippocampi were homogenized (10 % w/v) in phosphate buffer (pH 7.4), and the homogenates were used for the biochemical analyses. In addition, representative brains in each group were fixed in 10 % formalin for the histological examination.
Biochemical Analysis
The hippocampal contents of beta amyloid 1–42 (Aβ1–42), phosphorylated tau protein (p-τ), beta site amyloid peptide precursor cleaving enzyme 1 (BACE1), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), brain-derived neurotrophic factor (BDNF), mitochondrial transcription factor A (TFAM), inducible nitric oxide synthase (iNOS), caspase-3, phosphorylated signal transducer and activator of transcription-3 (p-STAT3), B cell leukemia/lymphoma-2 (Bcl-2), and nuclear factor-kappa B (NF-κB) were estimated using ELISA kits according to the manufacturer’s instructions.
Histopathological Examination
Whole brains of two rats, randomly selected from each group, were fixed in 10 % formalin for 24 h. The specimens were washed, dehydrated by alcohol, cleared in xylene, and embedded in paraffin at 56 °C in hot air oven for another 24 h. Sections of 3-μm thickness were stained with hematoxylin and eosin and examined under the light microscope fitted with camera. All histopathological processing and assessment of specimens were performed by an experienced observer unaware of the identity of the sample being examined to avoid any bias.
Statistical Analysis
Comparisons between different groups were performed using one-way analysis of variance (ANOVA), followed by Tukey-Kramer’s test for multiple comparisons. Statistical analysis was carried out using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). Values were expressed as mean ± SEM and the minimal level of significance was identified at p < 0.05.
Results
Behavioral Analysis
The results of the behavioral study revealed that the escape latency was increased in the aluminum-treated group as compared to control rats starting from the second day during the training course of Morris water maze test (Fig. 2a). Interestingly, ND administration afforded an improvement in reaching the platform with escape latency approaching the normal values during the third and the fourth days of training. These actions were similar to those exerted by the reference drug memantine (MEM). Twenty-four hours after the final trial, the probe test was performed without the platform and the latency to reach the target quadrant and the time spent in the target quadrant as well as the passing times across the target quadrant were presented in Fig. 2b–d. Aluminum-treated rats spent less time in the target quadrant as compared to normal rats, whereas the treatment with ND restored these values to normal. These effects were analogous to those afforded by MEM. The same results were observed for the latency to reach the target quadrant and the passing times across the target quadrant. Aluminum significantly lowered the SAP and increased the SARs in Y-maze test as compared to the normal group, events that were reversed by ND and MEM (Fig. 2e, f).

ND attenuates aluminum-induced learning and memory deficits in rats performing Morris water maze (a-d) and Y-maze (e-f) behavioral tests. In Morris water maze; a ND lowered the escape latency starting from the second day of the 4-days training course. The probe test (b-d) was carried out 24 h after the final trial with removal of the platform; b ND decreased the scape latency to the target quadrant, c ND increased the passing times through the target quadrant, d ND increased the time spent in the target quadrant. In Y-maze test; e ND increased the spontaneous alternation percentage (SAP%), f reduced the same arm returns (SAR). Values represent mean ± SEM (n=7). * p<0.05 compared to the normal group and # p<0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, MEM memantine
Biochemical Analysis
The results revealed that aluminum exposure up-regulated hippocampal levels of NF-κB by 6-fold and caspase-3 by 5.5-fold and down regulated p-STAT3 by 4.6-fold and Bcl-2 by 8.3-fold compared to the normal group. However, ND reduced hippocampal levels of NF-κB (39 %) and caspase3 (55 %) and boosted levels of p-STAT3 (42 %) and Bcl-2 (2.5-fold) as compared to aluminum chloride group, and these effects were more pronounced when it was combined with MEM. The effects of different treatments on NF-κB, phosphorylated STAT3, Bcl-2 protein, and caspase-3 are presented in Fig. 3.

