
Abstract
Alzheimer’s disease (AD) is the most common cause of progressive decline of memory function in aged humans. To study about a disease mechanism and progression, animal models for the specific disease are needed. For AD, although highly valid animal models exist, none of the existing models recapitulates all aspects of human AD. The pathogenic mechanisms involved in AD are diverse and thus it is difficult to recapitulate human AD in model organisms. Intracerebroventricular (ICV) injection of okadaic acid (OKA), a protein phosphatase 2A (PP2A) inhibitor, in rats causes neurotoxicity associated with neurofibrillary degeneration. However, this model lacks amyloid pathology as observed in AD. We aimed at combining two different treatments and hence producing a better animal model of AD which may mimic most of the neuropathological, neurobehavioral, and neurochemical changes observed in AD. For this, OKA (200 ng) was microinjected bilaterally into the hippocampus of male Wistar rats followed by exposure of same rats to hypoxic conditions (10%) for 3 days. The result of which, the combination model exhibited tau hyperphosphorylation along with Aβ upregulation as evident by western blotting and immunohistochemistry. The observed changes were accompanied with dysfunction of neurotransmitter system, i.e., decreased acetylcholine activity and expression. This combinatorial model also exhibited cognitive deficiency which was assessed by Morris water maze and avoidance tests along with enhanced oxidative stress which is thought to be a major player in AD pathogenesis. Taken together, we established an easily reproducible and reliable rat model for sporadic dementia of Alzheimer’s type in rats which allows effective testing of new therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder which is associated with loss of memory and cognition. Pathological features of AD include neurofibrillary tangles (NFT) composed of hyperphosphorylated tau and amyloid plaques consisting of beta amyloid 42 (Aβ) aggregates which lead to neuronal death associated with cerebral atrophy and gliosis. These pathological changes are most prominent in the hippocampus, entorhinal cortex, and association areas of the neocortex and are believed to be responsible for the clinical features of AD (Pratico et al. 2000). Apart from these pathological abnormalities in AD, neurotransmitter disorders do occur in AD patients. Impairment of neurotransmitter systems in AD is due to degeneration of cholinergic neurons with the ensuing cholinergic hypofunction which is seen by decreased choline acetyl transferase (ChAT) and acetyl cholinesterase (AChE) activity in the cerebral cortex and hippocampus (Francis et al. 1999). Cognitive and neural dysfunction occurs due to the oxidative damage to nucleic acid, protein, and mitochondria in the brain (Anand et al. 2012). Memory loss in AD is mimicked by severe depression (Squire 1992). This places an emotional toll on the patient’s caretakers, while placing a financial burden of approximately $214 billion in 2014 alone on the American society (Alzheimer’s Association 2015; Alzheimer’s Association 2016). The number of those affected is expected to reach 115 million worldwide by 2050. Thus, there is an ever-growing need to find biomarkers for the early diagnosis of AD as well as to predict disease progression.
To understand AD disease progression, animal studies can be useful. But, the known animal models for AD depict one or two pathological conditions of AD, instead of showing a full blown symptomatology. A model depicting most of the disease features would be more informative in AD research. In the recent past, transgenic animals attracted attention to serve as AD models. However, very few cases of AD are known to be genetic and majority of AD cases are known to be sporadic in nature. Although some of the sporadically developed models successfully mimic AD disease phenotypes, very few animal models for AD are known which can develop most of the neuropathological, neurobehavioral, and neurochemical symptoms of AD.
Among the environmental factors causing SAD, the damaging effects of hypoxia on neurodegeneration have been considered. It is reported that hypoxia lead to enhanced expression of amyloid precursor protein (APP) which is a substrate for Aβ, upregulates cleavage of APP by increasing β-site amyloid precursor protein cleaving enzyme 1 (BACE1) gene expression, reduces Aβ degradation by decreasing production of neprilysin, and also brings calcium (Ca2+) dysfunction (Choi and Zelig 1994). Chronic intermittent hypoxia (CIH) exposure to rats for 3 days resulted in significant increase in the production of Aβ peptides (Snyder et al. 2017; Zhang and Le 2010). Also hypoxia has been found to increase plaque production along with memory deficit in AD transgenic mouse model (Gao et al. 2013). Various such studies have mentioned the role of hypoxia in accelerating Aβ pathology in AD which led us to design hypoxic model for studying AD.
NFTs, the other major hallmark of AD, are mainly comprised of aggregates of a hyperphosphorylated microtubule-associated protein called tau (Braak and Braak 1996). An imbalance between tau phosphorylation and dephosphorylation is a critical event in AD (Zhang and Simpkins 2010). Okadaic acid (OKA) is a potent and selective inhibitor of protein phosphatases, PP2A, and protein phosphatase 1 (PP1). Inhibition of PP2A by OKA induces an Alzheimer-like hyperphosphorylation and accumulation of tau both in vivo and in vitro (Kamat et al. 2014; Zhang et al. 2008). Apart from inducing tau hyperphosphorylation, OKA also increases oxidative damage in various parts of the brain. Experimental studies have shown memory impairment induced by OKA administration in rats (Zhang et al. 2008).
Along with Aβ aggregation and intracellular NFTs accumulation, the brain of an individual with AD exhibits profound loss of basal forebrain cholinergic neurons that innervate the hippocampus and the neocortex (Kar et al. 2004; Price and Sisodia 1998). Biochemical investigations of tissues from biopsy and autopsy indicate that neurotransmitters and modulators like acetylcholine (ACh), serotonin, noradrenaline, and somatostatin are differentially altered in the AD brains. The early and most consistently reproduced finding is a profound reduction in the activity of the ACh synthesizing enzyme, choline acetyl transferase (ChAT), in the neocortex, which correlates positively with the severity of dementia (Ladner and Lee 1998). The oxidation of mitochondria and nuclear DNA has been observed in the parietal cortex of AD patients (Mecocci et al. 1994). Many studies have shown increased lipid peroxidation in the temporal lobe of AD patients, where histopathologic alterations are very noticeable (Marcus et al. 1998; Palmer and Burns 1994).
