
Abstract
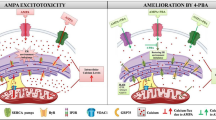
Excessive stimulation of ionotropic glutamate receptor is associated with glutamate-mediated excitotoxicity, thereby causing oxidative imbalance and mitochondrial dysfunction, resulting in the excitotoxic death of neurons. Eminent role of endoplasmic reticulum under glutamate-induced excitotoxicity has been highlighted in numerous literatures which have been observed to trigger endoplasmic reticulum stress (ER stress) as well as regulating oxidative stress. However, combating ER stress in excitotoxic neurons can provide a novel approach to alleviate the mitochondrial dysfunctioning and ROS generation. Therefore, we propose to investigate the cross-communication of α-amino-3-hydroxy-5-methyl-4-isoxzole-propionate (AMPA) excitotoxicity-induced oxidative injury with ER stress by employing ER stress inhibitor—4-phenlybutyric acid (4-PBA). Male SD rats were divided into four groups viz sham group (group 1), AMPA (10 mM)-induced excitotoxic group (group 2), curative group of AMPA-induced excitotoxic animals given 4-PBA at a dose of 100 mg/kg body weight (group 3), and alone 4-PBA treatment group (100 mg/kg body weight) (group 4). Animals were sacrificed after 15 days of treatment, and hippocampi were analyzed for histopathological examination, ROS, inflammatory markers, mitochondrial dysfunction, and ER stress markers. AMPA-induced excitotoxicity exhibited a significant increase in the levels of ROS, upregulated ER stress markers, inflammation markers, and compromised mitochondrial functioning in the hippocampus. However, 4-PBA administration significantly curtailed the AMPA-induced excitotoxic insult. This study suggests that targeting ER stress with a chemical chaperone can provide a better therapeutic intervention for neurological disorders involving excitotoxicity, and thus, it opens a new avenue to screen chemical chaperones for the therapeutic modalities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Prevalence of neurological diseases is growing at an alarming rate, and it is becoming one of the most imperative health problems faced by the modern society (Pereira et al. 2017). Glutamate is the most abundant excitatory amino acid neurotransmitter of the central nervous system (CNS), and over the past several years, studies have demonstrated that in cerebral cortex and hippocampus, glutamate serves a very crucial role in psychological functions such as learning and memory (Cao et al. 2014). Excessive release of glutamate causing an extreme excitability resulting in neuronal cell death is called excitotoxicity (Dong et al. 2009; Wu et al. 2017). Acute excitotoxicity has been suggested to be involved in conditions leading to stroke, ischemia, epilepsy, and traumatic brain injury; on the other hand, chronic excitotoxicity has been linked with neurodegenerative diseases like Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (Van Den Bosch et al. 2006).
Neurons exhibiting excitotoxicity have two types of receptors namely ionotropic receptors (N-methyl-d-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxzole-propionate (AMPA) receptors, and Kainate receptors) and metabotropic receptors (viz G-protein coupled glutamate receptors). Various studies have shown that exogenous administration of glutamatergic agonists such as AMPA, NMDA, and kainic acid to living animals or to cultured hippocampal primary neurons has been proven a useful model for studying the mechanism of glutamate-induced excitotoxic brain injury in hippocampal neurons as hippocampus and cerebral cortex predominantly expressed the subunits of the glutamate receptors (Liu et al. 2001; Hilton et al. 2006; Isaac et al. 2007). Uninterrupted/excessive activation of large number of AMPA/NMDA receptors by glutamate agonists leads to substantial accumulation of intracellular calcium and catabolic enzyme activities (proteases, phospholipase A, nitric oxide synthase, and endonucleases), which further elicit generation of the reactive oxygen species (ROS) and nitrogen oxide synthase (NOS) in the neurons (Rego and Oliveira 2003; Dong et al. 2009). Generation of oxidation stress (ROS) from various sources accelerates the damage of nucleic acids, oxidation of proteins, and peroxidation of lipids and potentially opens the mitochondrial permeability transition pores. Opening of these transition pores under this oxidative stress condition worsen the disruption of energy transport chain of the mitochondria which in turn further advances the ROS production (Nicholls 2004; Jung et al. 2009; Ndountse and Chan 2009).