ND reduces hippocampal levels of NF-κB and caspase-3, and increases p-STAT3 and Bcl-2 in aluminum-treated rats. a Nuclear factor-kappa B (NF-κB); b phosphorylated signal transducer and activator of transcription 3 (p-STAT3), c B cell leukemia/lymphoma 2 (Bcl-2), and d caspase-3. Values represent mean ± SEM (n = 7). *p < 0.05 compared to the normal group and # p < 0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
The present study also revealed that administration of aluminum increased the levels of BACE1 by 6.2-fold (Fig. 4a), Aβ42 by 4.3-fold (Fig. 4b), and the p-tau protein by 3.3-fold compared to the normal group (Fig. 4c). Interestingly, ND blunted the levels of Aβ42 (56 %), BACE1 (45 %), and p-tau (43 %) as compared to aluminum chloride group and afforded greater protection against aluminum-induced neurotoxicity when combined with MEM.

ND reduces the hippocampal levels of BACE1, Aβ42, p-tau in aluminum treated rats: a beta site amyloid peptide precursor cleaving enzyme 1 (BACE1); b amyloid beta 42 peptide (Aβ42), c phosphorylated tau protein (p-tau). Values represent mean ± SEM (n=7). * p<0.05 compared to the normal group and # p<0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, MEM memantine
The effects of different treatments on hippocampal glutamate neurotransmitter content revealed that aluminum administration resulted in a significant decrease in this neurotransmitter by 80 % as compared to the normal group (Fig. 5). However, ND succeeded to counteract aluminum-induced depletion of hippocampal glutamate levels (1.8-fold) as compared to the aluminum chloride group, and this improvement was more pronounced when combined with MEM.

ND reverses the aluminum-induced depletion of the hippocampal glutamate neurotransmitter in aluminum-treated rats. Values represent mean ± SEM (n = 7). *p < 0.05 compared to the normal group and # p < 0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
The results also showed that aluminum administration increased the hippocampus pro-inflammatory cytokines TNF-α by 3.8-fold, (Fig. 6a), IL-6 by 4-fold (Fig. 6b), and iNOS by 3.8-fold (Fig. 6c) compared to the normal control group. ND diminished these pro-inflammatory cytokines TNF-α (39 %) and IL-6 (49 %) and reduced iNOS (52 %) as compared to the aluminum chloride group; this decrement was more pronounced when ND was combined with MEM.

ND diminishes the hippocampal levels of TNF-α, IL-6, and iNOS in aluminum-treated rats. a Tumor necrosis factor-α (TNF-α). b Interleukin-6 (IL-6). c Inducible nitric oxide synthase (iNOS). Values represent mean ± SEM (n = 7). *p < 0.05 compared to the normal group and # p < 0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
On the other hand, aluminum chloride administration diminished the expression of TFAM which reached 64 % (Fig. 7a) and BDNF which reached 52 % (Fig. 7b) in the hippocampus compared to the normal control group. However, ND markedly enhanced expression of BDNF (45 %) and TFAM (54 %) as compared to the aluminum chloride group, and this enhancement was more pronounced when ND was combined with MEM.

ND enhances the hippocampal expression of a BDNF and b mitochondrial transcription factor A (TFAM). *p < 0.05 compared to the normal group and # p < 0.05 compared to the AlCl3 group. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
Histological Analysis
The biochemical results of the current study were further confirmed by the histological examination of the hippocampus. The microscopic examination of the sections obtained from the normal group revealed an intact architecture of the hippocampus with normal histological appearance and distribution of the neuronal cells (Fig. 8a). On the other hand, the hippocampus of the aluminum-treated group showed degenerative changes and atrophy of the neuronal cells besides gliosis (Fig. 8b). Administration of ND or MEM to non-aluminum treated rats did not alter the normal architecture of the hippocampus (Fig. 8c, d). Moreover, ND administration to aluminum-treated rats attenuated the pathological degenerative changes in the hippocampus area (Fig. 8e). These actions were analogous to those afforded by MEM which mitigated histological alterations in the hippocampus (Fig. 8f). The same results were observed in the hippocampus of animals that received the combined treatment of ND and MEM (Fig. 8g). These observations indicate that ND mitigated the Alzheimer-like histopathological alterations in the hippocampus of aluminum-treated rats.