The present study was planned to develop a dementia model of Alzheimer’s type in rats by combining two different treatments, viz. stereotaxic injection of OKA and exposure to hypoxic environment. We examined the effect of OKA injection (200 ng) and hypoxic exposure (10% O2) on male Wistar rats. The model was validated by examining the pathological, neurochemical, and neurobehavioral changes associated with AD along with oxidative stress generation.
Materials and Methods
Animals and Their Care
The study protocol was approved by Institutional Animal Ethical committee (IAEC 285 dated 27 July 2011). Male albino Wistar rats of approximately 2 months of age were used for the study (Maddahi and Edvinsson 2008; Richer et al. 2009; Unkruer et al. 2009). Animals were housed under standard conditions of light and dark cycle with free access to rat pellet diet (Ashirvad Industries, Chandigarh) and water ad libitum. Animals were acclimatized to laboratory conditions at room temperature prior to experimentation. OKA was obtained from Sigma Chemical Co., St. Louis, MO, USA (Cat no. 459620).
Intrahippocampal Infusion of OKA
During surgical procedures, the animals were anesthetized with ketamine/xylazine (90 and 4 mg/kg, respectively, intraperitoneally) and restrained in a stereotaxic apparatus. The head was positioned in a frame, and a midline sagittal incision was made in the scalp. Two holes were drilled in the skull for the placement of the injection cannula in both lateral cerebral ventricles. Coordinates for the i.c.v. cannula implantation were 0.8 mm posterior to bregma, 1.8 mm lateral to the sagittal suture, and 3.6 mm beneath the cortical surface. Rats were infused i.c.v. with 200 ng of OKA dissolved in artificial cerebrospinal fluid (aCSF) (147 mmol/L NaCl, 2.9 mmol/L KCl, 1.6 mmol/L MgCl2, 1.7 mmol/L CaCl2, and 2.2 mmol/L dextrose). Solution (10 μL) was injected using a Hamilton micro-syringe positioned in the injection cannula which was left in place for 3 min to allow diffusion of the drug (Watson and Paxinos 2007). The scalp was then closed with a suture. Gentamicin (5 mg/kg) was injected intraperitoneally into the animals in order to prevent sepsis.
Hypoxia Exposure
Rats were placed in a hypoxic chamber containing 10% oxygen for 22 h/day for a period of 3 consecutive days. The desired oxygen levels were established by a mixture of room air and nitrogen, which was monitored by an oxygen analyzer (Chavez et al. 2000; Pichiule and LaManna 2002).
Experimental Groups
The rats were divided into following five groups namely, control, sham, OKA, hypoxia, and OKA + hypoxia groups. Each group consisted of different number of animals (Table 1).
Control Group (C)
In this group, animals received no treatment.
Sham Group (S)
In this group, animals received aCSF intracerebroventricularly once.
Okadaic Acid-Treated Group (OKA)
In this group, animals received OKA (200 ng/10 μL) dissolved in freshly prepared aCSF intracerebroventricularly once with the help of stereotaxic apparatus.
Hypoxia-Treated Group (Hyp)
In this group, animals received hypoxia (10% oxygen) for 3 days.
Okadaic Acid + Hypoxia Group (OKA + Hyp)
In this group, animals received both OKA (200 ng/10 μL) and hypoxia (10% oxygen).
After the completion of treatment, animals were fasted overnight and sacrificed by decapitation using sodium pentothal. The brains were removed and rinsed in ice-cold physiological saline (0.9% NaCl). Brain regions were processed immediately for the assay of parameters related to oxidative stress. One separate set of animals were employed for behavioral studies. One set of animals was used for biochemical assays. One set of animals were used to study the gene and protein expression and another set of animals were used to study the histology and immunohistochemistry.
Assessment of Short-Term Memory by Shuttle Avoidance Tests
Shuttle box (Techno, India) was used to assess the conditioned avoidance learning tasks (Piala et al. 1959) with slight modifications (Sharma et al. 2009). In brief, a shuttle box apparatus consisting of a dark and an illuminated chamber separated by a door was used in the study. The rats were placed in the floor of the dark chamber, then the illuminated compartment and allowed to explore both chambers of the apparatus for 5 min. On the second day, each animal was placed into the illuminated chamber of the apparatus. As soon as the rat entered the dark chamber, 0.1 mA, 40 V was applied till the animal crossed the door and came back into the illuminated chamber. After the shock, animal was removed and returned to their home cage. On day 3, each animal was placed into the illuminated chamber, and the latency to enter into the dark chamber was measured, which served as measure of retention of avoidance response.
Active Avoidance Test
It was performed in the same way as passive avoidance test, with the change that in case of active avoidance test, no shock was applied and rats are placed in dark chamber and their latency to enter in light chamber was measured. The mean value of latency time of each session is shown in the results.
Assessment of Learning and Spatial Memory by Morris Water Maze Test
This test was carried out by the method given by Morris (Morris 1984) in which the animal is trained to escape from water by swimming to a hidden platform. The Morris water maze (MWM) is a circular pool filled with opaque water. Rats or mice are trained to use extra-maze visual cues to locate an escape platform hidden just below the surface of the opaque water. The hidden platform version of the MWM is a test of spatial memory which is sensitive to hippocampal damage. In navigation tank, the place of the platform is the same on each day but the starting point of the animal varies. This method requires long-term spatial memory and learning. In our laboratory, a video tracking system (Any-Maze software, USA), installed on a Dell PC computer connected to a video camera, was used to collect behavioral data. This test was performed for 6 days. The first 4 days were acquisition training with an invisible platform. Day 5 was reversal training, again with an invisible platform, and the number of entries and time spent in the quadrant containing hidden platform were recorded. On the sixth day, a probe trial was conducted with no escape platform and then the time spent in the target area was recorded.
Acquisition
During acquisition training, the water tank was filled with water up to the level so that the platform was covered by 1 cm of water (invisible platform). The platform was placed in the center of the northwest quadrant. Each animal received four trials of 60 s (max) per day. Rats were placed in all the four quadrants (e.g., west, north, east, or south) during trials per day. Each rat was taken out from its holding cage and then placed into the water tank at the appropriate start position, with its nose facing the center of the pool. The rat was then permitted to explore the pool and to search for the hidden escape platform for 60 s. Once on the platform, the rats were permitted to stay on the platform for 20 s to visually explore their surroundings and afterwards picked up and returned to their home cage. Each animal completed four trials per day over 4 days, for 16 trials of acquisition training. On 5th day the number of entries and time spent in quadrant containing hidden platform were recorded. The results were analyzed with the help of video tracking system and Any-Maze software.
