
Abstract
The mammalian brain is extremely sensitive to alterations in cellular homeostasis as a result of environmental or physiological insults. In particular, hypoxic/ischemic challenges (i.e. reduced oxygen and/or glucose delivery) cause severe and detrimental alterations in brain function and can trigger neuronal cell death within minutes. Unfortunately, as we age, oxygen delivery to cells and tissues is impaired, thereby increasing the susceptibility of neurons to damage. Thus, hypoxic (neuronal) adaptation is significantly compromised during aging. Many neurological diseases, such as stroke, Alzheimer’s disease (AD), Parkinson’s disease and diabetes, are characterized by hypoxia, a state that is believed to only exacerbate disease progression. However, the contribution of hypoxia and hypoxia-mediated pathways to neurodegeneration remains unclear. This review discusses current evidence on the contribution of oxygen deprivation to AD, with an emphasis on hypoxia inducible transcription factor-1 (HIF-1)-mediated pathways and the association of AD with the cytoskeleton regulator cyclin-dependent kinase 5. (Part of a multi-author review.)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proper neuronal activity is critical for the regulation of physiological functions throughout the entire organism, and any disruption of neuronal function has potentially devastating consequences. Neurons are extremely dependent on oxygen availability and demonstrate a high vulnerability during conditions that cause or result in alterations in oxygen supply. Even just a few minutes of oxygen deprivation initiates significant dysfunction, and longer episodes can ultimately result in the induction of cell death [1].
Unfortunately, as we age, oxygen delivery to cells and tissues is somewhat impaired, thereby increasing the susceptibility of neurons to damage. In addition, cellular adaptation to hypoxia is significantly compromised with increasing age and, at the same time, an ischemic episode has a more dramatic outcome in old versus young patients [2, 3]. In line with these findings, the risk of stroke doubles with each decade after the age of 55 years, while two-thirds of cerebral infarctions are reported in people over the age of 65 [4]. Increased neuronal vulnerability is also very evident in the sharp rise in the incidence of neurodegenerative diseases that occur in the later stages of life and as the lifespan continues to increase. Indeed, mortality as a result of neurological disorders constitutes 12% of total deaths globally, while nervous system diseases lead to more hospitalizations than any other disease group, including heart disease and cancer, resulting in a burden on both social and economic sectors of society.
A key characteristic of many neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease and frontotemporal dementias, is the accumulation of potentially toxic proteins which although present throughout life become neurotoxic as a result of environmental stimuli, including head injury, oxygen deprivation and oxidative stress. The precise contribution of such stimuli to neurodegeneration remains largely unknown, but reduced oxygen supply seems to play a key role in being a cause and/or consequence of neurodegeneration during the aging process.
This review discusses current evidence on the contribution of oxygen deprivation and HIF-1-mediated pathways to neurodegeneration, with particular emphasis on AD and the cytoskeleton regulator cyclin-dependent kinase 5 (Cdk5).
Alzheimer’s pathogenesis
Alzheimer’s disease is a complex, progressive neurodegenerative disorder. Individuals affected with this disease show severe memory and cognitive function impairment, which is believed to be the outcome of disrupted neuronal circuits and severe neuronal loss. The prevalence of AD is directly related to age [5, 6] and represents an enormous healthcare burden for society. Alzheimer's disease is a growing social and economic burden on the developed world, with the current cost of care for patients with AD around the world estimated at approximately $150 billion annually.
The major pathological hallmarks of AD are large extracellular β-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles (NFTs) composed of abnormally hyperphosphorylated forms of the microtubule-associated protein tau. Aβ is the cleaved product of the transmembrane amyloid precursor protein (APP). The cleavage of APP by β-secretase (β-site amyloid precursor protein cleavage enzyme, BACE) and subsequently by the γ-secretase complex results in the formation of two Aβ isoforms, Aβ40 and Aβ42, that have neurotoxic potential. Interestingly, the presence of Aβ in the cerebrospinal fluid (CSF) of healthy individuals and also in the media of neuronal cultures suggests an additional physiological role for Aβ. Currently available evidence indicates that during the initial phase of AD, both Aβ deposition and the formation of NFTs are the consequence of oxidative stress and help to protect neurons against injury [7, 8].
However, during disease progression it appears that this anti-oxidant capacity becomes pro-oxidant, promoting the generation of free radicals, the disruption of microtubule networks, disturbances in axoplasmic transport, synapse loss and, eventually, neuronal death. It is therefore argued that Aβ becomes toxic only when the balance between its production and degradation is disturbed as a result of stressful stimuli. The density of NFTs correlates with the severity of the AD symptoms, and a number of proteins, such as protein kinases Cdk5 and glycogen synthase kinase 3 (GSK3), have been shown to be involved in tau hyperphosphorylation [9, 10].