ND mitigates the histopathological damage in the hippocampus of aluminum-treated rats. Representative photomicrographs (H&E ×400) of sections obtained from the hippocampus harvested on the 43rd day of the study. a Normal group displayed intact structure of the hippocampus (hp) and normal distribution of the neuronal cells. b The neuronal cells in the hippocampus of aluminum-treated rats showed degeneration, atrophy, and gliosis. c ND group displayed normal architecture of the hippocampus. d MEM group showed normal neuronal appearance in the hippocampus. e AlCl3 + ND group revealed attenuation of the histopathological changes in the hippocampus. f AlCl3 + MEM group displayed mitigated neuronal degeneration of the hippocampus. g AlCl3 + ND + MEM group showed attenuated pathological changes in the hippocampus. AlCl 3 aluminum chloride, ND nanodiamond, and MEM memantine
Discussion
The current study evaluates the neurotoxic effects of aluminum chloride administration in adult rats over a 6-week period as well as the neuroprotective effects of ND and memantine. The study highlights the impact of aluminum on memory, provides further evidence for the health hazards associated with the aluminum food additives, and describes the mechanisms by which aluminum contributes in development of AD.
This study depicts an ameliorative effect of ND on aluminum-induced learning and memory impairment in MWM and Y-maze tests which correlates to modulation of biochemical biomarkers in the hippocampus. These behavioral tasks have been ordinarily used to assess the effect of various treatments on spatial recognition learning and memory [19–23]. Previous studies indicated that high levels of aluminum in the brain can impair long-term potentiation, which is thought to be the major physiological basis of learning and memory [12, 24].
A main finding of the present work is that ND hampered NF-κB activation and thereby alleviated the subsequent pathologic events. Up-regulation of NF-κB in aluminum treated rats has been formerly demonstrated [25]. Activated NF-kB translocates into the nucleus where it binds to DNA and induces expression of genes involved in inflammation including IL-8 and IL-6 [26–28]. Moreover, NF-κB promotes BACE1 expression and impairs microglial cell-mediated phagocytosis and clearance of Aβ42 peptide monomers which ultimately results in aggregation into higher-order amyloid species [25, 29]. Furthermore, activated NF-κB induces the expression of suppressor of cytokine signaling-3 (SOCS3) that acts as a negative regulator of JAK/STAT3 axis, preventing STAT3 activation [28]. Hence, NF-κB is likely the main upstream mediator of the neuronal aberrations reported in the present study including Aβ deposition, diminished STAT3 phosphorylation, overexpression of pro-inflammatory cytokines, and initiation of apoptosis. Noteworthy, NF-κB activation has been demonstrated to be mediated by glutamate NMDA ionotropic receptors and voltage-gated Ca2+ channels [30], and this provides a plausible evidence that ND reduced NF-κB activation via modulating NMDA receptors.
The current study revealed the ND induced up-regulation of phosphorylated STAT3, a key component of the cellular anti-apoptotic cascade, in the hippocampus of aluminum-treated rats. Phosphorylated STAT3 dimerizes and translocates into the nucleus where it induces transcription of target genes critical for promoting neuronal survival such as Bcl-2 [31, 32]. Moreover, phosphorylation of STAT3 has been linked with suppression of the transcription factor NF-κB and caspase-3 [33]. Down-regulation of this essential survival protein Bcl-2 in the hippocampus of aluminum-treated rats described in this study goes in line with previous findings [34] and is likely involved in the neuronal cell death induced by aluminum. Apoptosis is under heavy regulation by the two main subclasses of Bcl-2 family proteins, the anti-apoptotic members such as Bcl-2 and Bcl-xL and the pro-apoptotic members such as Bax, Bak, and BH3-domain-only proteins [35]. In response to apoptotic stimuli, BH3-domain-only proteins bind to and inactivate the anti-apoptotic Bcl-2 protein which normally inactivates the pro-apoptotic Bax/Bak proteins [36]. Activated Bax/Bak proteins oligomerize in the outer mitochondrial membrane and form pores leading to permeabilization of the mitochondrial wall with subsequent release of cytochrome c into the cytosol which triggers caspase-9 activation [37, 38] that acts on downstream targets including caspase-3 leading to apoptotic cell death [39]. Moreover, exposure to β-amyloid has been reported to be associated with activation of caspase-3 by reducing phosphorylation of STAT3, and this cascade is inhibited by activation of JAK2 [40]. Thus, aluminum-induced overexpression of caspase-3 may be attributed to blunting Bcl-2 and p-STAT3 levels. Of interest, ND administration triggered down-regulation of caspase-3 levels probably due to reinstating the Bcl2 and STAT3 signaling.