Retrieval Test
Reversal training began on day 5–6. The platform was removed from the water tank. Each animal was placed in the tank from one of the four possible start positions and allowed to explore the pool for 90 s. The time spent in the target area (where the platform had been positioned on days 1–5) was recorded.
Brain Tissue Collection
The rats were sacrificed with ether anesthesia at the end of behavioral studies on the 27th day. The brain was removed quickly and kept on an ice-cold plate immediately. The hippocampus and cortex were dissected as per guidelines (Glowinski and Iversen 1966) and proceeded for biochemical and molecular studies. For immunohistochemistry, rats were perfused intracardially with chilled normal saline followed by 10% paraformaldehyde and brains were removed (Fig. 1).

Schematic representation of experimental procedures and treatment schedule
Preparation of Mitochondria
Rat brain mitochondria were isolated using a method in which the hippocampal brain sections were placed in glass dounce homogenizer containing homogenizing medium, with 0.1 mM ethylene glycol tetraacetic acid (EGTA), 0.32 M of sucrose, and 10 mM of Tris HCl and pH adjusted to 7.4 with potassium hydroxide (KOH) (Partridge et al. 1994). The tissue homogenate was spun at 2100×g for 15 min in a microcentrifuge at 4 °C and the supernatant was transferred to new tubes. The resulting supernatant was again spun at 14,000×g for 15 min. The supernatant was discarded and the pellet containing mitochondria was washed with homogenizing medium by centrifuging at 14,000×g. The pellet thus collected was pure mitochondria which was further resuspended in 300 μL of homogenizing buffer, and incubated with protease inhibitor on ice to carry out various experiments.
Reactive Oxygen Species Generation
Mitochondrial reactive oxygen species (ROS) generation was assessed as described previously (Wasilewski and Wojtczak 2005) with modifications. In brief, mitochondria were added to respiration buffer containing 5 mM pyruvate, 2.5 mM malate, and 10 μM of dichlorodihydrofluorescein diacetate (H2DCFDA). Fluorescence was quantified after 20 min incubation using a Cary Eclipse fluorimeter (Varian, Palo Alto, USA) (excitation 488 nm, emission 525 nm) and related to total protein content.
Lipid Peroxidation
Lipid peroxidation was ascertained by the formation of malondialdehyde (MDA) and measured by thiobarbituric acid (TBA) method as described by Wills (1966). Brain tissue from the hippocampus (0.5 mL) was diluted to 1.0 mL using Tris HCl buffer (0.1 M, pH 7.4). The method involves heating of hippocampal samples with TBA reagent for 10 min in a boiling water bath. TBA reagent contained mixture of 20% TCA, 0.5% TBA, and 2.5 N HCl. After heating of these samples with TBA reagent, the mixture then is cooled at room temperature. The colored supernatant was used to read absorbance at 532 nm.
AChE Assay
AChE activity was assayed in brain homogenates (forebrain and midbrain sections) by the method of Ellman et al. wherein the hydrolysis of acetylthiocholine to thiocholine and acetate is measured. The thiocholine reacts with 5,5′-dithiobisnitrobenzoic acid (DTNB) to give a mixed disulfide and 5-mercapto-2-nitrobenzoic acid, a yellow compound which was measured spectrophotometrically at 412 nm.
Histological Staining
For hematoxylin and eosin (H&E) staining and immunohistochemistry, rats were transcardially perfused with 0.1 M phosphate-buffered saline (PBS) at pH 7.4, followed by 10% buffered formalin. Then the fixed brains were embedded in paraffin and then paraffin-embedded blocks were made. For histological experiments, the sectioning (4–5 μm) was carried out on freshly prepared poly-L-lysine-coated clean slides. H&E staining was performed and examined by light microscopy.
For immunohistochemistry, first deparaffinization was done followed by dipping of slides in freshly prepared blocking solution of 1% H2O2 in methanol for 20 min in order to quench endogenous peroxidase. Then slides were washed thrice in PBS for 5 min each. The slides were then incubated in primary antibodies overnight at 4 °C. After washing sections with PBS, slides were incubated with HRP-labeled secondary antibody (1:100) for 30 min. Sections were again washed three times with PBS and covered with 3,3′-diaminobenzidine (DAB) solution for 10 min at room temperature followed by washing in distilled water. Finally, slides were visualized by counterstaining with hematoxylin and visualized.
For IHC, evaluation of the sections was undertaken by assessing the intensity of staining (four grades) as follows: “0” indicates very low density of positive staining; “1” indicates moderate density of positive staining; “2” indicates a higher but submaximal density of positive staining; and “3” indicates the highest density of positive staining.
Protein Isolation and Western Blot Analysis
Brain tissue was homogenized with lysis buffer and pelleted down in cytoplasmic, nuclear, and mitochondrial fractions with the help of differential centrifugation. The rat brain hippocampal tissue (2 g) was lysed in 4 mL of buffer A (50 mM of 1 M Tris (7.8), 0.01% NP-40, 150 mM of NaCl, 2 mM of EDTA, 0.1% SDS, 1 mM PMSF, and protease inhibitor in 50 mL of water). Lysate was centrifuged at 3000 rpm for 5 min at 4 °C to pellet unlysed cells. The supernatant formed the extracellular fraction and was used for detection of amyloid beta plaques. Insoluble pellet (nuclear) were treated with TNT buffer (50 mM of Tris, 150 mM NaCl, 0.1% of Triton X-100, 1 mM PMSF, and protease inhibitor cocktail in 50 mL of water). The lysate was centrifuged at 13,000 rpm at 4 °C for 90 min to pellet mitochondria. The pellet formed the mitochondrial fraction and contained mitochondrial proteins. The supernatant contained the cytoplasmic fraction. Supernatant was then used for protein estimation by bicinchoninic acid assay (BCA) method.