Prominent mechanism involved in glutamate excitotoxicity is associated with excessive influx of ions into the cells resulting in calcium overload which further initiates a cascade of events leading to ROS generation and subsequently neuronal cell death (Dong et al. 2009; Wu et al. 2017). Endoplasmic reticulum (ER) is one such organelle which is responsible for maintaining the cytoplasmic calcium levels by acting as its sink. Also, reports suggest that glutamate-mediated excitotoxicity positively induces the disruption of endoplasmic reticulum environment leading to endoplasmic reticulum stress (ER stress) and subsequently activation of cascade of events known as unfolded protein response (UPR) (Sokka et al. 2007; Concannon et al. 2008).
Since glutamate-induced excitotoxicity is mediated by NMDA and/or AMPA receptors; therefore, targeting them would be the first choice to reduce excitotoxicity-induced neuronal cell death. But there are reports which state that the use of glutamate receptor inhibitors causes adverse effects on humans and rodents, which includes hallucinations, dizziness, paranoid delusions, confusion, difficulty in concentrating, agitation, alteration in the mood, ataxia, anesthesia, and learning and memory deficits (Kavirajan and Schneider 2007; Kovacic and Somanathan 2010).
So, if we could target intracellular ER stress in excitotoxic neurons, it is possible to reduce neuronal cell death without interfering in the normal functioning of excitatory neurotransmitters. The disruption in environment of ER is signified by loss of its protein folding capacity and is measured by a marker glucose regulated protein-78 (GRP78). Neurotoxicity induced by interleukin (IL)-1β via increased glutamate production in hippocampal neurons elicits ER stress response mediated by increased expression of molecular chaperone GRP78, which signifies accumulation of misfolded proteins (Dong et al. 2017). Therefore, using a chemical chaperone which could enhance the protein folding capacity of neurons could be a better treatment modality for excitotoxic insults.
So far, there are several chemical chaperones explored for their therapeutic potential in various diseases. 4-Phenylbutyric acid (4-PBA) is one such chaperone which has shown therapeutic potential which attenuates the amyloid plaques formation and rescues the cognitive behavior of AD transgenic mice (Wiley et al. 2011). In the current study, we focused our research on deciphering the efficacy of 4-PBA-a chemical chaperone to ameliorate AMPA-induced excitotoxic insult in hippocampal neurons and its possible association with mitochondrial dysfunction and ROS generation. Our study shall be an attempt to unveil the missing links via inhibition of ER stress which still remains unexplored regarding its subsequent involvement in curtailing excitotoxicity and its downstream effects on neuronal damage. This novel approach will open a new window to explore chemical chaperones for the therapeutic approaches of excitotoxicity involved in neuronal diseases.
Materials and Methods
Animals
Male adult Sprague-Dawley (SD) rats of body weight between 150 and 200 g were obtained from the inbred population of Central Animal House, Panjab University, Chandigarh. Animals were acclimatized to the control diet (rodent chow) and water ad libitum for at least 1 week before commencing any treatment. Animals were maintained as per the principles and guidelines of the Ethics Committee of the Animal Care of Panjab University in accordance with the Indian national law on animal care and use. The animals were housed five per cage in polypropylene cages with a wire mesh top and a hygienic bed of husk (regularly changed) in a well-ventilated animal room till the end of the experimental period. The animals were provided with a room cooler and a room heater during the summers and winter months, respectively, and also maintained under a 12-h photoperiod of light and darkness, alternatively. The animal experimental protocols were approved by Institutional Animal Ethics Committee (reference no. PU/IAEC/S/15/42) and conducted according to the Indian National Science Academy Guidelines for the use and care of experimental animals.
Experimental Design
Animals are randomly assorted into the four groups (six animals per group). Group 1 animal served as sham. They received single stereotactic microinjection of 0.5 μl of 50 mM phosphate buffer saline (PBS) pH 7.4. To induce excitotoxicity, group 2 animals were given single stereotactic microinjection of 0.5 μl of AMPA 10 mM, dissolved in 50 mM PBS pH 7.4 into hippocampus at the target site (Bregma AP 3.3 mm, ML 2.2 mm, DV − 2.9 mm) of the brain (Petegnief et al. 1999; Bernal et al. 2009). To study the therapeutic potential of chemical chaperone 4-PBA, another group 3 rats were subjected to intraperitoneal injections of 4-PBA (4-phenylbutyric acid) at dose of 100 mg/kg body weight for a period of 15 days followed by single stereotactic microinjection of AMPA into hippocampus at the target site (Bregma AP 3.3 mm, ML 2.2 mm, DV − 2.9 mm) as given in group 2 (Srinivasan and Sharma 2011). Group 4 rats were administered with 4-PBA (100 mg/kg body weight i.p) for a period of 15 days.