Elevated hippocampal Aβ levels were associated with increased expression of BACE1 in aluminum-treated rats. Indeed, Aβ is generated by two sequential endoproteolytic cleavages of the amyloid precursor protein by β-secretase (BACE1) and γ-secretase [41]. BACE1 cleavage is the initiating step and is a pre-requisite for Aβ42 generation, and BACE1 levels are elevated in both AD experimental models and in the AD patient brain [42–44]. Aluminum also impairs phagocytosis of Aβ peptides through NF-κB-mediated down-regulation of the sensing membrane receptor known as the triggering receptor expressed in myeloid cells 2 (TREM2) that ultimately contribute to Aβ42 peptide accumulation in the brain [45]. Downstream mechanisms of Aβ toxicity include phosphorylation of tau and breakdown of microtubule networks which are key underlying events for neuronal death and AD progression [46]. In the present study, augmented phosphorylation of tau protein in aluminum-induced AD rat model was elucidated. Aβ triggers tau phosphorylation, probably via Aβ-induced protease activation [47] and up-regulation of several tau-targeting kinases including glycogen synthase kinase-3 (GSK-3) [48]. Additionally, Aβ oligomers could be incorporated into neuronal cell membranes resulting in formation of ion channels with subsequent influx of Ca2+ through these amyloid channels which leads to phosphorylation of tau [49, 50]. ND reduced phosphorylated tau protein level evoked by aluminum administration. Resetting of the normal phosphorylation of tau protein is likely secondary to the suppression of Aβ levels.
The results described depletion of hippocampal glutamate neurotransmitter in response to aluminum exposure. Glutamate plays an essential role in the long-term potentiation which is thought to be the major physiological basis of learning and memory [12]. Aβ peptides trigger neurotoxicity partly via formation of ion channels into neuronal cell membranes with subsequent Ca2+ influx through these amyloid channels [49]. Calcium influx evokes excessive glutamate release resulting in stimulation of cascades of events culminating in neuronal cell death [51]. Elevated TNF-α levels reported herein also could provide an additional justification where it triggers glutamate release from microglia by up-regulating glutaminase and gap junction hemichannels [52, 53]. Notably, ND reversed glutamate aberration signifying the pleiotropic actions of ND to alleviate the cognitive deficits in AD.
Inflammation, a cardinal event in AD, is triggered by aluminum administration and accumulation of the toxic Aβ which activates the microglia and increase the number of astrocytes in the hippocampus [54]. Generation of inflammatory cytokines including TNF-α and IL-6 in the hippocampus of aluminum-treated rats ensued in this study is probably linked to Aβ deposition and tau phosphorylation [55], NF-kB activation [28], and diminished STAT3 phosphorylation [56]. Moreover, overexpression of iNOS during inflammatory response has been linked to NF-κB activation [57]. NO elevations were triggered by iNOS promote reactions between NO and superoxide resulting in accumulation of the highly reactive oxidant peroxynitrite metabolite that leads to oxidative DNA damage and prevent protein phosphorylation, therefore disrupting signal transduction mediated by tyrosine kinases [58, 59]. In the current study, ND lowered hippocampal TNF-α and IL-6 probably via boosting STAT3 phosphorylation and halting NF-kB which also hampered overexpression of iNOS.