For western blot analysis, 80 μg of protein from each sample was resolved by electrophoresis on a 10% polyacrylamide gel. The proteins on the gel were transferred on a nitrocellulose membrane electrically. After incubating with 5% bovine serum albumin (BSA) for 1 h, the membrane was probed with the primary antibodies listed in Table 1. Blots were developed using metal enhanced DAB substrate kit followed by quantification of blots using AlphaEase software, Version 6.0.0 (Alpha Innotech Corporation, USA).
Statistical Analysis
All values are expressed as mean ± SEM of six animals in each group. One-way ANOVA, with Holm-Sidak test was used for analysis of the data and values with p < 0.001, p < 0.01, p < 0.05 were considered statistically significant. All the calculations were carried out by Sigma Stat computer software program 3.5 (TE subsystems, Inc., Germany).
Results
OKA and Hypoxia Treatment in Rats Induce AD Type Neuropathological Changes
To validate our model, the first approach was to evaluate the levels of abnormally aggregated, amyloid beta, and tau proteins. Amyloid plaques are formed by aggregation of Aβ40 or 42, which being hydrophobic, self-assemble, and form oligomers and polymers. Western blotting and immunohistochemistry were performed to check for the amyloid deposition and phospho-tau (p-tau) accumulation using anti-Aβ42 antibody and paired helical filament 1 (PHF-1) antibody, respectively.
The protein expression of Aβ42 in the hippocampus of rat brain was determined in all the groups. As shown in Fig. 2a, b, densitometry analysis showed that hypoxia treatment significantly elevated amyloid protein levels by 127% (p < 0.01) in hippocampal region of hypoxia exposed rats as compared to control. Similarly, combined treatment of OKA and hypoxia in rats showed 112% (p < 0.01) increase in β-amyloid levels. OKA caused a small but significant upregulation (17%) in Aβ levels compared to control rats. Further to confirm the presence of amyloid deposition with OKA and hypoxia treatment, it was imperative to look for the expression of amyloid beta protein by immunohistochemistry. Immunohistochemistry for amyloid plaques was done by using anti-Aβ42 antibody. The area observed mainly was the hippocampus, followed by cortex. After 15 days of OKA injection, amyloid positivity was observed in OKA-treated rats in comparison to control rats. However, hypoxia-treated rats showed more number of distinct amyloid plaques. Similar to hypoxic rats, in combined group of OKA + hypoxia (OH), high immunoreactivity for Aβ was observed. As seen in the Fig. 2c, the amyloid plaques are denser and compact in hypoxia-treated rats as compared to OKA alone and OH.

Hypoxia and OKA + hypoxia significantly increases Aβ42 protein levels in the rat brain. a Western blot analysis of Aβ42 (87 kDa). b Graph represents densitometric analysis of Aβ42 protein levels done by ImageJ software 1.48e NIH, USA. Data are expressed as means ± SD of three separate experiments. **p < 0.01 vs. respected control, ##p < 0.01 vs. respected okadaic acid. c Representative IHC images (×100 and ×400) of the hippocampal region of rat brain. d Western blot analysis of p-tau (62 kDa) normalized with total tau (79 kDa) from the brains of control and treated rats. Data is plotted as mean ± S.D. of six animals. e Significant difference was found between control, sham vs. OKA, hypoxia, and OKA + hypoxia rats. f–h Western blotting and immunohistochemistry assays for PHF-1 (62 kDa) in the brains of control and treated rats. Magnifications are ×100 and ×400. C, control; S, sham; O, okadaic acid; H, hypoxia; OH, okadaic acid + hypoxia
We performed western blot analysis of phosphorylated tau (P-tau) as it is the component of paired helical filaments which subsequently form NFTs. Anti P-tau antibody was used to check for phosphorylation on tau protein (Fig. 2d). Densitometry of P-tau protein was done by normalizing it with unphosphorylated tau protein (Fig. 2e). Densitometry analysis showed that P-tau levels were increased significantly (p < 0.01) in OKA-injected rats by 96%, in hypoxic rats by 44%, and in OH-treated animals by 107% as compared to control (p < 0.001). The combined treatment of OH resulted in 5 and 43% increase in P-tau levels as compared to OKA and hypoxia, respectively.
In order to assess the effect of OKA and hypoxia treatment on PHF expression, western blotting was performed using PHF-1 antibody (Fig. 2f, g). A significant upregulation in the expression of PHF in OKA-, hypoxia-, and OH-treated rats was observed in comparison to control rats. The upregulation observed was 84% in OKA-injected rats, 29% in Hyp-exposed rats, and 111% in rats given both OKA and Hyp treatment as compared to control animals (p < 0.01). On comparison to OKA alone, PHF levels were further increased by 15% in OH-treated rats.
After confirmation of tau hyperphosphorylation with OKA and hypoxia treatment, we further checked PHF formation following OKA and hypoxia exposure by immunohistochemistry using anti-PHF-1 antibody (Fig. 2h). Immunohistochemistry showed increased PHF expression in OKA- and hypoxia-treated rats as compared to control animals. PHF-1 antibody intensely stained cytoplasm of hippocampal neurons in the treated groups, i.e., OKA, hypoxia, and OH group as compared to control rats. Along with the hippocampus, PHF positivity was also observed in the cortex of rat brains injected with OKA.
OKA and Hypoxia Treatment Affect Learning and Memory of Rats
The clinical outcome of AD is memory impairment which we checked in our designed animal model, using avoidance tasks and MWM test. Having observed AD like pathological and neurochemical alterations in our rat model, it was imperative to assess memory functions to check whether OKA and hypoxia treatment had any effect on memory.
The active avoidance response increased significantly in terms of latency time in OKA (11 s), hypoxia (11 s), and OH (10 s) treated rats in comparison to control animals (3 s). The latency period of the OKA, hypoxia, and OH animals was about 76, 75, and 73% higher than controls, respectively (p < 0.01) (Fig. 3a). The time taken to remember not to enter the chamber where rats experienced a foot pad shock was significantly less in OKA (70%), hypoxia (78%), and OH (78%) treated rats as compared to control rats (p < 0.01) (Fig. 3b).