Alterations in the actin cytoskeleton frequently accompany AD pathology. Actin-rich inclusions, known as Hirano bodies, as well as cofilin–actin rods have been described in a number of neurodegenerative disorders [11, 12]. Immunohistochemical studies have revealed that Hirano bodies are mainly composed of actin microfilaments, although tau, tubulin and APP are also present [13]. The number of Hirano bodies increases during physiological aging and is further increased in AD patients, indicating their participation in disease pathogenesis [14]. Cofilin–actin rods, much smaller aggregates that appear as separate units in AD brains, are also thought to give rise to Hirano bodies over time [15].
Along with progressive Aβ and NFT accumulation, cytoskeletal and synapse disruption, AD pathology is further characterized by inflammation as well as deficits in the expression of neurotrophic factors, transcription factors and antioxidant enzymes. Altogether these multi-faceted events render the disease complex and thus particularly difficult to treat.
Hypoxia and AD pathogenesis
Alzheimer’s disease is a multifactorial disease in which both genetic and environmental factors contribute to—and accelerate—disease progression. It is now apparent that genetic predisposition is accountable for only a small number of AD cases. Fully penetrant mutations leading to protein cleavage and consequent aggregation are responsible for only 5% of all AD cases and result in familial early onset AD (from 65 years) [16]. Although most cases have no known genetic basis, exposure to other pathogenic conditions, including chronic inflammation, traumatic brain injury, cerebrovascular disease, hypoxia/ischemia and exposure to pesticides [5, 16–18], could be contributing factors.
It has been shown that individuals who have suffered severe hypoxia or ischemia are more susceptible to developing AD [19, 20]. The risk of incident dementia is particularly elevated in association with illnesses that may cause cerebral hypoxia or ischemia, suggesting that cerebral hypoperfusion may serve as a basis for some cases of dementia after stroke [20]. Head injury also induces ischemic changes that induce a tau-like pathology subsequent to neuronal damage, indicating that such injuries/insults can lead to a relatively rapid induction of APP and amyloidogenic phenotypes even in the young brain [21]. In vitro, when cortical neurons are treated with Aβ concomitantly with hypoxic exposure, the number of apoptotic neurons increase dramatically compared to treatment with Aβ alone [22], indicating that hypoxia itself accelerates the neuronal death observed in AD brains. Furthermore, hypoxia has been found not only to contribute to plaque formation in an AD transgenic mouse model but also to increase the memory deficit characterizing these mice [23]. Thus, impairment of the proper oxygen supply has been shown to be involved in AD pathogenesis, thereby substantiating clinical observations that associate hypoxic episodes to AD.
Despite available evidence, the molecular mechanisms that link hypoxia to AD pathogenesis are not well defined. Hypoxia/ischemia upregulate APP at both the mRNA and protein level and lead to subsequent Aβ accumulation [19, 22, 24–27]. It is thought that an immediate increase in APP levels is a defense mechanism employed to induce the production of the cleaved soluble product APPα, which has known neuroprotective properties [19]. Concomitantly, however, these increased levels of APP provide the resource for the generation and accumulation of Aβ, which also increases dramatically as a result of hypoxic injury. Aβ neurotoxic effects are also known to involve alterations in Ca2+ homeostasis, a key event in AD pathogenesis, leading to cytoskeletal disruption and, eventually, neuronal death [28, 29]. Prolonged hypoxia selectively upregulates L-type Ca2+ channels, an event that requires Aβ formation, since in its absence these channels are no longer affected by the hypoxic insult [30]. Aβ clearly interacts directly with the α1 subunit, and hypoxia further promotes this interaction, leading to an increased expression of functional Ca2+ channels at the cell membrane and thus an increase in Ca2+ influx [31], probably mediated by Ca2+-permeable AMPA receptors [22]. This mechanism suggests that a feedback loop between Aβ and hypoxia may promote disease progression. Other evidence shows that brief periods of hypoxia can potentiate the Aβ-induced expression of the pro-inflammatory markers cyclooxygenase-2 and presenilin 1 (COX-2 and PS1, respectively), thus accelerating the inflammatory neuropathology that characterizes AD brains [32].
Taken together these data provide convincing evidence that hypoxia contributes to AD pathogenesis through amyloidogenesis and Ca2+ dependent mechanisms. However, it is clear that many more studies are needed to fully understand the impact of hypoxia-mediated changes on neurodegenerative pathologies.
HIF-1 signaling during AD: a double-edged sword?
