
Abstract
Nucleus accumbens (NAc) neurons are excited primarily by AMPA-type glutamate receptors (AMPAR). This is required for cocaine seeking in animal models of cocaine addiction, suggesting AMPAR transmission in the NAc as a key control point for cocaine-related behaviors. This review will briefly describe AMPAR properties and trafficking, with a focus on studies in NAc neurons, and then consider mechanisms by which cocaine may alter AMPAR transmission. Two examples will be discussed that may be important in two different stages of addiction: learning about drugs and drug-related cues during the period of drug exposure, and persistent vulnerability to craving and relapse after abstinence is achieved. The first example is drawn from studies of cultured NAc neurons. Elevation of dopamine levels (as would occur following cocaine exposure) facilitates activity-dependent strengthening of excitatory synapses onto medium spiny neurons, the main cell type and projection neuron of the NAc. This occurs because activation of D1-class dopamine receptors primes AMPAR for synaptic insertion. This may create a temporal window in which stimuli related to cocaine-taking are more efficacious at eliciting synaptic plasticity and thus being encoded into memory. The second example involves rat models of cocaine addiction. Cell surface and synaptic expression of AMPAR on NAc neurons is persistently increased after withdrawal from repeated cocaine exposure. We hypothesize that this increases the reactivity of NAc neurons to glutamate inputs from cortex and limbic structures, facilitating the ability of these inputs to trigger cocaine seeking and thus contributing to the persistent vulnerability to relapse that characterizes addiction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to AMPA-Type Glutamate Receptors and Plasticity
Scope of the Review
It is now well accepted that drug addiction involves activity-dependent plasticity at glutamate synapses within neuronal circuits important for motivated behaviors (Wolf et al. 2004; Kauer and Malenka 2007). This conclusion is based on evidence that drugs of abuse can influence fundamental cellular processes that regulate synaptic strength. Much of this work has focused on the nucleus accumbens (NAc), which occupies a key position in the neural circuitry related to motivation and drug addiction (Kelley 2004; Kalivas and Volkow 2005). The majority of NAc neurons (~90%) are medium spiny GABA neurons (MSN; Meredith and Totterdell 1999). From both anatomical and functional perspectives, the NAc MSN serve as an interface between limbic and cortical brain regions important for regulating motivated behaviors (including drug seeking) and motor regions important for the execution of motivated behaviors (Mogenson 1987; Groenewegen et al. 1999; Kelley 1999). Indeed, considerable evidence indicates that activation of NAc MSN, by glutamate inputs originating from limbic and cortical regions, is the final common pathway for drug seeking in many animal models of addiction (Kalivas and Volkow 2005), although motor circuitry involving the dorsal striatum becomes more important as drug use becomes habitual (Everitt and Robbins 2005).
The activation of NAc MSN by glutamate inputs is mediated primarily via AMPAR (Pennartz et al. 1994; Hu and White 1996). Therefore, it is not surprising that drug seeking, elicited by cocaine or cues previously paired with cocaine, depends upon AMPAR transmission in the NAc (Cornish et al. 1999; Cornish and Kalivas 2000; Di Ciano and Everitt 2001, 2004; Suto et al. 2004; Kruzich and Xi 2006; Bäckstrom and Hyytiä 2007; Conrad et al. 2008; Famous et al. 2008; Ping et al. 2008). Furthermore, several groups have shown that enhanced AMPAR transmission in the NAc is associated with enhanced drug seeking (Suto et al. 2004; Anderson et al. 2008; Conrad et al. 2008). These results and others suggest that the strength of AMPAR transmission in the NAc represents a critical determinant of the intensity of drug seeking (for a comprehensive review, see Wolf and Ferrario 2010).
The present review focuses on specific examples of how cocaine, which acts initially by elevating extracellular dopamine (DA) levels, may enhance AMPAR transmission in the NAc. The examples are drawn from our own work (presented at the 2009 Congress of the Fundación Cerebro y Mente) and work from other laboratories. The section titled “Introduction to AMPA-Type Glutamate Receptors and Plasticity” will briefly review the role of AMPAR in synaptic plasticity. Most studies on this topic have been conducted in hippocampal neurons, but similar results have been obtained in NAc neurons. In “Facilitation of AMPAR Synaptic Insertion by DA May Contribute to the Strength of Drug-Related Learning” and “AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking”, the focus will be on cocaine- or DA-mediated mechanisms that may be important in two different stages of addiction: (1) learning about drugs and drug-related cues during the period of drug exposure and (2) persistent vulnerability to craving and relapse after abstinence is achieved. Specifically, the section titled “Facilitation of AMPAR Synaptic Insertion by DA May Contribute to the Strength of Drug-Related Learning” will discuss how cocaine administration, by elevating DA levels, may facilitate synaptic plasticity in NAc neurons. This may help explain why drug-related learning is exceptionally powerful and persistent. The section titled “AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking” will discuss studies of in vivo AMPAR plasticity after withdrawal from repeated cocaine exposure. Based on these studies, we hypothesize that upregulation of AMPAR at excitatory synapses onto NAc neurons enhances their response to glutamate inputs that trigger drug craving and relapse. This in turn may contribute to persistent vulnerability to relapse even after long periods of abstinence.