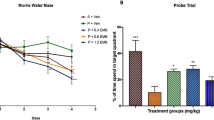
Impairment of spatial learning and memory in rats with OKA and hypoxia treatment. a Time of memory retention in the passive avoidance test carried out in rats at day 15 after OKA i.c.v. injection and hypoxia treatment. b Different groups of animals treated with vehicle (i.c.v.), OKA (i.c.v.), hypoxia, and OKA + hypoxia. The results are expressed as medians of n = 6 animals. c Plot curves showing the path followed by animals of different groups in the Morris water maze test performed over 6 days. d During acquisition, learning was expressed as the time spent in the quadrant containing platform. e Overall average speed was calculated to check for any motor impairment. f Spatial memory was assessed during the probe test on day 6 by measuring the time spent into the target and the opposite quadrants. g The latency to first cross the platform zone. i The overall speed of animals. h, j The number of entries and total distance traveled in the platform zone (Values are mean ± SEM, n = 6 animals per group)
This experiment conveyed that OKA and hypoxia treatment induced short-term memory loss as rats were taking less time in entering the dark chamber where they had experienced foot shock earlier.
The results indicated that the time spent in the quadrant containing platform in the water maze by OKA- and hypoxia-treated rats was comparatively less, when compared to control. However, the overall speed of all the treated and control groups remain almost same at 0.20 ± 0.005 m/s. The plot curve (Fig. 3c) shows the path followed by animals of different groups, which depicts that rats from OKA, hypoxia, and OH groups are confused and are not able to locate the hidden platform. Thus, the animals from OKA, hypoxia, and OH groups demonstrated significant memory impairments as shown by reduced ability to perform the task as compared to controls.
After 24 h of acquisition test, time spent, number of entries, and total distance traveled in the target quadrant (without platform) were decreased in case of OKA- and hypoxia-treated rats as compared to control rats (Fig. 3d, e). On the final day, the time spent, number of entries, and distance traveled in target quadrant for OKA-treated rats were 10 ± 0.43 s, 4.3 ± 0.17, 1.9 ± 0.22 m/s, respectively, and for hypoxia-treated rats, it was 9.3 ± 0.12 s, 4.6 ± 0.34, 2.45 ± 0.29 m/s and for OH it was 7.6 ± 0.41 s, 5 ± 0.22, 1.89 ± 0.23 m/s (p < 0.01) (Fig. 3g–j). The OKA and hypoxia treatments alone and in combination resulted in decreased ability of rats in recognizing the area where platform was placed as compared to control animals.
OKA and Hypoxia Treatment Resulted in Decreased Activity and Expression of AChE in Rats
To detect cholinergic deficits occurring in the rat brain following OKA and hypoxia exposure, AChE activity was assessed separately in forebrain and midbrain regions of rats. The effect of OKA and hypoxia on AChE activity was observed only in forebrain region and not in midbrain. In forebrain region, OKA, hypoxia, and OH induced significant decrease of 33% (p < 0.05), 52% (p < 0.01), and 56% (p < 0.01) in AChE activity respectively as compared to controls (Fig. 4a). However, in midbrain tissue, there was no change in the AChE activity (Fig. 4b).

AChE activity and expression was decreased in rat forebrain in response to OKA injection and hypoxic environment. a AChE activity in rat forebrain and midbrain separately using Ellman’s spectrophotometric method for control and treated rat groups. There was no significant change in AChE activity in midbrain tissue for all groups. b Immunohistochemistry images showing AChE expression in the hippocampal region of all rat groups. Magnification ×100, ×400. In the OKA- and hypoxia-treated animals, hippocampal cells exhibited diffuse and weak staining for AChE. Results are from three independent experiments (n = 6)
In the next experiment, AChE immunohistochemistry was performed to confirm the above spectrophotometric findings. We determined AChE expression in hippocampus region by staining sections with antibody against AChE. Immunohistochemistry showed weak (1+) AChE positivity in neurons of rat brain in OKA- and hypoxia-treated animals as compared to controls which showed high (3+) cytoplasmic positivity (Fig. 4c).
OKA and Hypoxia Exposure Induces Oxidative Stress in Rat Brain
For estimation of pro-oxidant status of cell, we estimated ROS levels and lipid peroxidation in the control and treated rat brains. As compared to controls, the mitochondria isolated from the hippocampus of rat brains of OKA- and hypoxia-treated rats showed significant increase (p < 0.01) in signal fluorescence suggesting higher ROS accumulation in mitochondria of these treated rats. ROS accumulation was seen to be increased by 48% in OKA, 50% in hypoxia, and 51% in OKA + Hyp-treated groups as compared to control (p < 0.01) (Fig. 5a).

Oxidative stress parameters of rat brains. ROS levels, lipid peroxidation, and protein carbonylation were evaluated after OKA and hypoxia treatment. a ROS levels in normal and treated rat brains; 200 ng of OKA and 3 days of hypoxia increased ROS production when compared to control rats; b MDA levels were significantly increased with both OKA and hypoxia treatments compared to either of treatments alone. c The effects of OKA and hypoxia exposure on protein oxidation were assessed from protein carbonyl contents in brain homogenates. OKA and hypoxia treatment sharply increased carbonyl contents in brain (p < 0.01). With combined treatment of OKA and hypoxia, carbonyl content increased even further (p < 0.01 vs. OKA and hypoxia alone treatment), indicating that combined treatment inflicted appreciable oxidative damage on brain proteins. **p < 0.01 vs. control group, ##p < 0.01 vs. respected okadaic acid, $$p < 0.01 vs. respected hypoxia. Data are shown as means ± SE (n = 6)
We measured lipid peroxidation by measuring MDA accumulation in brains of treated rats and found that MDA levels were significantly increased in the hippocampal region of brains of OKA-injected rats by 46%, in hypoxia-exposed animals by 90%, and after OKA + Hyp exposure by 110% (p < 0.01, Fig. 5b) in comparison to control rat hippocampal brain. Similar to lipid peroxidation levels, protein carbonylation levels were also found to be increased with both OKA and hypoxia treatment (Fig. 5c).
OKA and Hypoxia Treatment Resulted in Neurodegeneration/Neuronal Death in Rats
Western blotting was performed to detect protein levels of caspase 3 and procaspase 3 by using antibody against caspase 3 (Fig. 6a, b). We found that procaspase 3 levels were decreased in OKA, Hyp, and OH groups. Simultaneously, caspase 3 levels were significantly higher in OKA, Hyp, and OH groups respectively as compared to control, showing procaspase cleavage on OKA and hypoxia and combination treatment.