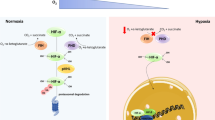
At the molecular level, a large percentage of hypoxic responses are controlled by hypoxia-inducible transcription factor-1 (HIF-1, reviewed by Webb et al. in this issue). The HIF complex is a heterodimer composed of a constitutively expressed HIF-1β subunit (also known as the aryl hydrocarbon receptor nuclear translocation, ARNT) and a HIF-1α oxygen-regulated subunit [33–37]. The regulation of HIF-1α activity occurs at different levels, with such processes as protein stability, phosphorylation and nuclear translocation and activity, among others, all being influenced by alterations in oxygen levels. HIF-1α is degraded under normoxic conditions. In contrast, under hypoxic conditions, HIF-1α is stabilized and translocated to the nucleus where it dimerizes with ARNT and subsequently binds to hypoxic binding sites (HBS) of target genes. The HBS is a conserved consensus sequence (A/G)CGTG within the hypoxic response element (HRE) present in oxygen-regulated target genes involved in cell survival, glycolysis, angiogenesis, erythropoiesis, and iron metabolism [38]. The degradation of HIF-1α is triggered by the oxygen-dependent hydroxylation of proline residues located in the oxygen-dependent degradation domain mediated by a family of prolyl hydroxylases, namely PHD1, PHD2, and PHD3 [39–41]. These enzymes are specific HIF-1α proline hydroxylases that require Fe(II) as a co-factor as well as oxygen and 2-oxoglutarate as co-substrates [42, 43]. Prolyl hydroxylation promotes the recruitment of the tumor suppressor protein von Hippel–Lindau, which is part of the E3 ligase ubiquitination complex and primes HIF-1α for degradation in the proteosomes.
Since hypoxia may mediate AD progression, is there evidence to suggest that HIF-1 has a role in AD pathology? It was apparent over a decade ago that the iron chelator desferoxamine, which also induces and stabilizes HIF-1 expression, protects cultured cells from Aβ [44] and inhibits AD progression in patients [45]. A second metal chelator, clioquinol, was subsequently shown to reverse AD-like pathologies in transgenic mice [45] and induce HIF-1 expression [35]. These studies provided the first clues to the possible involvement of HIF-1 in neurodegenerative disorders, which is now becoming more widely accepted. However, the precise role of the pathway, albeit positive or negative, remains a matter of debate.
Notably, HIF-1-mediated signaling has been implicated in both cell survival and cell death pathways [46], and the modulation of the HIF pathway seems to be very complex. During aging, the HIF induction and signaling pathways are down-regulated [47]. This impairment of important HIF-1 signaling pathways in the aged brain may render the individual more susceptible to neurodegeneration. Soucek et al. [35] showed that Aβ directly induces HIF-1α expression and activity in vitro. Interestingly, low levels of Aβ protect neurons from a more aggressive insult via the induction of HIF-1α pathways and, crucially, over-expression of HIF-1α has been found to be sufficient to protect neurons from Aβ neurotoxicity. A very recent study also demonstrated that HIF-1 levels are further reduced in AD brains in comparison to age-matched controls [48], again pointing to the possibly detrimental downregulation of important HIF target genes. In line with this, the observed decreased expression of two major brain glucose transporters, GLUT1 and GLUT3, in the AD brain was most probably the result of reduced levels of HIF-1, their main regulator. The decrease in GLUT1 and GLUT3 levels is also negatively correlated with tau hyperphosphorylation and with an increased density of NFTs, providing evidence for the involvement of glucose transporters in abnormal tau hyperphosphorylation and the formation of NFTs, both events being tightly linked to neurodegeneration [48]. Providing further support for the idea of a detrimental down-regulation of HIF-1 target genes during disease, Chong et al. [49] demonstrated a neuroprotective role of erythropoietin (EPO), a well-described HIF-1 target, against Aβ toxicity in hippocampal neurons in vitro. In this study, EPO was both necessary and sufficient to prevent Αβ-induced apoptotic death in both the early and later stages of neurodegeneration via the expression and translocation of NF-kB p65. Similarly, levels of EPO in the CSF of AD patients have been found to be reduced, or in the same range, as those in control brains [50], a surprising finding in light of upregulation of EPO in adults with traumatic brain injury or depression. This once more indicates that a deficiency of protective endogenous EPO in dementia, again presumably due to decreased HIF expression, may contribute to disease progression.
The lack of, or insufficient neuroprotective effects of vascular endothelial growth factor (VEGF), another infamous HIF target gene, has also been proposed to accelerate neurodegenerative disorders [51]. The treatment of motor neuron degeneration by intracerebroventricular delivery of VEGF in a rat model of amyotrophic lateral sclerosis had therapeutic effects [52], hinting perhaps that VEGF may have an analogous role in AD. Polymorphisms in the promoter region of VEGF were also suggested to be associated with an increased risk of developing AD [53], although this finding was not confirmed in subsequent studies [54, 55]. Notably, the precise contribution of this important growth factor to AD progression remains as yet elusive.
Overall, it is perhaps not so surprising that, as a result of the above studies, some scientists have proposed hypoxic preconditioning as an alternative therapy to prevent the deterioration of neuronal cells in AD [56]. However, since contradictory results have also been published (reviewed below) and it has also been shown that the manipulation of hypoxic pathways can have many different outcomes, much more evidence is needed to provide sufficient backing to such potential therapies.