In light of the central role of AMPAR in many forms of experience-dependent plasticity (Kessels and Malinow 2009), cocaine-induced alterations in AMPAR function are expected to profoundly alter the responsiveness of NAc neurons and their subsequent plasticity. However, it should be emphasized that many other neuroadaptations are critically important in regulating the excitability and output of NAc MSN after cocaine withdrawal. We touch upon a few of these, but the reader is referred to recent reviews for more comprehensive coverage (Kauer and Malenka 2007; Thomas et al. 2008; Goto and Grace 2008; Kalivas 2009; Wheeler and Carelli 2009).
AMPAR Trafficking and Synaptic Plasticity
The importance of AMPAR for plasticity was first demonstrated approximately 10 years ago by studies showing that trafficking of AMPAR in and out of synapses determines the level of excitatory synaptic strength and contributes to LTP and LTD (Malinow and Malenka 2002). It is now appreciated that AMPAR trafficking involves receptor endocyotosis and exocytosis, occurring in specialized regions outside of the synapse, coupled with receptor lateral diffusion along the surface of the plasma membrane between synaptic and extrasynaptic compartments (Groc and Choquet 2006).
Functional AMPAR are tetramers (dimers of dimers) (Mano and Teichberg 1998; Rosenmund et al. 1998; Greger and Esteban 2007). They are composed of GluA1–4 subunits (formerly termed GluR1–4; see Collingridge et al. 2009). Early information about AMPAR subunit composition was provided by landmark co-immunoprecipitation (co-IP) studies using adult rat hippocampus (CA1/CA2) as starting material (Wenthold et al. 1996). These studies showed that about 80% of GluA1 was pulled down with GluA2/A3 antibody. Together with other results, this indicated that GluA1A2 receptors are an important population. About 30% of the GluA2 was not pulled down by GluA1 antibody. This could represent GluA2 present in GluA2A3 receptors (Wenthold et al. 1996). However, subsequent studies have shown that GluA2 in cultured neurons and rat brain is largely retained in the endoplasmic reticulum (ER) in dimer form, whereas GluA1 is mainly found in tetrameric receptors in post-ER compartments (Greger et al. 2003). These findings suggest that “left-over” GluA2 may represent partially assembled receptors rather than a significant GluA2A3 population. If this is the case, the co-IP results (Wenthold et al. 1996) indicate a predominant role for GluA1A2 receptors, which is consistent with more recent results obtained using a single-cell genetic approach combined with electrophysiological recordings (Lu et al. 2009). This latter study, conducted in hippocampal slices from young mice, found that GluA1A2 receptors comprise ~81% of AMPAR in synapses on CA1 pyramidal neurons and ~95% of receptors in extrasynaptic regions of their soma, whereas GluA2A3 receptors represented ~16% of the synaptic pool and were absent from the extrasynaptic somatic pool. As described in the section titled “AMPAR in the NAc”, we have recently obtained biochemical results in the NAc (and other regions) that are very similar to those reported by Wenthold et al. (1996) and Greger et al. (2003).
Subunit composition plays a critical role in determining biophysical properties of the AMPAR (Dingledine et al. 1999; Palmer et al. 2005). For example, absence of the GluA2 subunit results in Ca2+ permeability of the AMPAR channel (see below). Subunit composition is also important in determining “rules” that govern AMPAR trafficking. According to the leading theory, AMPAR comprised of subunits with short C-termini (GluA2 or GluA3) cycle constitutively in and out of synapses, whereas AMPAR containing a subunit with a long C-terminus (GluA1 or GluA4; GluA4 is important mainly during early development) are inserted into synapses in an activity-dependent manner, e.g., during LTP (Malinow 2003). This may reflect differences in regulatory phosphorylation events and protein–protein interactions between subunits with long and short C-terminal tails (Song and Huganir 2002; Derkach et al. 2007). AMPAR gating and trafficking are also regulated by AMPAR auxiliary subunits (transmembrane AMPA receptor regulatory proteins, or TARPs, and cornichon proteins) (Milstein and Nicoll 2008; Schwenk et al. 2009; Tigaret and Choquet 2009).