BDNF affords neuroprotective actions. In particular, the hippocampus area displays the greatest abundance of BDNF which plays essential roles in neuronal proliferation and differentiation, as well as protection against variety of cellular insults [60]. BDNF promotes neuronal survival through activation of tyrosine receptor kinase B (TrkB) which induces JAK2/STAT3 signaling [61, 62]. In the present study, aluminum administration down-regulated BDNF levels in conformity with previous results [63]. Notably, ND exerted neuroprotective actions, at least partly, via elevating its hippocampal levels.
TFAM is responsible for maintenance and transcription of mitochondrial DNA and protects cells against oxidative stress [64]. It has been shown that overexpression of TFAM protects mitochondria against Aβ-induced oxidative damage in neurons [65], inhibits mitochondrial ROS generation, and improves mitochondrial respiratory function [66, 67]. The current results showed that while aluminum down-regulated TFAM, ND enhanced its hippocampal level. Thus, these data suggest that boosting of the hippocampal levels of TFAM, which will likely reverse the mitochondrial dysfunction, is partly implicated in ND protection against cognitive impairment (Fig. 9).

Diagram depicting the alleviating actions of ND against aluminum-induced Alzheimer-like pathologic aberrations. ND attenuates NF-κB and boosts BDNF and TFAM levels. Aβ 42 amyloid beta 1–42, BACE1 beta site amyloid precursor protein cleaving enzyme-1, BDNF brain-derived neurotrophic factor, Bcl2 B cell leukemia/lymphoma 2, Bax Bcl-2-associated X protein, Bak Bcl-2-antagonist/killer-1, iNOS inducible nitric oxide synthase, GSK-3 glycogen synthase kinase-3, IL-6 interleukin-6, ND nanodiamond, NF-κB nuclear factor kappa-B, NOS nitrogen reactive species, ROS reactive oxygen species, STAT3 signal transducer and activator of transcription 3, p-tau phosphorylated tau protein, TFAM mitochondrial transcription factor A, TNF-α tumor necrosis factor alpha, and TrKB tyrosine receptor kinase B
Conclusions
The study highlights, for the first time, the beneficial effects of ND against aluminum-induced AD-like pathological perturbations. ND reversed the histopathological changes and mitigated the abnormal BACE1, Aβ, and p-tau overexpression. It also reversed aluminum-evoked glutamate depletion, suppressed neuronal inflammation, curbed neuronal oxidative stress, depressed caspase-3 activation, and augmented BDNF and TFAM levels. ND probably exerted protective effects by modulating NF-kB activation which further regulates STAT3 and Bcl2 signaling, effects mediated likely by modulating NMDA receptors.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ42:
-
Amyloid beta 1–42
- AlCl3 :
-
Aluminum chloride
- BACE1:
-
Beta site amyloid precursor protein cleaving enzyme-1
- BDNF:
-
Brain-derived neurotrophic factor
- Bcl2:
-
B cell leukemia/lymphoma 2
- Bax:
-
Bcl-2-associated X protein
- Bak:
-
Bcl-2-antagonist/killer-1
- Casp-3:
-
Caspase-3
- GSK-3:
-
Glycogen synthase kinase-3
- iNOS:
-
Inducible nitric oxide synthase
- IL-6:
-
Interleukin-6
- MEM:
-
Memantine
- MWM:
-
Morris water maze
- ND:
-
Nanodiamond
- NF-κB:
-
Nuclear factor kappa-B
- NMDA:
-
N-methyl-D-aspartate
- p-STAT3:
-
Phosphorylated signal transducer and activator of transcription-3
- p-tau:
-
Phosphorylated tau protein
- TFAM:
-
Mitochondrial transcription factor A
- TNF-α:
-
Tumor necrosis factor alpha
- TrKB:
-
Tyrosine receptor kinase B
References
Querfurth HW, Laferla FM (2010) Alzheimer’s disease. N Engl J Med 362:329–344
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766
Braak H, Braak E, Bohl J (1993) Staging of Alzheimer-related cortical destruction. Eur Neurol 33:403–408
Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6:85