In complete contrast to the aforementioned data, conflicting evidence suggests that the induction of HIF pathways as a consequence of AD could be detrimental to neuronal survival by inducing apoptotic genes and pro-inflammatory proteins. Recent evidence has shown a definite link between ischemia/hypoxia, HIF-1 and APP processing/Aβ production. Notably, Zhang et al. [57] showed that during acute hypoxia, HIF-1 binding to the BACE1 promoter induced BACE1 expression and resulted in increased Aβ production in neuroblastoma cells. Moreover, HIF-1α deficiency reduced BACE1 expression in the cortex and hippocampus of transgenic mice, pointing to HIF-1 as the primary mediator of the hypoxic induction of BACE1. Wang et al. [58] showed that APH-1A expression and subsequent γ-secretase-mediated Aβ and Notch generation are also regulated by HIF-1 binding to the APH-1A promoter during hypoxia. Since inhibiting or reducing the levels of secretase activity decreases the production of Aβ in the absence of severe detrimental phenotypes [59, 60], it could be speculated that HIF-1 inhibition may provide an alternative approach for therapeutic targeting.
This is further substantiated by the fact that brief periods of hypoxia, which can potentiate the Aβ-induced neural expression of the pro-inflammatory markers COX-2 and PS1 and accelerate the inflammatory neuropathology that characterizes AD brains, most probably also occurs throughout HIF signaling pathways [32]. In addition, potential HIF binding sites identified on presenilin promoter regions further imply that HIF-1 may be involved in stress-mediated PS1 transcriptional regulation [61]. Thus, episodic hypoxia during aging may drive the expression of COX-2, PS1 and related genes by way of HIF-1 signaling that ultimately contributes, cumulatively, to inflammatory responses and amyloidogenic neuropathology.
Another AD event linked to HIF-1 is the oxidative stress-induced accumulation of Aβ and subsequent activation of the pro-death gene BNIP3 in primary cortical neurons [62]. The knockdown of HIF-1α by RNA interference not only inhibits BNIP3 induction but also reduces Aβ-induced neuronal death. Thus, HIF-1 seems to mediate a potentially pathological role of BNIP3 in AD etiology. That hypoxia/HIF-1 signaling is involved in amyloidogenic processing of APP and subsequent downstream events also provides a good rationale for the increased incidence of AD and neurodegeneration after cerebral ischemia and stroke. However, despite this body of data suggesting that HIF-1 inhibition may also be a good therapeutic strategy, more evidence is needed to understand whether the role of HIF-1 in neurodegeneration, especially in vivo, is beneficial or detrimental.
Thus, the overall contribution of HIF signaling pathways per se to the progression of neurodegenerative disease is still very hazy. On the one hand, it would appear that aged neurons with attenuated HIF signaling neuroprotective pathways are more susceptible to neurodegeneration. On the other hand, sustained activation of HIF also seems to be harmful and can accentuate undesirable disease characteristics. Based on current research, it seems likely that the role of HIF in neurodegeneration may greatly depend on whether it is the cause or the consequence of disease progression.
Oxygen deprivation-mediated cytoskeletal alterations and AD
Uncertainty still surrounds the sequence of events that lead to AD pathogenesis. Nevertheless, cytoskeletal abnormalities in postmortem AD brains, ranging from the formation of NFTs to axonal and dendritic atrophy, and the deregulation of cytoskeletal–regulatory proteins suggest that the cytoskeleton is an important player in the disease. Alterations in the actin cytoskeleton have been linked with both hypoxic injury and neurodegeneration [63]. In neurons, the dynamic regulation of actin microfilament polymerization is implicated in maintaining dendritic structure and, notably, the number of dendritic spines is significantly decreased in Alzheimer’s neurons, indicating that actin plays an active role in disease progression [64]. Similarly, oxygen deprivation in the brain also causes neurons to undergo morphological changes, including process retraction and a loss of shape, which are partially attributable to the depolymerization of actin filaments [65]. Hypoxia causes decreased neuritic sprouting, impaired mitochondrial function, reduced expression of the proteins required to maintain synaptic connections and, ultimately, neuronal death [66]. Thus, similar cytoskeletal abnormalities typify both oxygen-deprived and AD neurons.
Cofilins are actin-binding proteins regulating the assembly and disassembly of actin filaments [67]. Following a stressful stimulus, cofilins undergo changes in phosphorylation, leading to the formation of rod-shaped actin bundles, commonly known as rods, which contribute to AD pathogenesis by inhibiting vesicle transport and disrupting synaptic function [15]. Interestingly, rods are also rapidly formed in response to anoxia [68], indicating that alterations in oxygen levels may be one underlying event contributing to the synaptic deficits responsible for memory impairment in AD patients. Independent studies have further documented a direct effect of oxygen deprivation on neurofilament breakdown and tau hyperphophosphorylation both in vivo and in vitro [69, 70]. Notably, cytoskeletal alterations are also documented in a number of other (neuro)pathogenic diseases and disorders characterized by oxygen deprivation [71].