OKA and hypoxia induces apoptosis in rat brain. a Representative detection of caspase 3 protein by western blot. Procaspase 3 (35 kDa) cleavage was increased in OKA- and hypoxia-treated rats compared to control and sham rats. Western blotting was used to determine the protein level of procaspase 3 and cleaved caspase 3 (17 kDa) in all groups. b We examined caspase 3 protein by western blotting and conducted densitometry on the blots. We found that OKA and hypoxia treatment, alone and in combination, significantly activated caspase 3. Results shown are from six rats in each group. c Representative hematoxylin and eosin images from the hippocampus of rat brain. Low magnification, 100 μM; high magnification, 200 μM
We performed H&E staining to check the pathological changes at the neuronal level. In order to distinguish between degenerating neuron and dark neuron artifact, all rat brains were perfused. Histological examination of the hippocampal region of different treatment groups demonstrated marked neuronal damage as depicted by eosinophilic neurons (swollen cytoplasm) and shrunken and pyknotic hyperchromatic nuclei (darkened nuclei) in the OKA-, hypoxia-, and OH-treated groups when compared to the control group (Fig. 6c).
Discussion
OKA was used to induce tau pathology. OKA inhibits protein phosphatases 1 and 2A and thus induces hyperphosphorylation of tau protein (Broetto et al. 2016; Kamat et al. 2013b). OKA-treated rats also show memory impairment, oxidative stress, and mitochondrial dysfunction, thereby sharing some features of AD (Kamat et al. 2014). Hyperphosphorylation of tau protein was evident by western blot analysis of p-tau protein and immunohistochemical analysis for PHF in the combinatorial model. Previous studies on rats have also shown that OKA induces tauopathic features (Kamat et al. 2013a; Kamat et al. 2013b) in rats. Cognitive defects were noted in the model along with NFTs-like conformational changes in both the cortex and hippocampal regions (Kamat et al. 2013a; Kamat et al. 2013b). In the present study, OKA treatment caused a significant increase in protein expression of phosphorylated tau and PHF levels in the cerebral cortex and hippocampus.
Amyloid plaques deposition occurs either due to an overproduction or decreased clearance of Aβ peptides. To induce Aβ aggregation, various studies have suggested i.c.v. injection of Aβ peptide in various forms to induce amyloid plaque formation in rats (Lecanu et al. 2006; Nakamura et al. 2001). Aβ injection does induce cognitive deficits in animals with amyloid plaque formation (Gordon et al. 2009). However, the pathology develops primarily at the site of injection; frequently, amyloid deposits were observed in the hippocampus and neocortex but not cerebellum and brainstem (Forny-Germano et al. 2014). Stroke is a well-known risk factor for AD (Honig et al. 2003) and one of the pathological mechanisms is neuronal hypoxia (Ogunshola and Antoniou 2009). Hence, hypoxia was chosen since amyloid generation by this pathway could closely mimic the pathophysiology of SAD. An earlier study where 3-day chronic intermittent hypoxia was given to rats resulted in increased beta amyloid generation (Ng et al. 2010). We adopted this strategy in our study to target amyloidogenesis. Rats were kept in hypoxic conditions, following which they were sacrificed and brain tissue was examined for the presence of amyloid plaques. Three-day acute exposure of 10% oxygen in rats resulted in upregulation of amyloid beta levels as shown by western blotting and amyloid plaque formation as evident by immunohistochemistry in the present study.
xApart from amyloid plaques and NFTs, other neuropathological alterations observed in AD are neuronal and synaptic loss (Serrano-Pozo et al. 2011). Neuronal loss is the causative factor behind clinical manifestation of AD (Cotman and Su 1996). In the present study, as depicted by H&E staining, OKA- and hypoxia-treated sections showed significant degeneration of neurons. With OKA and hypoxia treatment, CA1 neurons appeared intensely hypereosinophilic with extensive clumping of chromatin, particularly in the combined group of OKA and hypoxia compared to the OKA and hypoxia treatments alone. The final outcome of such pathological and neurochemical changes along with enhanced oxidative stress in rat brain is neuronal and synapse loss which leads to memory impairment in rats. As depicted by H&E staining, OKA- and hypoxia-treated sections showed marked cell distortion with high level of degeneration in the neurons as compared to control.
The result of OKA and hypoxia treatment thus is neurodegeneration in the subregions of the hippocampus (CA1, CA3, and DH). However, the extent of degeneration was almost similar for OKA and hypoxia treatments alone and in combination. Thus, it can be inferred that combined treatment of OKA and hypoxia resulted in pathological alterations of amyloid plaques, NFTs, and neuronal degeneration. Neuronal and hence synaptic loss results in cognitive dysfunction in AD. It has also been suggested that in some cases, behavioral changes may be more sensitive than neurochemical alterations, as indicators of neurodegeneration and may be observed early during disease progression.
To check, if significant neuronal shrinkage and degenerated nuclei in the OKA and hypoxia rats lead to memory impairment, we checked memory deficits in terms of short-term and spatial memory. OKA- and hypoxia-treated rats showed decrease in the number of avoidances in the learning session and retention which has been reported by others also (Dimitrova and Getova-Spassova 2006; Dwivedi et al. 2013). MWM has been performed to evaluate learning and memory deficits in AD animals (Bromley-Brits et al. 2011). In the current study, the effect of OKA on memory impairment was tested by the avoidance and Morris water maze. The memory deficit in OKA-treated rats was evident by significant changes in the latency time and path length to reach the hidden platform. Deficits in cholinergic neurotransmission are not the only cause of memory loss but this deficit clearly impairs memory in AD. So we studied cholinergic neurotransmission in our AD model by evaluating AChE activity and expression in rat brain. Animal models of AD have been established based on experimental cholinergic denervation (Muir et al. 1993). We observed significant decrease in AChE activity in rat forebrain after OKA and hypoxia exposure. This finding is in conformity with previous study showing OKA induced cholinergic dysfunction by decreasing AChE activity and mRNA expression (Dwivedi et al. 2013). OKA also resulted in generation of oxidative stress which reduces cerebral blood flow (CBF) in the brain. Decreased CBF impairs glucose and energy metabolism which lead to increased cellular dysfunction (Olanow 1993). Disturbed glucose and energy metabolism impairs cholinergic system, thus resulting in decreased level of choline acetyl transferase and acetyl cholinesterase (Areosa and Sherriff 2003). The role of hypoxia in decreasing AChE activity has also been previously studied (Udayabanu et al. 2008). The cholinergic system is involved in many physiological processes, including synaptic plasticity, learning, and memory. Cholinergic agonists can facilitate memory, whereas cholinergic antagonists can impair memory. Thus, OKA and hypoxia affect AChE and ChAT enzyme activity and expression and interfere with cholinergic neurotransmission, affecting learning, memory, and cognition functions.