Synaptic strength can also be regulated by switching between AMPAR containing the GluA2 subunit (e.g., GluA1A2) and AMPAR lacking the GluA2 subunit (homomeric GluA1, homomeric GluA3, or GluA1A3) (Cull-Candy et al. 2006; Thiagarajan et al. 2007; Liu and Zukin 2007; Isaac et al. 2007). GluA2-lacking AMPAR are permeable to Ca2+ and therefore have higher conductance than GluA2-containing AMPAR. Their higher conductance means that insertion or removal of only a small number of these receptors markedly alters synaptic strength (Liu and Cull-Candy 2000; Plant et al. 2006; Guire et al. 2008), whereas their Ca2+ permeability permits AMPAR transmission to be directly coupled to the many important Ca2+-dependent signaling pathways that govern subsequent plasticity (Thiagarajan et al. 2007). GluA2-lacking AMPAR can be identified experimentally based on inward rectification due to voltage-dependent block by polyamines as well as sensitivity to antagonists that do not block GluA2-containing AMPAR.
Although the cellular mechanisms underlying LTP and LTD vary at different synapses, the best studied form of LTP requires strong synaptic activation, Ca2+ entry through the NMDA receptor (NMDAR), and activation of protein kinase-dependent cascades that recruit additional AMPAR to the synapse. Synaptic activation leading to LTD is associated with less Ca2+ entry and selective activation of protein phosphatases, leading to AMPAR removal from synapses (Shepherd and Huganir 2007; Pi and Lisman 2008). These events occur in the early phase of LTP. There is also a late phase of LTP, in which protein synthesis is required to stabilize the increase in synaptic strength (see Abraham and Williams 2008 for a recent review).
Recently, there has been considerable interest in a homeostatic form of plasticity termed synaptic scaling, in which prolonged activity blockade (typically 1–3 days) leads to augmented excitatory synaptic transmission, whereas prolonged increases in activity produce opposite effects (Turrigiano and Nelson 2004; Turrigiano 2008). Synaptic scaling is believed to stabilize the activity of neurons and neuronal circuits. For example, scaling up would prevent rundown of synaptic strength after repeated LTD. Although many mechanisms contribute to the expression of synaptic scaling, including presynaptic changes in transmitter release, the major postsynaptic mechanism involves increases in synaptic AMPAR levels in response to activity blockade and decreased AMPAR levels after prolonged increases in activity. Synaptic scaling is a promising candidate mechanism for slowly developing plasticity that occurs after drug withdrawal (Sun and Wolf 2009; see “Long-Term Effects of DA Receptor Stimulation”).
AMPAR in the NAc
Excitatory synapses in the NAc undergo NMDAR-dependent LTP and several forms of LTD, a topic that has been reviewed recently (Kauer and Malenka 2007) and therefore will not be addressed here. Instead, this review will focus on studies of AMPAR subunit composition and trafficking in NAc neurons.
To determine the subunit composition of NAc AMPAR, we conducted quantitative co-IP studies identical to those of Wenthold et al. (1996). This section will discuss data obtained using NAc membrane preparations from untreated adult rats as starting material (Reimers et al. 2007 and unpublished findings), but similar results were found in the NAc of rats with saline self-administration experience (Conrad et al. 2008; see also Boudreau et al. 2007) and in other brain regions (Reimers et al. 2007 and unpublished findings). In the NAc of untreated rats, we found that ~90% of the GluA1 is physically associated with GluA2 or GluA3, and the majority of this represents GluA1A2. GluA1A3 complexes also exist, but this is a minor population (only ~6% of the GluA3 is pulled down by GluA1 antibody). It is possible that there is also a very small population of homomeric GluA1 receptors (Reimers et al. 2007 and unpublished findings). These findings are consistent with earlier studies suggesting that most GluA1-containing AMPAR in the striatum and other forebrain regions also contain GluA2 or GluA3 (Bernard et al. 1997; Gold et al. 1997). Turning to the question of GluA2A3 receptors, we found that ~50% of GluA2 was not physically associated with GluA1 and therefore could represent GluA2A3. However, using blue native electrophoresis to assess AMPAR assembly state, we found that a substantial portion of GluA2 was present in dimers or monomers, whereas relatively more GluA1 was present in tetramers (Reimers et al. 2007 and unpublished findings). These findings are consistent with prior work in cultured neurons and rat brain (Greger et al. 2003; see “AMPAR Trafficking and Synaptic Plasticity”) and argue that most of the “left-over” GluA2 probably represents partially assembled receptors. Overall, our results suggest a predominant role for GluA1A2 receptors in the NAc and thus fall into line with single-cell genetic/electrophysiological assessments of AMPAR subunit composition in hippocampus (Lu et al. 2009). However, a role for GluA2A3 or GluA1A3 receptors in the NAc should not be ruled out. GluA3 is expressed on the cell surface in the adult rat NAc (Boudreau et al. 2007) and this measure is altered after cocaine self-administration (Conrad et al. 2008). GluA4 is not present in medium spiny neurons (Bernard et al. 1997; Stefani et al. 1998).