Tissue Collection
After the completion of all treatment, animals were sacrificed under overdose of anesthesia and brain was extracted. A coronal section of brain was fixed in buffered formaldehyde for histological analysis, a portion of isolated hippocampus for RNA extraction and other mitochondrial studies.
Histopathological Studies
Histological analysis of brain tissue was done with hematoxylin and eosin staining. Brain tissue was fixed in 10% formaldehyde and embedded in paraffin wax. Transverse sections of 5 μm thickness were cut and picked over albumin coated slides. Sections were then stained with hematoxylin and eosin and viewed under polarized light using Leica DM3000 light microscope. The method used for the neuronal count is a classical method used with thin brain sections to quantify undamaged hippocampal cells. The neuronal count in the CA1, CA2, CA3, and DG region was performed with the ImageJ 1.48 software. Neurons were picked up by the software pointer, and resulting number was used for further analysis (Sharma et al. 2016). Their survival index was calculated as follows: survival index (%) = (number of surviving neurons/total number of neurons) × 100% (Chen et al. 2018).
RNA Isolation and mRNA Expression
Total RNA from hippocampus of brain tissue was isolated using TRI-reagent (Sigma). Two micrograms of total RNA was retro-transcribed to synthesize cDNA by cDNA synthesis kit (BIORAD). Two microliters of cDNA was used for polymerase chain reaction (PCR) amplification using 5× FIRE Pol Master Green PCR reaction mix (Biodyn from Bio-Rad). The value obtained for each specific product was normalized to GAPDH. Band intensity was measured densitometrically using ImageJ 1.48 software. The primers used are given in Table 1.
Mitochondrial Isolation
Isolation of the mitochondria was done by standardized method described by Dhanda et al. (2018). The extracted rat brain was weighed and homogenized (10% w/v) in a homogenizing buffer A containing 10 mM Tris-HCl, 0.44 M Sucrose, 10 mM EDTA, and 0.1% BSA, pH 7.4. The homogenates were centrifuged at 2100×g for 15 min at 4 °C to remove the cell debris. The resultant supernatants were again centrifuged at 14,000×g for 15–20 min at 4 °C. The pellets obtained were re-suspended in 0.5 ml of homogenizing buffer A and again centrifuged at 7000×g for 15 min at 4 °C. Thus, the final pellets were obtained and re-dissolved in suspension buffer B containing 10 mM Tris-HCl and 0.44 M Sucrose, pH 7.4.
Mitochondrial ROS Estimation
Mitochondrial ROS (mtROS) were determined using dichlorodihydrofluorescein diacetate (DCFH-DA) which is oxidized into fluorescent DCF by mitochondrial ROS. Briefly, isolated mitochondria were incubated with DCFH-DA (1.25 mM stock solution in methanol) at 37 °C for 30 min in the dark. Fluorescence of samples was estimated using PerkinElmer Fluorescence spectrophotometer at an excitation and emission wavelength 488 and 525 nm, respectively. The results were expressed as relative fluorescence intensity (Best et al. 1999).
Mitochondrial Membrane Potential
For measuring mitochondrial membrane potential (MMP), isolated mitochondria were incubated with the 2 ml of reaction buffer containing 150 mM sucrose, 4 mM MgCl2, 5 mM K2HPO4, and 30 mM HEPES-KOH, pH 7.4 at 37 °C for 5 min. After that, the reaction was initiated by adding 5 μM Rhodamine 123 and fluorescence intensity was monitored at excitation and emission wavelength of 507 and 527 nm, respectively (Varbiro et al. 2001).
Mitochondrial Cardiolipin Content
Cardiolipin content (CL Content) of mitochondria was estimated by cardiolipin specific dye nonyl-acridine orange (Petit et al. 1992). One hundred microliters of mitochondrial sample was incubated with buffer containing 150 mM sucrose, 4 mM MgCl2, 5 mM K2HPO4, and 30 mM HEPES-KOH, pH 7.4 for 1 h at 37 °C. The samples were centrifuged at 15,000×g for 15 min. The pellets obtained were re-suspended in the same buffer and incubated at 37 °C for 45 min in the presence of 5 μM nonyl-acridine orange. The excess dye was washed out by same centrifugation. The final mitochondrial pellet was appropriately diluted in buffer containing 0.44 M sucrose and 10 mM Tris-HCl, pH 7.4, and fluorescence intensity was measured with excitation wavelength at 485 nm and emission wavelength at 535 nm.