Hampel H, Prvulovic D, Teipel S, Jessen F, Luckhaus C, Frolich L, Riepe MW, Dodel R, Leyhe T, Bertram L, Hoffmann W, Faltraco F (2011) The future of Alzheimer’s disease: the next 10 years. Prog Neurobiol 95(4):718–728
Nazem A, Mansoori GA (2008) Nanotechnology solutions for Alzheimer’s disease: advances in research tools, diagnostic methods and therapeutic agents. J Alzheimers Dis 13(2):199–223
Kaur R, Badea I (2013) Nanodiamonds as novel nanomaterials for biomedical applications: drug delivery and imaging systems. Int J Nanomedicine 8:203–220
Zhang XY, Fu CK, Feng L, Ji Y, Tao L, Huang Q, Li SX, Wei Y (2012) PEGylation and polyPEGylation of nanodiamond. Polymer 53(15):3178
Mansoori GA (2007) Diamondoid molecules. Adv Chem Phys 136:207–258
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348(14):1333–1341
Lipton SA (2004) Paradigm shift in NMDA receptor antagonist drug development: molecular mechanism of uncompetitive inhibition by memantine in the treatment of Alzheimer’s disease and other neurologic disorders. J Alzheimers Dis 6(6):S61–S74
Shuchang H, Qiao N, Piye N, Mingwei H, Xiaoshu S, Feng S, Sheng W, Opler M (2008) Protective effects of gastrodia elata on aluminium-chloride-induced learning impairments and alterations of amino acid neurotransmitter release in adult rats. Restor Neurol Neurosci 26(6):467–473
Eidi H, David MO, Crépeaux G, Henry L, Joshi V, Berger MH, Sennour M, Cadusseau J, Gherardi RK, Curmi PA (2015) Fluorescent nanodiamonds as a relevant tag for the assessment of alum adjuvant particle biodisposition. BMC Med 13:144
Huang YA, Kao CW, Liu KK, Huang HS, Chiang MH, Soo CR, Chang HC, Chiu TW, Chao JI, Hwang E (2014) The effect of fluorescent nanodiamonds on neuronal survival and morphogenesis. Sci Rep 4:6919
Chen YC, Lee DC, Hsiao CY, Chung YF, Chen HC, Thomas JP, Pong WF, Tai NH, Lin IN, Chiu IM (2009) The effect of ultra-nanocrystalline diamond films on the proliferation and differentiation of neural stem cells. Biomaterials 30(20):3428–3435
Thalhammer A, Edgington RJ, Cingolani LA, Schoepfer R, Jackman RB (2010) The use of nanodiamond monolayer coatings to promote the formation of functional neuronal networks. Biomaterials 31(8):2097–2104
Wu MS, Sun DS, Lin YC, Cheng CL, Hung SC, Chen PK, Yang JH, Chang HH (2015) Nanodiamonds protect skin from ultraviolet B-induced damage in mice. J Nanobiotechnol 13:35
Walton JR (2014) Chronic aluminum intake causes Alzheimer’s disease: applying Sir Austin Bradford Hill’s causality criteria. J Alzheimers Dis 40(4):765–838
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11(1):47–60
Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1(2):848–858
Nunez J (2008) Morris water maze experiment. J Vis Exp 19:897
Van der Borght K, Havekes R, Bos T, Eggen BJ, Van der Zee EA (2007) Exercise improves memory acquisition and retrieval in the Y-maze task: relationship with hippocampal neurogenesis. Behav Neurosci 121(2):324–334
Hidaka N, Suemaru K, Takechi K, Li B, Araki H (2011) Inhibitory effects of valproate on impairment of Y-maze alternation behavior induced by repeated electroconvulsive seizures and c-Fos protein levels in rat brains. Acta Med Okayama 65(4):269–277
Llansola M, Minana MD, Montoliu C, Saez R, Corbalan R, Manzo L, Felipo V (1999) Prenatal exposure to aluminum reduces expression of neuronal nitric oxide synthase and of soluble guanylate cyclase and impairs glutamatergic neuro-transmission in rat cerebellum. J Neurochem 73:712–718
Zhao Y, Hill JM, Bhattacharjee S, Percy ME, Pogue AI, Lukiw WJ (2014) Aluminum-induced amyloidogenesis and impairment in the clearance of amyloid peptides from the central nervous system in Alzheimer’s disease. Front Neurol 5:167
Karin M (2009) NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1:a000141
Brasier AR (2010) The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 86(2):211–118