Although the co-IP studies described above suggest that a small pool of GluA2-lacking AMPAR is present in NAc neurons (Reimers et al. 2007; Conrad et al. 2008; Boudreau et al. 2007), electrophysiological studies indicate that they play a minimal role in synaptic transmission. Thus, whole-cell patch clamp recordings in the NAc core of adult rats with previous saline self-administration experience found linear AMPAR current–voltage relationships and only a small reduction (~5%) in the evoked excitatory postsynaptic current (EPSC) amplitude after bath application of 1-naphthylacetylsperimine (Naspm), a selective antagonist of GluA2-lacking AMPAR (Conrad et al. 2008). Linear AMPAR current–voltage relationships have also been found in the NAc shell of young mice given saline injections (Kourrich et al. 2007, P24-28; Mameli et al. 2009, P16-35; P, postnatal day), although a small contribution of GluA2-lacking AMPAR to synaptic transmission in the NAc shell was found in untreated adult mice (Campioni et al. 2009, P56-70).
To study AMPAR trafficking in the NAc, we have used primary cultures prepared from the NAc of postnatal day 1 rats and co-cultures in which prefrontal cortex (PFC) neurons are included to restore excitatory synapses onto NAc MSN. Despite the fact that NAc MSN are GABA neurons, AMPAR trafficking was similar to what has been described in hippocampal pyramidal neurons. Thus, brief exposure to glutamate agonists produces a decrease in AMPAR surface expression in cultured NAc neurons (Mangiavacchi and Wolf 2004b) akin to that implicated in LTD in other brain regions (Carroll et al. 2001). We have also described the activity-dependent synaptic insertion of GluA1-containing AMPAR in cultured NAc neurons. This occurs through a two-step process that involves externalization onto the cell surface at extrasynaptic sites followed by translocation into the synapse (Fig. 1). The first step is accelerated by protein kinase A (PKA), most likely via phosphorylation of GluA1 at serine 845 (a PKA phosphorylation site), whereas the second step requires NMDAR stimulation (Chao et al. 2002a, b; Mangiavacchi and Wolf 2004a; Sun et al. 2008). A similar two-step mechanism occurs in other cell types (Passafaro et al. 2001; Esteban et al. 2003; Sun et al. 2005; Gao et al. 2006; Oh et al. 2006; Man et al. 2007; Yudowski et al. 2007). Recent studies have provided additional information about this process. Petrini et al. (2009) demonstrated that AMPAR recycling via postsynaptic endocytic zones is crucial for maintaining a mobile pool of extrasynaptic AMPAR that can be inserted into synapses in an activity-dependent manner to increase synaptic strength. Furthermore, in addition to the important priming role noted above for PKA phosphorylation of serine 845, it has been demonstrated that protein kinase C phosphorylation of serine 816 and serine 818 of GluA1 enhances the interaction with its binding partner 4.1 N and thus enhances insertion of GluA1 into surface extrasynaptic pools (Lin et al. 2009).

AMPAR enter synapses through a two-step process, indicated by numbered boxes in the figure: (1) exocytosis onto the extrasynaptic cell surface (this step is accelerated by PKA activation) and (2) translocation into the synapse following activation of NMDAR and CaMKII. In the NAc and several other regions (e.g., PFC and hippocampus), spines of principal neurons receive convergent inputs from glutamate and DA terminals; thus, DA receptors are well-positioned to modulate AMPAR trafficking at glutamate synapses. In primary cultures from these brain regions, we have found that brief (5–15 min) treatment with a D1-class receptor agonist, leading to PKA activation, increases the rate of AMPAR externalization at extrasynaptic sites. This in turn primes AMPAR for synaptic insertion in response to subsequent NMDAR stimulation. This schematic is based on data presented in Sun et al. (2005), Gao et al. (2006), and Sun et al. (2008)
While most studies of synaptic plasticity in NAc neurons have focused on LTP or LTD, we showed recently that NAc MSN also exhibit bidirectional synaptic scaling. Prolonged activity blockade [1–3 days of incubation with tetrodotoxin (TTX) or the AMPAR antagonist CNQX] leads to increased levels of GluA1A2-containing AMPAR on the surface of MSN, both at synaptic and extrasynaptic locations, whereas prolonged increases in activity (produced by incubating with the GABAA receptor antagonist bicuculline) lead to opposite effects. Increased AMPAR surface expression after prolonged activity blockade requires protein synthesis and is occluded by inhibition of the ubiquitin–proteasome system, perhaps indicating a requirement for new receptors as well as slowing of the turnover of existing receptors, or, more generally, a stabilization of newly translated proteins (Sun and Wolf 2009). It is possible that synaptic scaling contributes to the increased AMPAR surface and synaptic expression that is observed in the rodent NAc after withdrawal from repeated cocaine exposure (see “AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking”). Imaging studies in humans and primates have demonstrated persistent metabolic hypoactivity after cocaine exposure in cortical areas that send glutamate projections to the NAc (Goldstein and Volkow 2002; Porrino et al. 2007), which could translate into decreased excitatory transmission onto NAc neurons after cocaine withdrawal. We have speculated that this leads to synaptic scaling in NAc neurons, resulting in the accumulation of AMPAR at excitatory synapses onto these neurons and explaining in vivo results indicating AMPAR upregulation after cocaine withdrawal (see “AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking”). For discussion of rodent data that support this hypothesis, see Sun and Wolf (2009).