Cdk5, hypoxic signaling and HIF-1
Neurofilaments (NFs) are cytoskeletal intermediate filaments exclusively expressed in neurons that provide rigidity and cell shape and facilitate intracellular transport and guidance of axons and dendrites. An abnormal accumulation of NFs is frequently detected in neurodegenerative disorders, and this accumulation is considered to be a major hallmark of neuronal dysfunction. NFTs are composed of abnormally modified and hyperphosphorylated tau, NFs and other cytoskeletal proteins. The cause of this aberrant transformation is not fully understood, but it has been attributed to perturbations in the normal balance of kinases and phosphatases. In particular, the kinases GSK3 and Cdk5, both of which have been co-purified with microtubules, play significant roles in cytoskeletal protein hyperphosphorylation and NFT generation [72, 73].
Cyclin-dependent kinase 5, a proline-directed serine/threonine kinase, is an important regulator of the neuronal cytoskeleton and yet another key player in neurodegeneration and AD pathogenesis [5]. Under normal conditions, membrane-bound Cdk5 is critical for the maintenance of neuronal survival. It exerts its function through the phosphorylation of a number of target proteins, including cytoskeletal proteins, signal transduction kinases and transcription factors [74]. Cdk5 is associated with two non-cyclin activators, p35 and p39, both of which restrict Cdk5 activity to neurons, keep the protein attached to the membrane and activate the protein via phosphorylation [75]. However under pathological conditions, Cdk5 activity is deregulated. First, an increase in Ca2+ influx stimulates the activation of calpains, a family of proteases with a key role in maintaining the cytoskeletal architecture [76]. Calpain then cleaves p35 and p39, the main regulators of Cdk5, resulting in the generation of truncated products, p25 and p29, respectively. This cleavage leads to loss of the protein complex membrane attachment, mislocalization of Cdk5 and deregulation of its activity [77]. The hyperactivation of Cdk5 is tightly linked to the Alzheimer’s pathogenesis observed in postmortem brains and in neurons exhibiting NFT. Cdk5 has also been shown to physically interact and phosphorylate tau in vivo [78, 79].
However, it appears that Cdk5 regulation is also complex since some recent data have suggested that transient upregulation of Cdk5 activity in the adult brain as a result of DNA damage is beneficial and protects neurons from death. Studies have shown that Cdk5 phosphorylates and inhibits c-Jun N-terminal kinase, thereby countering the induction of apoptotic signals [80]. Furthermore, O’Hare et al. [81] showed that cytoplasmic Cdk5 is neuroprotective in models of excitotoxicity and DNA damage in vitro, whereas nuclear Cdk5 is harmful and contributes to neuronal death. Thus, localization may also play a key role in determining the pro-survival or pro-death functions of this kinase. The molecular mechanisms that regulate the beneficial versus detrimental effects of Cdk5 are still to be determined.
It is now apparent that Cdk5/p35 may be involved in the regulation of oxidative stress-mediated pathways [82]. The first evidence of a role for the Cdk5/p35 complex in neuronal responses to oxygen deprivation came from research on Drosophila almost a decade ago [83]. Since then, a number of studies have reported upregulation of the complex in models of ischemia both in vivo and in vitro [84–87], additionally implicating modulation of this pathway during ischemic injury. Interestingly, similar to HIF-1 regulation, oxidative stress induces the activity of Cdk5, and overall expression is downregulated with age in the human brain [88]. Tamada et al. [89] demonstrated that p35 is cleaved during hypoxia as a result of calpain activation in mouse retina neurons. Vartianen et al. [90] also showed that Cdk5 activity and p35 expression play an important role in aspirin-induced tolerance in rat neurons against hypoxia/reoxygenation damage.
Very recent results from our group show that oxygen deprivation alone induces cytoskeletal rearrangement and distinctly modulates both components of the complex (Antoniou et al., unpublished data); these results are in agreement with those of another study showing increased activation of Cdk5 in neuroblastoma cells subjected to chemical oxidative stress [87]. Thus, hypoxia and oxidative stress both significantly alter Cdk5 activity, and this could have important consequences for neuronal function and survival since the localization of Cdk5 has been suggested to determine neuronal fate [81]. In agreement with this suggestion, hypoxia has been observed to cause the redistribution of Cdk5 to mainly the neuronal soma, signifying an altered function for the kinase and possible interaction with aberrant proteins. The nuclear translocation of Cdk5 can modulate transcription factors that are involved in cell death pathways [81] and, importantly, we have observed that Cdk5 function can regulate the expression of HIF-1 in primary cortical neurons during hypoxic exposure. Notably, pharmacological inhibition of Cdk5 by roscovitine attenuated HIF-1α induction in a time-dependent manner and additionally induced dramatic downregulation of EPO mRNA hypoxic induction in primary cortical neurons (Antoniou et al., unpublished data). In contrast, the surprising inability of roscovitine treatment to alter VEGF mRNA induction suggests that Cdk5 differentially modulates hypoxic-induced signaling pathways. This observation could be of importance in terms of improving our understanding of the mechanisms of neuronal survival and the progression of neurodegenerative disorders in general as well as of treatment therapies as a whole. Roscovitine treatment has been found to exacerbate cell death during chronic hypoxia, suggesting the requirement of Cdk5 function in the modulation of neuronal survival pathways during long-term injury/insult, presumably via the modulation of HIF-1 signaling. As such, these pathways may be intertwined with pathogenetic disease progression that occurs over extended time periods. Unfortunately, the mechanism of Cdk5-regulated HIF induction is still unclear, and how specificity for target gene induction is determined remains perplexing. These and other important questions are now being fully addressed in our ongoing studies. Nevertheless, our current evidence supports the notion that Cdk5, a regulator of AD pathogenesis, does play a beneficial role during hypoxic events and interacts with HIF-1 to modulate neuronal fate during neurodegenerative-related processes. Taken together, these findings underline the importance of obtaining a better understanding of the overlap and contribution of HIF-1 pathways to AD.