Hence, we validated our induced model of AD on neuropathological, neurobehavioral, and neurochemical parameters and inferred that OKA and hypoxia treatment in the rat brain resembles the changes observed in AD patients.
In AD, brain is under increased oxidative stress which may have a role in neuronal degeneration and death. Therefore, we looked for the presence of oxidative markers viz. reactive oxygen species and lipid peroxidation levels. As compared to control rats, increased mitochondrial ROS levels in rat brain after OKA and hypoxia treatment were observed when given alone and in combination. Several biological markers of oxidative stress were found to be elevated in AD animal models in different studies (Melov 2002). MPTP-treated mice demonstrate elevated levels of ROS and lipid peroxidation (Sriram et al. 1997). Evidence for the involvement of oxidative stress in AD has also been obtained from AD patients. Increased lipid peroxidation and oxidative damage to DNA and proteins have been observed in hippocampus of AD patients (Reist et al. 1998). The ROS molecules generated in AD might cause neuronal damage by reacting with nucleic acids, proteins, and lipids. Our data indicated increased ROS levels and lipid peroxidation in brains of OKA- and hypoxia-treated rats. With OKA and hypoxia treatment, MDA levels were found to be significantly upregulated. Increased ROS generation predisposes rats to enhanced oxidative stress and consequently neuronal death.
Conclusion
To summarize, the present study proposes the use of combined treatment of okadaic acid and hypoxia for developing a dementia model of Alzheimer’s type in rats. The dementia model described displays neurobehavioral impairments along with pathological and neuorchemical changes as observed in AD. Thus, a picture emerges that depicts a similar pathological mechanisms between the experimental rat model and clinical conditions. This novel rat model reliably simulates the main pathological outcomes during Alzheimer disease. It represents a promising and relevant preclinical model for testing new therapeutic approaches.
Abbreviations
- Aβ:
-
Beta amyloid
- NFTs:
-
Neurofibrillary tangles
- OKA:
-
Okadaic acid
- ROS:
-
Reactive oxygen species
- PHF:
-
Paired helical filament
- H2-DCFHDA:
-
2′,7′-Dichlorodihydrofluorescein diacetate
- SOD:
-
Superoxide dismutase
References
Alzheimer’s Association (2015) 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 11:332–384
Alzheimer’s Association (2016) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12:459–509
Anand R, Kaushal A, Wani WY, Gill KD (2012) Road to Alzheimer’s disease: the pathomechanism underlying. Pathobiology 79:55–71
Areosa SA, Sherriff F (2003) Memantine for dementia Cochrane Database Syst Rev:CD003154
Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 165:3–12
Broetto N, Hansen F, Brolese G, Batassini C, Lirio F, Galland F, dos Santos JPA, Dutra MF, Gonçalves CA (2016) Intracerebroventricular administration of okadaic acid induces hippocampal glucose uptake dysfunction and tau phosphorylation. Brain Res Bull 124:136–143
Bromley-Brits K, Deng Y, Song W (2011) Morris water maze test for learning and memory deficits in Alzheimer’s disease model mice. J Vis Exp 20:2920
Chavez JC, Agani F, Pichiule P, LaManna JC (2000) Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol (1985) 89:1937–1942
Choi PM, Zelig MP (1994) Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut 35:950–954
Cotman CW, Su JH (1996) Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol 6:493–506
Dimitrova DS, Getova-Spassova DP (2006) Effects of galantamine and donepezil on active and passive avoidance tests in rats with induced hypoxia. J Pharmacol Sci 101:199–204
Dwivedi S, Nagarajan R, Hanif K, Siddiqui HH, Nath C, Shukla R (2013) Standardized extract of Bacopa monniera attenuates okadaic acid induced memory dysfunction in rats: effect on Nrf2 pathway. Evid Based Complement Alternat Med 2013:294501
Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, Martinez AMB, Klein WL, Houzel JC, Ferreira ST, Munoz DP, de Felice FG (2014) Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J Neurosci 34:13629–13643
Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 66:137–147
Gao L, Tian S, Gao H, Xu Y (2013) Hypoxia increases Abeta-induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J Mol Neurosci 51:138–147
Glowinski J, Iversen L (1966) Regional studies of catecholamines in the rat brain. 3. Subcellullar distribution of endogenous and exogenous catecholamines in various brain regions. Biochem Pharmacol 15:977–987
Gordon RY, Mugantseva EA, Khutzian SS, Podolski IY (2009) Cycloheximide-induced inhibition of protein synthesis in hippocampal pyramidal neurons is time-dependent: differences between CA1 and CA3 areas. Neurosci Lett 461:249–251
Honig LS, Tang MX, Albert S, Costa R, Luchsinger J, Manly J, Stern Y, Mayeux R (2003) Stroke and the risk of Alzheimer disease. Arch Neurol 60:1707–1712
Kamat PK, Rai S, Nath C (2013a) Okadaic acid induced neurotoxicity: an emerging tool to study Alzheimer's disease pathology. Neurotoxicology 37:163–172
Kamat PK, Rai S, Swarnkar S, Shukla R, Ali S, Najmi AK, Nath C (2013b) Okadaic acid-induced Tau phosphorylation in rat brain: role of NMDA receptor. Neuroscience 238:97–113
Kamat PK, Rai S, Swarnkar S, Shukla R, Nath C (2014) Molecular and cellular mechanism of okadaic acid (OKA)-induced neurotoxicity: a novel tool for Alzheimer’s disease therapeutic application. Mol Neurobiol 50:852–865
Kar S, Slowikowski SPM, Westaway D, Mount HTJ (2004) Interactions between β-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci 29:427–441
Ladner CJ, Lee JM (1998) Pharmacological drug treatment of Alzheimer disease: the cholinergic hypothesis revisited. J Neuropathol Exp Neurol 57:719–731