Mitochondrial Respiratory Chain Complexes
NADH co-enzyme Q oxidoreductase (complex I) activity was measured spectrophotometrically as described by King and Howard (1967). The activity of succinate dehydrogenase (complex II) was assayed by following the method described by King et al. (1976). Mitochondrial cytochrome oxidase (complex IV) was assayed according to Sottocasa et al. (1967).
Statistical Analysis
Data was analyzed using one-way ANOVA and post-hoc analysis with least significant difference (LSD) using GraphPad Prism (version 6.0; San Diego, USA). The values are expressed as mean ± SD. Results were considered significant if p ˂ 0.05.
Results
In our current study, the implications of AMPA-induced excitotoxicity were investigated on the extent of endoplasmic reticulum stress and inflammation with generation of reactive oxygen species via mitochondria. By the end of the treatment, histopathological analysis was assessed in all the groups.
Coronal sections of the brain tissue were stained with hematoxylin and eosin to visualize the extent and remediation of neurodegeneration as a part our objective (Fig. 1a). Sham-treated rats exhibited a normal histoarchitecture pattern in terms of intact and no neuronal loss. CA1, CA2, CA3, and DG (dentate gyrus) regions of the AMPA-treated rat brain displayed a loss of structure, increased degeneration of the neurons, and pyknosis (Fig. 1a). However, AMPA-induced excitotoxic animals when subjected to 4-PBA treatment showed significant (p < 0.05) shielding of neurons against neurodegeneration with the signatures of less number of pyknotic nuclei in all the four regions of the hippocampus (Fig. 1b–e), whereas animals exposed to 4-PBA alone revealed a normal histoarchitecture similar to the sham group animals.

Effect of 4-PBA on the light micrographs of hematoxylin and eosin stained sections of hippocampus of rat brain. a Hippocampal section (× 50 and × 400) of sham-, AMPA-, AMP + PBA-, and PBA-treated animals. Red arrow heads represent the degenerating or pyknotic nuclei, and black arrow heads represent the normal neuronal structure. b–e Survival index (%) of the CA1, CA2, CA3, and DG region of the hippocampus sections. Values are expressed as mean ± S.D., n = 3 (*p < 0.05 represents significance difference from AMPA-treated group)
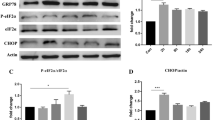
To evaluate the effects of AMPA on ER functionality, mRNA expression of GRP78 a hallmark of ER stress was evaluated and was found to be significantly (p < 0.05) upregulated in the AMPA-treated group when compared to the sham group (Fig. 2b). However, co-treatment of potent ER stress inhibitor 4-PBA with AMPA significantly (p < 0.05) downregulated the expression of GRP78 as compared to AMPA-treated group (Fig. 2b). Furthermore, mRNA expressions of unfolded protein response intermediates were assessed. mRNA expression of activation transcription factor-4 (ATF4) was found to be significantly (p < 0.05) elevated in AMPA-treated group as compared to sham group (Fig. 2d), whereas counteraction of 4-PBA under excitotoxic conditions was successful in diminishing the levels of ATF4 significantly (p < 0.05) when compared to the AMPA-treated group (Fig. 2d). Splicing of X-box binding protein-1 (XBP1) mRNA expression in AMPA-treated group further concreted the presence of ER stress in response to the AMPA-induced excitotoxicity (Fig. 2a). Apoptotic marker CHOP expression in response to ER insult was studied in all the groups. AMPA- and AMPA + 4-PBA-treated group demonstrated an upregulation of mRNA expression of C/EBP homologous protein (CHOP) when compared to sham group (Fig. 2c).