McFarland BC, Gray GK, Nozell SE, Hong SW, Benveniste EN (2013) Activation of the NF-κB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Mol Cancer Res 11(5):494–505
Tamagno E, Guglielmotto M, Monteleone D, Vercelli A, Tabaton M (2012) Transcriptional and post-transcriptional regulation of β-secretase. IUBMB Life 64(12):943–950
Shen W, Zhang C, Zhang G (2002) Nuclear factor kappaB activation is mediated by NMDA and non-NMDA receptor and L-type voltage-gated Ca(2+) channel following severe global ischemia in rat hippocampus. Brain Res 933(1):23–30
Guo Z, Jiang H, Xu X, Duan W, Mattson MP (2008) Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem 283:1754–1763
Park KW, Nozell SE, Benveniste EN (2012) Protective role of STAT3 in NMDA and glutamate-induced neuronal death: negative regulatory effect of SOCS3. PLoS One 7(11), e50874
Marrero MB, Lucas R, Salet C, Hauser TA, Mazurov A, Lippiello PM, Bencherif M (2010) An alpha7 nicotinic acetylcholine receptor selective agonist reduces weight gain and metabolic changes in a mouse model of diabetes. J Pharmacol Exp Ther 332:173–180
Ghribi O, Herman MM, Forbes MS, DeWitt DA, Savory J (2001) GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis 8(5):764–773
Fiandalo MV, Kyprianou N (2012) Caspase control: protagonists of cancer cell apoptosis. Exp Oncol 34(3):165–175
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) Bcl-2, Bcl-X(L) sequester BH3 domain-only molecules preventing Bax- and Bak-mediated mitochondrial apoptosis. Mol Cell 8:705–711
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292:727–730
Ohtsuka T, Buchsbaum D, Oliver P, Makhija S, Kimberly R, Zhou T (2003) Synergistic induction of tumor cell apoptosis by death receptor antibody and chemotherapy agent through JNK/p38 and mitochondrial death pathway. Oncogene 22:2034–2044
Shaw S, Bencherif M, Marrero MB (2002) Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against abeta-(1–42) amyloid. J Biol Chem 277:44920–44924
Vardy ER, Catto AJ, Hooper NM (2005) Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease. Trends Mol Med 11:464–472
Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y (2004) Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A 101:3632–3637
Harada H, Tamaoka A, Ishii K, Shoji S, Kametaka S, Kametani F, Saito Y, Murayama S (2006) Beta-site APP cleaving enzyme 1 (BACE1) is increased in remaining neurons in Alzheimer’s disease brains. Neurosci Res 54:24–29
Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R (2007) Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci 27:3639–3649
Alexandrov PN, Zhao Y, Jones BM, Bhattacharjee S, Lukiw WJ (2013) Expression of the phagocytosis-essential protein TREM2 is down-regulated by an aluminum-induced miRNA-34a in a murine microglial cell line. J Inorg Biochem 128:267–269
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402(6762):615–622
Park SY, Ferreira A (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates beta amyloid-induced neurodegeneration. J Neurosci 25(22):5365–5375
Reifert J, Hartung-Cranston D, Feinstein SC (2011) Amyloid beta-mediated cell death of cultured hippocampal neurons reveals extensive tau fragmentation without increased full-length tau phosphorylation. J Biol Chem 286(23):20797–20811
Kawahara M (2010) Neurotoxicity of β-amyloid protein: oligomerization, channel formation, and calcium dyshomeostasis. Curr Pharm Des 16(25):2779–2789
Kawahara M, Kato-Negishi M (2011) Link between aluminum and the pathogenesis of Alzheimer’s disease: the integration of the aluminum and amyloid cascade hypotheses. Int J Alzheimers Dis: 276393
Revett TJ, Baker GB, Jhamandas J, Kar S (2013) Glutamate system, amyloid ss peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 38(1):6–23