Summary
It is blatantly clear that the increasing incidence of neurodegenerative diseases is a growing problem in all the world's societies. Although the precise mechanisms of age-dependent neuronal susceptibility are unknown, a decreased expression of cellular defense genes in the aged brain make the latter less able to withstand injury. The current challenge is to find ways to increase the adaptive reserve of the organism to combat such age-related deficits and support or promote normal neuronal function. Oxygen deprivation seems to characterize most pathological processes and diseases, and in this regard neurodegeneration is no exception. How oxygen deprivation affects the progression of such disorders remains unclear, but it seems likely that it can only serve to exhaust the adaptive reserves of the brain. Thus, it is more and more crucial to explore the contribution of hypoxic-mediated pathways to age-related pathogenesis in order to gain more knowledge on how to intervene during disease progression. In the future, it may be that the manipulation of hypoxia and/or HIF-modulated signaling will provide new hope for the development of novel therapies urgently needed to combat neurodegenerative disease.
References
Siesjo BK (1988) Mechanisms of ischemic brain damage. Crit Care Med 16:954–963
Yager JY, Wright S, Armstrong EA, Jahraus CM, Saucier DM (2006) The influence of aging on recovery following ischemic brain damage. Behav Brain Res 173:171–180
Kolb B (2003) Overview of cortical plasticity and recovery from brain injury. Phys Med Rehabil Clin N Am 14:S7–S25
Marini AM, Choi J, Labutta R (2001) Synaptic deprivation and age-related vulnerability to hypoxic–ischemic neuronal injury. A hypothesis. Ann N Y Acad Sci 939:238–253
Jellinger KA, Attems J (2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J Neurol Sci 229–230:37–41
Joseph J, Shukitt-Hale B, Denisova NA, Martin A, Perry G, Smith MA (2001) Copernicus revisited: amyloid beta in Alzheimer’s disease. Neurobiol Aging 22:131–146
Tamaoka A, Sawamura N, Fukushima T, Shoji S, Matsubara E, Shoji M, Hirai S, Furiya Y, Endoh R, Mori H (1997) Amyloid beta protein 42(43) in cerebrospinal fluid of patients with Alzheimer’s disease. J Neurol Sci 148:41–45
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB et al (1992) Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359:322–325
Steinhilb ML, Dias-Santagata D, Mulkearns EE, Shulman JM, Biernat J, Mandelkow EM, Feany MB (2007) S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res 85:1271–1278
Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E (1993) Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett 336:417–424
Galloway PG, Perry G, Gambetti P (1987) Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol 46:185–199
Hirano A (1994) Hirano bodies and related neuronal inclusions. Neuropathol Appl Neurobiol 20:3–11
Anderton BH (2002) Ageing of the brain. Mech Ageing Dev 123:811–817
Mitake S, Ojika K, Hirano A (1997) Hirano bodies and Alzheimer’s disease. Kaohsiung J Med Sci 13:10–18
Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR (2000) Neurodegenerative stimuli induce persistent ADF/cofilin–actin rods that disrupt distal neurite function. Nat Cell Biol 2:628–636
Coppede F, Mancuso M, Siciliano G, Migliore L, Murri L (2006) Genes and the environment in neurodegeneration. Biosci Rep 26:341–367
Aliev G, Seyidova D, Neal ML, Shi J, Lamb BT, Siedlak SL, Vinters HV, Head E, Perry G, Lamanna JC, Friedland RP, Cotman CW (2002) Atherosclerotic lesions and mitochondria DNA deletions in brain microvessels as a central target for the development of human AD and AD-like pathology in aged transgenic mice. Ann N Y Acad Sci 977:45–64
Baldi I, Lebailly P, Mohammed-Brahim B, Letenneur L, Dartigues JF, Brochard P (2003) Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol 157:409–414
Mattson MP (1997) Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 77:1081–1132
Desmond DW, Moroney JT, Sano M, Stern Y (2002) Incidence of dementia after ischemic stroke: results of a longitudinal study. Stroke 33:2254–2260
Kalaria RN (2000) The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging 21:321–330
Egashira N, Iwasaki K, Ishibashi M, Hatip-Al-Khatib I, Wolozin B, Mishima K, Irie K, Fujiwara M (2002) Hypoxia enhances beta-amyloid-induced apoptosis in rat cultured hippocampal neurons. Jpn J Pharmacol 90:321–327
Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W (2006) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci USA 103:18727–18732
Chen GJ, Xu J, Lahousse SA, Caggiano NL, de la Monte SM (2003) Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: potential strategies for neuroprotection. J Alzheimers Dis 5:209–228