Effect of 4-PBA on mRNA expression of unfolded protein response (UPR) intermediates. a Representative gel of mRNA expression of GRP78, CHOP, ATF4, and XBP1 (spliced and unspliced). b Relative intensity of GRP78 in all the groups (sham-, AMPA-, AMPA + 4-PBA-, and 4-PBA-treated groups). c Relative intensity of CHOP in all the four groups. d Relative intensity of ATF4 in all the groups. Values are expressed as mean ± S.D. (*p < 0.05 represents significance difference from the AMPA-treated group)
Inflammation marker nuclear factor kappa light chain enhancer of activated B cells (NFkB) and monocyte chemoattractant protein-1 (MCP1) were also investigated. mRNA expression of NFkB and MCP1 was significantly (p < 0.05) enhanced in AMPA-treated group when compared to the sham group (Fig. 3a, b). However, co-supplementation of 4-PBA in AMPA-induced excitotoxic animals depicted a significant (p < 0.05) decline in the mRNA levels of NFkB and MCP1 when compared to the AMPA-treated group.

Effect of 4-PBA treatment on expression of inflammation markers Monocyte Chemoattractant Protein-1 (MCP1) and nuclear factor kappa light chain enhancer of activated B cells (NFkB). a Representative gel of MCP1 and Relative intensity of sham-, AMPA-, AMPA + 4-PBA-, and 4-PBA-treated groups. b Representative gel of NFkB and relative intensity of all the four groups. Values are expressed as mean ± S.D. (*p < 0.05 represents significance difference from AMPA-treated group)
In addition to the ER stress induction, mitochondrial ROS was also assessed and it was found to be upregulated in AMPA-treated group (Fig. 4a). However, upon treatment with 4-PBA, significantly (p < 0.05) diminished levels of mitochondrial ROS levels were found when compared to the AMPA-treated group (Fig. 4a). Mitochondrial membrane potential was also observed in all the groups. A significantly (p < 0.05) declined mitochondrial membrane potential was estimated in the AMPA-treated group which was mitigated by the 4-PBA treatment after AMPA excitotoxic insult (Fig. 4b). Similarly, another stress marker, cardiolipin content, was also observed to be significantly (p < 0.05) downregulated in AMPA-treated animals when compared to the sham group (Fig. 4c). However, a significant (p < 0.05) upregulated level of cardiolipin content was observed when subjected to the 4-PBA co-treatment as compared to the AMPA-treated group.

Effect of 4-PBA administration on mitochondrial reactive oxygen species (mtROS), mitochondrial membrane potential (MMP), and cardiolipin content (CL Content). a Significant reduction in the mitochondrial ROS was observed after 4-PBA administration to AMPA-treated group. b Protective effects of 4-PBA represent a significant increase in mitochondrial membrane potential. c Significant upregulation in the cardiolipin content was observed in 4-PBA-treated animals. Values are expressed as mean ± S.D. (*p < 0.05 represents significance difference from AMPA-treated group)
Moreover, the functioning stress of ER was compared with mitochondrial respiratory chain complexes (mitochondrial complexes) in all the four groups. Significant reduction (p < 0.05) in the levels of complexes I, II, and IV was observed in the AMPA-treated group. However, with 4-PBA supplementation, levels of complexes I, II, and IV were significantly (p < 0.5) alleviated when compared to the AMPA excitotoxic group (Fig. 5a–c).

Effect of 4-PBA supplementation on the mitochondrial activity of complexes I, II, and IV. a Significant alleviation in the levels of complex I activity was observed post-treatment of 4-PBA when compared to the AMPA-treated group. b Post-treatment of 4-PBA significantly upregulated the activity of complex II when compared to the AMPA-treated group. c Similarly, 4-PBA supplementation significantly restored the complex IV activity when compared to the AMPA-treated group. Values are expressed as mean ± S.D. (*p < 0.05 represents significance difference from AMPA-treated group)
Discussion
Neurodegenerative diseases have been suggested to be a consequence of certain neuronal dysfunction leading to cell death or change in histoarchitecture of the brain. Malfunctioning of particular group of neurons often exhibits some characteristic features comprising excitotoxicity, oxidative stress, and mitochondrial dysfunction. Literature has cemented the processes like protein misfolding and aggregation as prime suspects responsible for the occurrence and progression of neurodegenerative diseases. Most of the neurological disorders are associated with impairment of the proteasome system which further triggers protein misfolding (Hartl et al. 2011). ER is a major site for the folding and processing of proteins, and as stated before, glutamate-induced excitotoxicity distorts calcium homeostasis which jeopardizes the normal functioning of ER, resulting into accumulation of unfolded proteins (Paschen and Mengesdorf 2005). Therefore, alteration in ER environment is apparently an important event in excitotoxicity related neurological disorders. The present work aimed to study the efficacy of a chemical chaperone 4-PBA (which could restore ER environment) in reducing AMPA-induced excitotoxicity exhibition in neurons.