Facilitation of AMPAR Synaptic Insertion by DA May Contribute to the Strength of Drug-Related Learning
D1-Class DA Receptors Prime AMPAR for Synaptic Insertion
The insertion of new AMPAR into synapses is responsible for increasing synaptic strength in many forms of experience-dependent plasticity and learning (Kessels and Malinow 2009). As discussed in more detail in the section titled “Significance of the Priming Effect of D1-Class Receptors for Motivated Behavior”, one important role of DA is to facilitate learning about natural rewards and drugs of abuse. A very simple way to accomplish this would be to directly facilitate AMPAR synaptic insertion at relevant synapses. This mechanism is plausible in several brain regions—including the dorsal striatum, NAc (ventral striatum), PFC and hippocampus—where dendrites of the principal neurons receive convergent DA and glutamate inputs, forming a synaptic triad (Sesack et al. 2003; Fig. 1). Within the triad, DA receptors are well-positioned to modulate plasticity mechanisms, including AMPAR trafficking, at nearby glutamate synapses.
We tested this idea in cultured neurons. Our initial studies used primary cultures prepared from postnatal NAc neurons. These studies revealed that stimulation of D1-class receptors, which are positively coupled to adenylyl cyclase, increases the rate of externalization of GluA1-containing AMPAR to the cell surface, in concert with stimulation of GluA1 phosphorylation at serine 845 (Chao et al. 2002a, b; Mangiavacchi and Wolf 2004a; we are using the term D1-class because pharmacological agents used in these studies do not distinguish between D1 and D5 receptors). We could not study AMPAR synaptic targeting in “pure” NAc cultures, because the NAc does not contain intrinsic glutamate neurons and thus cultures prepared from the NAc do not contain glutamate synapses. To overcome this, we restored excitatory synapses onto the NAc neurons by co-culturing them with PFC neurons (Sun et al. 2008). We also studied primary cultures prepared from postnatal rat hippocampus (Gao et al. 2006) and PFC (Sun et al. 2005), brain regions that contain glutamate neurons and therefore yield cultures containing glutamate synapses. In all neurons studied (NAc MSN, hippocampal pyramidal neurons, and PFC pyramidal neurons), we found that brief (5–15 min) stimulation of D1-class receptors increased GluA1 surface expression through a mechanism requiring PKA activation (Sun et al. 2005, 2008; Gao et al. 2006) and the secretory pathway (Gao et al. 2006). Protein synthesis was not required in our studies (Gao et al. 2006; Sun et al. 2008), although D1-class agonists can also increase synaptic GluA1 levels in hippocampal neurons through a mechanism that involves stimulation of dendritic protein synthesis (Smith et al. 2005). Interestingly, in all cases, we found that D1-class receptor stimulation increased AMPAR surface expression exclusively at extrasynaptic sites; no changes in synaptic AMPAR levels were observed (Sun et al. 2005, 2008; Gao et al. 2006). These results indicated that stimulation of D1-class receptor/PKA signaling is not sufficient for AMPAR synaptic insertion, at least not under our experimental conditions (see next paragraph). However, we hypothesized that D1-class receptor stimulation, by increasing extrasynaptic AMPAR levels, might prime AMPAR for synaptic insertion. To test this, we took advantage of the ability of glycine, an obligatory co-agonist at the NMDAR, to elicit AMPAR synaptic insertion and LTP (Lu et al. 2001). We identified a concentration of glycine that was subthreshold for this effect. If cultures were incubated briefly with a D1 agonist followed immediately by 3 min of incubation with glycine, we observed that this subthreshold concentration of glycine was able to produce AMPAR synaptic incorporation. These results demonstrate that D1-class receptors can facilitate activity-dependent synaptic plasticity (Sun et al. 2005; Gao et al. 2006; Sun et al. 2008).