Jendroska K, Hoffmann OM, Patt S (1997) Amyloid beta peptide and precursor protein (APP) in mild and severe brain ischemia. Ann N Y Acad Sci 826:401–405
Shi J, Yang SH, Stubley L, Day AL, Simpkins JW (2000) Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res 853:1–4
Hall ED, Oostveen JA, Dunn E, Carter DB (1995) Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp Neurol 135:17–27
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3:862–872
Pearson HA, Peers C (2006) Physiological roles for amyloid beta peptides. J Physiol 575:5–10
Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C (2006) Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging 27:439–445
Scragg JL, Fearon IM, Boyle JP, Ball SG, Varadi G, Peers C (2005) Alzheimer’s amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J 19:150–152
Bazan NG, Lukiw WJ (2002) Cyclooxygenase-2 and presenilin-1 gene expression induced by interleukin-1beta and amyloid beta 42 peptide is potentiated by hypoxia in primary human neural cells. J Biol Chem 277:30359–30367
Wenger RH, Gassmann M (1997) Oxygen(es) and the hypoxia-inducible factor-1. Biol Chem 378:609–616
Wang GL, Semenza GL (1993) General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA 90:4304–4308
Soucek T, Cumming R, Dargusch R, Maher P, Schubert D (2003) The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron 39:43–56
Semenza GL (1998) Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev 8:588–594
Ratcliffe PJ, O’Rourke JF, Maxwell PH, Pugh CW (1998) Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol 201:1153–1162
Hofer T, Wenger H, Gassmann M (2002) Oxygen sensing, HIF-1alpha stabilization and potential therapeutic strategies. Pflugers Arch 443:503–507
Acker T, Acker H (2004) Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol 207:3171–3188
Maxwell PH, Ratcliffe PJ (2002) Oxygen sensors and angiogenesis. Semin Cell Dev Biol 13:29–37
Semenza GL (2001) HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107:1–3
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472
Schubert D, Chevion M (1995) The role of iron in beta amyloid toxicity. Biochem Biophys Res Commun 216:702–707
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337:1304–1308
Vangeison G, Carr D, Federoff HJ, Rempe DA (2008) The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci 28:1988–1993
Chavez JC, LaManna JC (2003) Hypoxia-inducible factor-1alpha accumulation in the rat brain in response to hypoxia and ischemia is attenuated during aging. Adv Exp Med Biol 510:337–341
Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX (2008) Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett 582:359–364
Chong ZZ, Li F, Maiese K (2005) Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr Neurovasc Res 2:387–399
Brettschneider J, Widl K, Ehrenreich H, Riepe M, Tumani H (2006) Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci Lett 404:347–351
Storkebaum E, Lambrechts D, Carmeliet P (2004) VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays 26:943–954
Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P (2005) Treatment of motor neuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8:85–92
Del Bo R, Scarlato M, Ghezzi S, Martinelli Boneschi F, Fenoglio C, Galbiati S, Virgilio R, Galimberti D, Galimberti G, Crimi M, Ferrarese C, Scarpini E, Bresolin N, Comi GP (2005) Vascular endothelial growth factor gene variability is associated with increased risk for AD. Ann Neurol 57:373–380
Mateo I, Llorca J, Infante J, Rodriguez-Rodriguez E, Sanchez-Quintana C, Sanchez-Juan P, Berciano J, Combarros O (2006) Case–control study of vascular endothelial growth factor (VEGF) genetic variability in Alzheimer’s disease. Neurosci Lett 401:171–173
Chapuis J, Tian J, Shi J, Bensemain F, Cottel D, Lendon C, Amouyel P, Mann D, Lambert JC (2006) Association study of the vascular endothelial growth factor gene with the risk of developing Alzheimer’s disease. Neurobiol Aging 27:1212–1215
Malyshev IY, Wiegant FA, Mashina SY, Torshin VI, Goryacheva AV, Khomenko IP, Kruglov SV, Pokidyshev DA, Popkova EV, Pshennikova MG, Vlasova MA, Zelenina OM, Manukhina EB (2005) Possible use of adaptation to hypoxia in Alzheimer’s disease: a hypothesis. Med Sci Monit 11:HY31–HY38
Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW (2007) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282:10873–10880
Wang R, Zhang YW, Zhang X, Liu R, Zhang X, Hong S, Xia K, Xia J, Zhang Z, Xu H (2006) Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J 20:1275–1277
Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4:233–234
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41:27–33
Cui JG, Fraser PE, St George-Hyslop P, Westaway D, Lukiw WJ (2004) Potential roles for presenilin-1 in oxygen sensing and in glial-specific gene expression. Neuroreport 15:2025–2028