Due to the accumulation of unfolded proteins in the ER lumen, a distress signal is generated by the ER stress sensing proteins (PERK, ATF6 and IRE1) which evokes the unfolded protein response (UPR) in an attempt to restore the ER homeostasis. However, under prolonged/severe ER stress condition, all three pathways upregulates the transcription factor CHOP to induce cell apoptosis by altering the levels of pro-apoptotic Bcl2 family (Prentice et al. 2015). As depicted by the results of present study, significant upregulation of the ER stress markers (GRP78, ATF4, and XBP1) led to UPR, thereby compromising the normal physiological and pathological conditions in the AMPA-induced excitotoxic neurons. Chemical chaperone 4-PBA dosage appreciably counter-acted UPR and maintained ER environment physiology, evident by maintaining levels of GRP78, ATF4, and suppressing the splicing of XBP1 in hippocampus of rat brain. Any other implication of 4-PBA on AMPA-induced neurotoxicity will strengthen the concept that ER environment is the major contributor of excitotoxic insult.
The role of mitochondrial dysfunctioning has been well documented in glutamate-induced excitotoxicity, leading to the progression of neuronal cell death (Ruiz et al. 2010). Alteration in the cardiolipin content (phospholipid of inner mitochondrial membrane) and mitochondrial membrane potential influenced by mtROS levels causes havoc in otherwise disciplined functioning of cellular machinery. Consistent with the findings, our results depicted a decreased mitochondrial membrane potential and elevation in the levels of mtROS, suggesting the burden of AMPA-induced excitotoxicity. Mitochondria and endoplasmic reticulum forms junctions, through which endoplasmic reticulum mediates calcium signal propagation to the mitochondria, which is important for both adenosine tri-phosphate (ATP) production and mitochondrial cell death under stress (Kaufman and Malhotra 2014). During ER stress, leakage of calcium from the ER and its subsequent uptake by mitochondria perturbs the normal cellular functioning, thereby resulting in the generation of ROS (Zeeshan et al. 2016). Therefore, stress in ER leading to mtROS was curbed by administration of 4-PBA which further controlled the extent of oxidation of cardiolipin content as well as lowered the burden of mtROS.
Calcium signaling is an important aspect of interaction between ER and mitochondria for normal physiology and functioning of these two organelles. Under the stress conditions, excess calcium release from the ER is taken up by the mitochondria, which inhibits the complex I activity and releases the cytochrome c that further blocks the respiratory chain complex III and initiates ROS generation (Chuang et al. 2004; Racay et al. 2009). With high production of mtROS, activity of complex IV also gets considerably reduced which further initiates the feedback loop leading to the further alarming production of the ROS (Bhandary et al. 2013). In our study, 4-PBA treatment in AMPA-induced excitotoxic environment efficiently restored the activity of the mitochondrial complexes and reduced the ROS production in the mitochondria, thus indicating that ER stress communicating with mitochondria through calcium signaling is also maintained by 4-PBA. These findings strengthen the concept that ER as an organelle of paramount importance is involved in supervising mitochondrial functioning and keeps a vigil on the production of ROS through it.
Production of ROS from mitochondria further mediates cellular stress responses inducing apoptosis, necrosis, or inflammation (Emerit et al. 2004; Chen et al. 2011). Therefore, it is suggested here that cross-talk between ER stress and inflammation is mediated via reactive oxygen species (ROS). We also found that AMPA-induced excitotoxicity stimulated the inflammation in the neurons and subsequently evoked the pathways leading to neurodegeneration. Several experimental studies have demonstrated that inflammatory markers (NFkB and MCP-1) and oxidative stress are strictly interconnected in excitotoxic neurodegenerative and epileptic disorders (Vezzani and Granata 2005; Salminen et al. 2009). Interestingly, 4-PBA treatment successfully ameliorated the impact of inflammation, which is a clear indicator that ER stress is a prime source of neuronal stress, which, if curbed by any chemical chaperone, can reduce inflammation.
Since, 4-PBA reduced inflammation and ROS which are the major contributors of neuronal cell damage; therefore, its impact on neuronal histoarchitecture was also studied. Histopathological examination also revealed improvisation in the degenerating neurons with less distortion and shrinkage of cell body of neurons after administration of 4-PBA in AMPA-induced excitotoxic animals. This can be considered as the foremost evidence of 4-PBA effectively preventing the neurons from excitotoxic insult induced by the AMPA.