Lecanu L, Greeson J, Papadopoulos V (2006) Beta-amyloid and oxidative stress jointly induce neuronal death, amyloid deposits, gliosis, and memory impairment in the rat brain. Pharmacology 76:19–33
Maddahi A, Edvinsson L (2008) Enhanced expressions of microvascular smooth muscle receptors after focal cerebral ischemia occur via the MAPK MEK/ERK pathway. BMC Neurosci 9:85
Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML (1998) Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol 150:40–44
Mecocci P, MacGarvey U, Beal MF (1994) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36:747–751
Melov S (2002) Animal models of oxidative stress, aging, and therapeutic antioxidant interventions. Int J Biochem Cell Biol 34:1395–1400
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60
Muir JL, Page KJ, Sirinathsinghji DJ, Robbins TW, Everitt BJ (1993) Excitotoxic lesions of basal forebrain cholinergic neurons: effects on learning, memory and attention. Behav Brain Res 57:123–131
Nakamura S, Murayama N, Noshita T, Annoura H, Ohno T (2001) Progressive brain dysfunction following intracerebroventricular infusion of beta(1-42)-amyloid peptide. Brain Res 912:128–136
Ng KM, Lau CF, Fung ML (2010) Melatonin reduces hippocampal beta-amyloid generation in rats exposed to chronic intermittent hypoxia. Brain Res 1354:163–171
Ogunshola OO, Antoniou X (2009) Contribution of hypoxia to Alzheimer’s disease: is HIF-1alpha a mediator of neurodegeneration? Cell Mol Life Sci 66:3555–3563
Olanow CW (1993) A radical hypothesis for neurodegeneration. Trends Neurosci 16:439–444
Palmer AM, Burns MA (1994) Selective increase in lipid peroxidation in the inferior temporal cortex in Alzheimer’s disease. Brain Res 645:338–342
Partridge RS, Monroe SM, Parks JK, Johnson K, Parker WD Jr, Eaton GR, Eaton SS (1994) Spin trapping of azidyl and hydroxyl radicals in azide-inhibited rat brain submitochondrial particles. Arch Biochem Biophys 310:210–217
Piala JJ, High JP, Hassert GL Jr, Burke JC, Craver BN (1959) Pharmacological and acute toxicological comparisons of triflupromazine and chlorpromazine. J Pharmacol Exp Ther 127:55–65
Pichiule P, LaManna JC (2002) Angiopoietin-2 and rat brain capillary remodeling during adaptation and deadaptation to prolonged mild hypoxia. J Appl Physiol (1985) 93:1131–1139
Pratico D, Clark CM, Lee VM, Trojanowski JQ, Rokach J, FitzGerald GA (2000) Increased 8,12-iso-iPF2alpha-VI in Alzheimer’s disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol 48:809–812
Price DL, Sisodia SS (1998) Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci 21:479–505
Reist M, Marshall KA, Jenner P, Halliwell B (1998) Toxic effects of sulphite in combination with peroxynitrite on neuronal cells. J Neurochem 71:2431–2438
Richer MJ, Lavallee DJ, Shanina I, Horwitz MS (2009) Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One 4:e4127
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189
Sharma DR, Sunkaria A, Bal A, Bhutia YD, Vijayaraghavan R, Flora SJ, Gill KD (2009) Neurobehavioral impairments, generation of oxidative stress and release of pro-apoptotic factors after chronic exposure to sulphur mustard in mouse brain. Toxicol Appl Pharmacol 240:208–218
Snyder B, Shell B, Cunningham JT, Cunningham RL (2017) Chronic intermittent hypoxia induces oxidative stress and inflammation in brain regions associated with early-stage neurodegeneration. Physiol Rep 5(9):e13258
Squire LR (1992) Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 99:195–231
Sriram K, Pai KS, Boyd MR, Ravindranath V (1997) Evidence for generation of oxidative stress in brain by MPTP: in vitro and in vivo studies in mice. Brain Res 749:44–52
Udayabanu M, Kumaran D, Nair RU, Srinivas P, Bhagat N, Aneja R, Katyal A (2008) Nitric oxide associated with iNOS expression inhibits acetylcholinesterase activity and induces memory impairment during acute hypobaric hypoxia. Brain Res 1230:138–149
Unkruer B et al (2009) Cellular localization of Y-box binding protein 1 in brain tissue of rats, macaques, and humans. BMC Neurosci 10:28
Wasilewski M, Wojtczak L (2005) Effects of N-acylethanolamines on the respiratory chain and production of reactive oxygen species in heart mitochondria. FEBS Lett 579:4724–4728
Watson C, Paxinos G (2007) The rat brain in stereotaxic coordinates. Academic Press, San Diego
Wills ED (1966) Mechanisms of lipid peroxide formation in animal tissues. Biochem J 99:667–676
Zhang X, Le W (2010) Pathological role of hypoxia in Alzheimer’s disease. Exp Neurol 223:299–303
Zhang Z, Simpkins JW (2010) Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res 1345:176–181
Zhang QG, Wang R, Khan M, Mahesh V, Brann DW (2008) Role of Dickkopf-1, an antagonist of the Wnt/beta-catenin signaling pathway, in estrogen-induced neuroprotection and attenuation of tau phosphorylation. J Neurosci 28:8430–8441
Funding
This study was funded by Council of Scientific and Industrial Research (CSIR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
No conflicts of interest are declared by the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Okadaic acid and hypoxia induce plaque and tangle pathology in this rat model of AD
• Combination of okadaic acid and hypoxia produced enhanced oxidative stress and associated neurodegeneration in rat brain
• Okadaic acid and hypoxia exposure to rats mimic the neurobehavioral and neurochemical alterations as observed in AD.
Rights and permissions
About this article

Cite this article
Kaushal, A., Wani, W.Y., Bal, A. et al. Okadaic Acid and Hypoxia Induced Dementia Model of Alzheimer’s Type in Rats. Neurotox Res 35, 621–634 (2019). https://doi.org/10.1007/s12640-019-0005-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-0005-9