Tsunoda M, Sharma RP (1999) Modulation of tumor necrosis factor alpha expression in mouse brain after exposure to aluminum in drinking water. Arch Toxicol 73(8–9):419–426
Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A (2006) Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281(30):21362–21368
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934
Rubio-Perez JM, Morillas-Ruiz JM (2012) A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 756357
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6(8):844–851
Jones E, Adcock IM, Ahmed BY, Punchard NA (2007) Modulation of LPS stimulated NF-kappaB mediated nitric oxide production by PKCε and JAK2 in RAW macrophages. J Inflamm (Lond) 4:23
Parathath SR, Parathath S, Tsirka SE (2005) Nitric oxide mediates neurodegeneration and breakdown of the blood–brain barrier in tPA-dependent excitotoxic injury in mice. J Cell Sci 119:339–349
Poderoso JJ (2009) The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide. Arch Biochem Biophys 484(2):214–220
Diniz BS, Teixeira AL (2011) Brain-derived neurotrophic factor and Alzheimer’s disease: physiopathology and beyond. Neuromol Med 13(4):217–222
Miranda C, Fumagalli T, Anania MC, Vizioli MG, Pagliardini S, Pierotti MA, Greco A (2010) Role of STAT3 in in vitro transformation triggered by TRK oncogenes. PLoS One 5(3), e9446
Park SJ, Shin EJ, Min SS, An J, Li Z, Hee Chung Y, Hoon Jeong J, Bach JH, Nah SY, Kim WK, Jang CG, Kim YS, Nabeshima Y, Nabeshima T, Kim HC (2013) Inactivation of JAK2/STAT3 signaling axis and downregulation of M1 mAChR cause cognitive impairment in klotho mutant mice, a genetic model of aging. Neuropsychopharmacology 38(8):1426–1437
Chen TJ, Cheng HM, Wang DC, Hung HS (2011) Nonlethal aluminum maltolate can reduce brain-derived neurotrophic factor-induced Arc expression through interrupting the ERK signaling in SH-SY5Y neuroblastoma cells. Toxicol Lett 200(1–2):67–76
Kang D, Kim SH, Hamasaki N (2007) Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 7:39–44
Xu S, Zhong M, Zhang L, Wang Y, Zhou Z, Hao Y, Zhang W, Yang X, Wei A, Pei L, Yu Z (2009) Overexpression of Tfam protects mitochondria against β-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS J 276:3800–3809
Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H (2005) Overexpression of mitochondrial transcription factor A ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation 112:683–690
Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, Kanki T, Kang D, Sunagawa K, Tsutsui H, Nakanishi H (2008) Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci 28:8624–8634
Acknowledgments
The authors are grateful to Dr. Adel Kholoussy (Department of Pathology, Faculty of Veterinary Medicine, Cairo University, Egypt) for his assistance in histopathology and Dr. Waleed Ali (Department of Biochemistry, Faculty of Medicine, Cairo University, Egypt) for his assistance in biochemical analysis. Author Shawqi Alawdi would also like to thank Thamar University, Yemen, for supporting his PhD study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All animal procedures were performed in accordance with the ethical procedures and policies approved by the Research Ethics Committee for Experimental and Clinical Studies of Faculty of Pharmacy, Cairo University, Egypt, and comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996). All efforts were exerted to minimize animal suffering.
Conflict of Interest
The authors declare that there are no conflicts of interest in this work.
Rights and permissions
About this article

Cite this article
Alawdi, S.H., El-Denshary, E.S., Safar, M.M. et al. Neuroprotective Effect of Nanodiamond in Alzheimer’s Disease Rat Model: a Pivotal Role for Modulating NF-κB and STAT3 Signaling. Mol Neurobiol 54, 1906–1918 (2017). https://doi.org/10.1007/s12035-016-9762-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9762-0