Interknitting the aforementioned observations, it would be worth theorizing that AMPA-induced excitotoxicity derails the normal functioning of endoplasmic reticulum aftermath, which can be observed in the form of impairment in mitochondrial workability and enhanced ROS production. Till date, emphasis has always been on rectifying the mitochondrial dysfunction and/or ROS production as a reparative alternative for excitotoxicity-induced neuronal insult. Contrary to the above mentioned approach, our study has plausibly hypothesized that alteration in the normal functioning of ER initiates the cascade of events leading to neuronal damage. Therefore, it is suggested that targeting ER stress with chemical chaperones can be proposed a better therapeutic alternative to neutralize the afflictions of excitotoxicity involved neurodegenerative disorders.
References
Bernal F, Petegnief V, Rodríguez MJ, Ursu G, Pugliese M, Mahy N (2009) Nimodipine inhibits TMB-8 potentiation of AMPA-induced hippocampal neurodegeneration. J Neurosci Res 87:1240–1249
Best TM, Fiebig R, Corr DT, Brickson S, Ji L (1999) Free radical activity, antioxidant enzyme, and glutathione changes with muscle stretch injury in rabbits. J Appl Physiol 87:74–82
Bhandary B, Marahatta A, Kim HR, Chae HJ (2013) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 14:434–456
Cao J, Wang Z, Mi W, Zuo Z (2014) Isoflurane unveils a critical role of glutamate transporter type 3 in regulating hippocampal GluR1 trafficking and context-related learning and memory in mice. Neuroscience 11:58–64
Chen C, Li B, Cheng G, Yang X, Zhao N, Shi R (2018) Amentoflavone ameliorates Aβ1-42-induced memory deficits and oxidative stress in cellular and rat model. Neurochem Res 43:857–868
Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC (2011) Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci 12:7199–7215
Chuang Y-C, Chang AYW, Lin J-W, Hsu SP, Chan SH (2004) Mitochondrial dysfunction and ultrastructural damage in the hippocampus during kainic acid-induced status epilepticus in the rat. Epilepsia 45:1202–1209
Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, Woods I, Engel T, Henshall DC, Prehn JH (2008) NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J Neurochem 105:891–903
Dhanda S, Sunkaria A, Halder A, Sandhir R (2018) Mitochondrial dysfunctions contribute to energy deficits in rodent model of hepatic encephalopathy. Metab Brain Dis 33:209–223
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin Acta Pharmacol Sin 30:379–387
Dong Y, Kalueff AV, Song C (2017) N-methyl-d-aspartate receptor-mediated calcium overload and endoplasmic reticulum stress are involved in interleukin-1beta-induced neuronal apoptosis in rat hippocampus. J Neuroimmunol 307:7–13
Emerit J, Edeas M, Bricaire F (2004) Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 58:39–46
Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475:324–332
Hilton GD, Nunez JL, Bambrick L, Thompson SM, McCarthy MM (2006) Glutamate-mediated excitotoxicity in neonatal hippocampal neurons is mediated by mGluR-induced release of Ca++ from intracellular stores and is prevented by estradiol. Eur J Neurosci 24:3008–3016
Isaac JTR, Ashby MC, McBain CJ (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54:859–871
Jung KH, Chu K, Lee ST, Park HK, Kim JH, Kang KM, Kim M, Lee SKRJ (2009) Augmentation of nitrite therapy in cerebral ischemia by NMDA receptor inhibition. Biochem Biophys Res Commun 378:507–512
Kaufman RJ, Malhotra JD (2014) Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim Biophys Acta 1843:2233–2239
Kavirajan H, Schneider LS (2007) Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: a meta-analysis of randomised controlled trials. Lancet Neurol 6:782–792
King TE, Howard RL (1967) [52] Preparations and properties of soluble NADH dehydrogenases from cardiac muscle. Methods Enzymol 10:275–294
King TE, Ohnishi T, Winter DB, Wu JT (1976) Biochemical and EPR probes for structure-function studies of iron sulfur centers of succinate dehydrogenase. Adv Exp Med Biol 74:182–227
Kovacic P, Somanathan R (2010) Clinical physiology and mechanism of dizocilpine (MK-801): electron transfer, radicals, redox metabolites and bioactivity. Oxidative Med Cell Longev 3:13–22
Liu W, Liu R, Chun JT, Hoe W, Schreiber SS, Baudry M (2001) Kainate excitotoxicity in organotypic hippocampal slice cultures: evidence for multiple apoptotic pathways. Brain Res 916:239–248