An implication of these results is that D1-class receptor agonists will only facilitate AMPAR synaptic insertion under conditions of sufficient NMDAR tone. This may explain some electrophysiological results in NAc neurons that do not support this mechanism. For example, Nicola et al. (1996) found no evidence for D1-class receptor mediated enhancement of postsynaptic AMPAR transmission, but their recordings were conducted in brain slices at membrane potentials that prohibit NMDAR transmission. In contrast, when NAc are activated by excitatory inputs in vivo, it is likely that that NMDAR tone would be sufficient to observe the priming effect. However, many factors are no doubt important in determining the in vivo consequences of D1-class receptor stimulation in the NAc, including effects on voltage-gated conductances (e.g., Nicola et al. 2000; Zhang et al. 1998, 2002) and the existence of presynaptic D1-class receptors in the NAc that dampen excitatory synaptic transmission (e.g., Pennartz et al. 1992; Harvey and Lacey 1996; Nicola et al. 1996).
In conclusion, our results in cultured NAc, PFC, and hippocampal neurons provided some of the first direct support for the two-step process of GluA1 synaptic incorporation described in “AMPAR in the NAc” and demonstrated that D1-class receptors prime AMPAR for synaptic insertion by accelerating the first step (PKA-dependent externalization onto extrasynaptic regions of the cell surface) and thus increasing the number of AMPAR available for translocation into synapses when NMDAR are subsequently stimulated (see Fig. 1 for summary). Facilitation of AMPAR synaptic insertion by D1-class receptors was blocked by KN-62, an inhibitor of Ca2+-calmodulin-dependent protein kinases (Gao et al. 2006). This is consistent with the fact that AMPAR synaptic insertion during LTP ultimately requires activation of Ca2+-calmodulin-dependent protein kinase II (CaMKII) (Lisman et al. 2002). Overall, our results indicate that the D1-class receptor/PKA pathway works cooperatively with the NMDAR/CaMKII pathway to induce AMPAR synaptic incorporation, thereby facilitating early phases of synaptic plasticity. Consistent with this, activation of D1-class receptors has been shown to augment the magnitude of early LTP in CA1 slices (Otmakhova and Lisman 1996). This role of D1-class receptor/PKA signaling is distinct from, but complementary to, its well established role in the late protein synthesis-dependent phase of LTP (Frey et al. 1990, 1991, 1993; Huang and Kandel 1995; Matthies et al. 1997; Swanson-Park et al. 1999; Duffy and Nguyen 2003; Morris et al. 2003).
D1-class receptor stimulation also increased GluA1 synaptic incorporation in VTA DA neurons co-cultured with PFC neurons to restore excitatory synapses, although the mechanism differed from that described above for NAc, PFC, and hippocampus (Gao and Wolf 2007). The VTA DA neurons responded to acute PKA activation with increased GluA1 surface expression, as expected from our prior results (above). However, D1-class receptors are not present on their surface (at least in our culture system), and thus D1-class receptor agonists did not reproduce this effect of PKA activation. Instead, we found that brief stimulation with a D1-class agonist (10 min) increased GluA1 and GluA2 surface expression through a glutamate receptor-dependent pathway, apparently by acting on D1-class receptors on the PFC neurons in the co-culture and thereby altering their excitatory transmission onto VTA DA neurons. We also tested the effect of a longer incubation with DA (1 h), to mimic the duration of elevated DA levels produced by systemic cocaine administration. In this case, we observed an increase in surface and synaptic GluA1 but not GluA2 on VTA DA neurons, suggesting an increase in GluA2-lacking AMPAR. Surface GluA3 and the area of GluA1A3 colocalization were also increased (Gao and Wolf 2007). Due to the higher conductance of GluA2-lacking AMPAR, their addition to VTA DA neurons would be expected to significantly enhance DA neuronal activation by glutamate inputs. These studies provide an in vitro model for the cocaine-induced potentiation of excitatory transmission onto VTA DA neurons that contributes to the initiation of behavioral sensitization (Ungless et al. 2001). GluA2-lacking AMPAR have been shown to contribute to this potentiation (Bellone and Lüscher 2006; Argilli et al. 2008).