Zhang S, Zhang Z, Sandhu G, Ma X, Yang X, Geiger JD, Kong J (2007) Evidence of oxidative stress-induced BNIP3 expression in amyloid beta neurotoxicity. Brain Res 1138:221–230
Mendoza-Naranjo A, Gonzalez-Billault C, Maccioni RB (2007) Abeta1–42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J Cell Sci 120:279–288
Lippa CF, Hamos JE, Pulaski-Salo D, DeGennaro LJ, Drachman DA (1992) Alzheimer’s disease and aging: effects on perforant pathway perikarya and synapses. Neurobiol Aging 13:405–411
Friedman JE, Chow EJ, Haddad GG (1998) State of actin filaments is changed by anoxia in cultured rat neocortical neurons. Neuroscience 82:421–427
de la Monte SM, Neely TR, Cannon J, Wands JR (2000) Oxidative stress and hypoxia-like injury cause Alzheimer-type molecular abnormalities in central nervous system neurons. Cell Mol Life Sci 57:1471–1481
McGough A, Pope B, Chiu W, Weeds A (1997) Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J Cell Biol 138:771–781
Maloney MT, Bamburg JR (2007) Cofilin-mediated neurodegeneration in Alzheimer’s disease and other amyloidopathies. Mol Neurobiol 35:21–44
Liu R, Pei JJ, Wang XC, Zhou XW, Tian Q, Winblad B, Wang JZ (2005) Acute anoxia induces tau dephosphorylation in rat brain slices and its possible underlying mechanisms. J Neurochem 94:1225–1234
Stys PK, Jiang Q (2002) Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett 328:150–154
Magin TM, Reichelt J, Hatzfeld M (2004) Emerging functions: diseases and animal models reshape our view of the cytoskeleton. Exp Cell Res 301:91–102
Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K (1993) Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett 325:167–172
Hosoi T, Uchiyama M, Okumura E, Saito T, Ishiguro K, Uchida T, Okuyama A, Kishimoto T, Hisanaga S (1995) Evidence for cdk5 as a major activity phosphorylating tau protein in porcine brain extract. J Biochem 117:741–749
Cheung ZH, Ip NY (2004) Cdk5: mediator of neuronal death and survival. Neurosci Lett 361:47–51
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2:749–759
Liu X, Van Vleet T, Schnellmann RG (2004) The role of calpain in oncotic cell death. Annu Rev Pharmacol Toxicol 44:349–370
Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 97:2910–2915
Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF (1998) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res 797:267–277
Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F (2001) Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis 8:32–44
Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC (2002) Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J 21:324–333
O’Hare MJ, Kushwaha N, Zhang Y, Aleyasin H, Callaghan SM, Slack RS, Albert PR, Vincent I, Park DS (2005) Differential roles of nuclear and cytoplasmic cyclin-dependent kinase 5 in apoptotic and excitotoxic neuronal death. J Neurosci 25:8954–8966
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405:360–364
Ma E, Haddad G (1999) A Drosophila CDK5alpha-like molecule and its possible role in response to O(2) deprivation. Biochem Biophys Res Commun 261:459–463
Hayashi T, Warita H, Abe K, Itoyama Y (1999) Expression of cyclin-dependent kinase 5 and its activator p35 in rat brain after middle cerebral artery occlusion. Neurosci Lett 265:37–40
Mitsios N, Pennucci R, Krupinski J, Sanfeliu C, Gaffney J, Kumar P, Kumar S, Juan-Babot O, Slevin M (2007) Expression of cyclin-dependent kinase 5 mRNA and protein in the human brain following acute ischemic stroke. Brain Pathol 17:11–23
Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, Bland RJ, Slack RS, During MJ, Park DS (2005) Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci USA 102:14080–14085
Strocchi P, Pession A, Dozza B (2003) Up-regulation of cDK5/p35 by oxidative stress in human neuroblastoma IMR-32 cells. J Cell Biochem 88:758–765
Wu DC, Yu YP, Lee NT, Yu AC, Wang JH, Han YF (2000) The expression of Cdk5, p35, p39, and Cdk5 kinase activity in developing, adult, and aged rat brains. Neurochem Res 25:923–929
Tamada Y, Nakajima E, Nakajima T, Shearer TR, Azuma M (2005) Proteolysis of neuronal cytoskeletal proteins by calpain contributes to rat retinal cell death induced by hypoxia. Brain Res 1050:148–155
Vartiainen N, Keksa-Goldsteine V, Goldsteins G, Koistinaho J (2002) Aspirin provides cyclin-dependent kinase 5-dependent protection against subsequent hypoxia/reoxygenation damage in culture. J Neurochem 82:329–335
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ogunshola, O.O., Antoniou, X. Contribution of hypoxia to Alzheimer’s disease: is HIF-1α a mediator of neurodegeneration?. Cell. Mol. Life Sci. 66, 3555–3563 (2009). https://doi.org/10.1007/s00018-009-0141-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0141-0