Ndountse LT, Chan HM (2009) Role of N-methyl-D-aspartate receptors in polychlorinated biphenyl mediated neurotoxicity. Toxicol Lett 184:50–55
Nicholls DG (2004) Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med 4:149–177
Paschen W, Mengesdorf T (2005) Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 38:409–415
Pereira EPL, Braga-De-Souza S, Santos CC, Amparo J, Short Ferreira R, Nuñez-Figueredo Y, Gonzaga Fernandez L, Ribeiro PR, Braga-de-Souza S, Amaral da Silva VD, Lima Costa S (2017) Amburana cearensis seed extract protects brain mitochondria from oxidative stress and cerebellar cells from excitotoxicity induced by glutamate. Rev Bras 27:199–205
Petegnief V, Saura J, Dewar D, Cummins DJ, Dragunow M, Mahy N (1999) Long-term effects of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate and 6-nitro-7-sulphamoylbenzo(f)quinoxaline-2,3-dione in the rat basal ganglia: calcification, changes in glutamate receptors and glial reactions. Neuroscience 94:105–115
Petit JM, Maftah A, Ratinaud MH, Julien R (1992) 10N-nonyl acridine orange interacts with cardiolipin and allows the quantification of this phospholipid in isolated mitochondria. Eur J Biochem 209:267–273
Prentice H, Modi JP, Wu JY (2015) Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev 2015:7 pages
Racay P, Tatarkova Z, Chomova M, Hatok J, Kaplan P, Dobrota D (2009) Mitochondrial calcium transport and mitochondrial dysfunction after global brain ischemia in rat hippocampus. Neurochem Res 34:1469–1478
Rego AC, Oliveira CR (2003) Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem Res 28:1563–1574
Ruiz A, Matute C, Alberdi E (2010) Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis 1:e54
Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J (2009) ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation 6:1–13
Sharma S, Verma S, Kapoor M, Saini A, Nehru B (2016) Alzheimer’s disease like pathology induced six weeks after aggregated amyloid-beta injection in rats: increased oxidative stress and impaired long-term memory with anxiety-like behavior. Neurol Res 38:838–850
Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L (2007) Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 27:901–908
Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A (1967) An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J Cell Biol 32:415–438
Srinivasan K, Sharma SS (2011) Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav Brain Res 225:110–116
Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta Mol basis Dis 1762:1068–1082
Varbiro G, Veres B, Sumegi B (2001) Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic Biol Med 31:548–558
Vezzani A, Granata T (2005) Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46:1724–1743
Wiley JC, Pettan-Brewer C, Ladiges WC (2011) Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 10:418–428
Wu J, Gao L, Shang L, Wang G, Wei N, Chu T, Chen S, Zhang Y, Huang J, Wang J, Lin R (2017) Ecdysterones from Rhaponticum carthamoides (Willd.) Iljin reduce hippocampal excitotoxic cell loss and upregulate mTOR signaling in rats. Fitoterapia 119:158–167
Zeeshan HMA, Lee GH, Kim HR, Chae HJ (2016) Endoplasmic reticulum stress and associated ROS. Int J Mol Sci 17:327
Funding Source
This work was supported by the University Grant Commission-Basic Scientific Research (UGC-BSR) (F.25-1/2013-14(BSR)/7-209/2009(BSR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Animals were maintained as per the principles and guidelines of the Ethics Committee of the Animal Care of Panjab University in accordance with the Indian national law on animal care and use. The animal experimental protocols were approved by Institutional Animal Ethics Committee (reference no. PU/IAEC/S/15/42) and conducted according to the Indian National Science Academy Guidelines for the use and care of experimental animals.
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
ESM 1
(PNG 1437 kb)
Rights and permissions
About this article

Cite this article
Bhardwaj, A., Bhardwaj, R., Dhawan, D.K. et al. Exploring the Effect of Endoplasmic Reticulum Stress Inhibition by 4-Phenylbutyric Acid on AMPA-Induced Hippocampal Excitotoxicity in Rat Brain. Neurotox Res 35, 83–91 (2019). https://doi.org/10.1007/s12640-018-9932-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-018-9932-0