Significance of the Priming Effect of D1-Class Receptors for Motivated Behavior
This section addresses the possible behavioral significance of the ability of D1-class receptors to prime AMPAR for synaptic insertion in principal neurons of the PFC (Sun et al. 2005), hippocampus (Gao et al. 2006), and NAc (Sun et al. 2008). Although the role of DA in motivation and reward is very complex, one idea is that DA transmission facilitates associative learning about natural rewards and drugs of abuse that is important for shaping future behavior (Di Chiara 1998; Berke and Hyman 2000; Schultz 2000; Kelley and Berridge 2002; Lisman and Grace 2006; Wheeler and Carelli 2009). Our results suggest that DA may enhance learning about reward-related stimuli at least in part by priming AMPAR for synaptic insertion and thereby facilitating synaptic plasticity. Thus, when a stimulus is presented within a temporal window in which DA levels are elevated, that stimulus is more likely to be encoded as an increase in the strength of excitatory synapses. This mechanism may help explain many results obtained in NAc, PFC, and hippocampal neurons indicating that D1-class receptors facilitate LTP and learning (Jay 2003; O’Donnell 2003; Lisman and Grace 2006). For example, Li et al. (2003) found that brief exposure to a novel environment, leading to D1 receptor-PKA signaling, enhanced the ability of a weak tetanus to induce LTP in hippocampal CA1 neurons. This observation is readily explained by a priming effect of D1-class receptor stimulation (Fig. 1), although it is important to note that DA also modulates other cellular mechanisms that regulate neuronal excitability and plasticity, including NMDAR transmission (Cepeda and Levine 2006) and voltage-gated ion channels (e.g., Seamans and Yang 2004) (see “D1-Class DA Receptors Prime AMPAR for Synaptic Insertion” for specific citations related to D1-class receptors in the NAc).
A similar priming effect has been demonstrated after stimulating hippocampal β-adrenergic receptors (which like D1-class receptors are positively coupled to adenylyl cyclase) with norepinephrine, a transmitter important for stress responses. Norepinephrine produced a robust increase in GluA1 phosphorylation at serine 845 (PKA phosphorylation site), and to a lesser extent at serine 831 (CaMKII or protein kinase C phosphorylation site), an effect that was reproduced by emotional stress (Hu et al. 2007). Through this mechanism, norepinephrine facilitated GluA1 synaptic delivery and LTP (Hu et al. 2007). This is proposed to contribute to stress-related learning (Krugers and Hoogenraad 2009). More broadly, the ability to promote plasticity by facilitating AMPAR synaptic delivery may be a shared property of neuromodulator receptors that are positively coupled to adenylyl cyclase (see Soel et al. 2009).
In contrast to natural rewards, psychomotor stimulants like cocaine, by blocking the DA transporter, produce robust and unregulated increases in extracellular DA levels. During repeated cocaine exposure, unregulated DA receptor signaling may lead to inappropriate modulation of AMPAR trafficking and abnormal synaptic plasticity. These processes may contribute to the rewiring of neuronal circuits that underlies the transition to compulsive drug use. More specifically, we speculate that unregulated DA release promotes very strong learning about stimuli that are present while the subject is under the influence of the drug, when DA levels are high, including cues and context that are associated with the drug-taking experience. As cues are important triggers for drug craving, this pathologically strong learning may explain the persistent vulnerability to relapse that makes addiction so difficult to treat. It is interesting to speculate that learning related to the negative consequences that follow the act of drug taking (e.g., family disapproval or loss of employment) is much less effective because DA levels are low when the negative consequences are experienced.
Long-Term Effects of DA Receptor Stimulation
The studies described in “Significance of the Priming Effect of D1-Class Receptors for Motivated Behavior” focused on acute effects of DA receptor stimulation on AMPAR trafficking. We wondered whether long-lasting effects also occur. This is difficult to address using primary neuronal cultures because there is a limited time window for experimentation. Typically, at least a week in vitro is required for synaptic connections to be established, but cultures deteriorate after about 3 weeks (although this depends on cell density and other factors). Within this window, we attempted to mimic repeated cocaine exposure by treating NAc/PFC co-cultures repeatedly with DA (30 min per day on days 7, 9, and 11 in vitro). Two interesting effects were observed 4 days after the last DA exposure (day 15). First, D1-class agonist treatment no longer increased AMPAR surface expression. This refractoriness was associated with decreased D1 receptor surface expression (Sun et al. 2008). Second, upregulation of GluA1 and GluA2 surface expression on NAc MSN was detected on day 15 (Sun et al. 2008; Sun and Wolf 2009). This effect required CaMK activity during the 4 day “withdrawal” period and was associated with CaMKII activation (Sun et al. 2008). Obviously cultures do not reproduce the circuitry of the brain and they are prepared from young neurons, which differ from adult neurons in many respects. For example, unlike adult rodent NAc neurons (see “AMPAR in the NAc”), cultured NAc neurons, prepared from P1 rats, express a substantial number of GluA2-lacking AMPAR under basal conditions (Sun and Wolf 2009). Therefore, caution must be used when extrapolating these and other in vitro results to the adult brain. However, our results in NAc/PFC co-cultures indicate that repeated DA treatment has effects on plasticity mechanisms that outlast the period in which DA levels are elevated, perhaps via changes in excitatory activity that are triggered by prior DA receptor overstimulation. Specifically, these results are reminiscent of the increased GluA1 and GluA2 surface expression that we have observed in the NAc of cocaine-sensitized rats (see “AMPAR Plasticity in Behavioral Sensitization”). These rats show a transient elevation of CaMKII levels in the NAc on withdrawal day 7 (Boudreau et al. 2009), suggesting another parallel to the in vitro findings discussed here.
Interestingly, we found that the upregulation of surface GluA1 and GluA2 in cultured NAc neurons produced by repeated DA treatment occluded subsequent synaptic scaling induced by prolonged activity blockade, i.e., no further increase in AMPAR surface expression was produced by activity blockade when it was preceded by repeated DA treatment (Sun and Wolf 2009). If this occlusion occurs in vivo, it would argue against the hypothesis that synaptic scaling is responsible for AMPAR upregulation in the NAc after withdrawal from repeated in vivo cocaine exposure (see “AMPAR in the NAc”). On the other hand, if the occlusion of scaling occurs in vivo but is transient, it could help explain the delayed onset of AMPAR upregulation after cocaine withdrawal (see “AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking”). Interestingly, a different kind of homeostatic plasticity, in which MSN adjust their membrane excitability to functionally compensate for changes in the level of excitatory synaptic input, shows a prolonged impairment in the NAc of cocaine-sensitized rats (Ishikawa et al. 2009).
AMPAR Upregulation in the NAc After Cocaine Withdrawal May Enhance Drug Seeking
Overview
AMPAR transmission in the NAc has been studied after both non-contingent and contingent cocaine administration. When cocaine is administered by the experimenter, this is referred to as “non-contingent” because drug administration is not dependent upon the animal’s behavior. In cocaine self-administration procedures, drug administration is dependent on the rat’s response, such as a nose poke or lever press, and is therefore “contingent.” As described below, we and others have observed increases in AMPAR surface and synaptic expression in the NAc following withdrawal from repeated non-contingent cocaine injections leading to behavioral sensitization (see “AMPAR Plasticity in Behavioral Sensitization”) and cocaine self-administration leading to the incubation of cocaine craving (“AMPAR Plasticity in the Incubation Model”), although the AMPAR subtype involved differs in these two situations. It is important to note that these increases do not occur during cocaine exposure but require a withdrawal period to become evident. Thus, they do not result from direct effects of elevated DA levels on mechanisms that control AMPAR trafficking. Instead, just as changes in the level of presynaptic activity trigger LTP, LTD, or synaptic scaling (see “AMPAR Trafficking and Synaptic Plasticity”), we speculate that withdrawal-dependent changes in the activity of glutamate pathways projecting to the NAc are responsible for postsynaptic AMPAR plasticity in the NAc after cocaine withdrawal. At present, the nature of such changes is not known. Candidate mechanisms include altered presynaptic glutamate transmission due to impaired cystine–glutamate exchange (Kalivas 2009) or hypoactivity of cortical areas that send glutamate projections to the NAc (Goldstein and Volkow 2002; Porrino et al. 2007). Another factor that cannot be neglected is “whole-cell plasticity” related to drug-induced changes in voltage-gated ion channels (Zhang et al. 1998, 2002; Hu et al. 2004, 2005; Dong et al. 2006; Ishikawa et al. 2009; Kourrich and Thomas 2009; Mu et al. 2010). For discussion of possible interactions between these phenomena and postsynaptic AMPAR plasticity, see Wolf and Ferrario (2010).
AMPAR Plasticity in Behavioral Sensitization
Increased AMPAR surface expression in the NAc after withdrawal from repeated cocaine exposure was first observed in the behavioral sensitization model. Behavioral sensitization refers to the progressive augmentation of behavioral responses to a drug that develops during repeated drug treatment and then persists long after drug exposure is discontinued. Sensitization of locomotor stimulatory effects is commonly studied. In addition, sensitization develops to the incentive-motivational properties of drugs, in other words, the properties that make them “wanted” (Robinson and Berridge 2008). Thus, rats previously sensitized to psychomotor stimulants will subsequently show enhanced motivation to obtain drugs compared to control rats (Vezina 2004), just as an addict will continue to seek drugs even in the face of adverse consequences.
Our studies were conducted using adult rats and initially employed a cocaine regimen that produces locomotor sensitization in approximately half the cocaine-treated rats. Using a protein crosslinking assay that can distinguish between cell surface and intracellular pools of AMPAR subunits (Boudreau and Wolf 2005), we found that sensitized rats, but not cocaine-treated rats that failed to sensitize, exhibit increased AMPAR surface expression in the NAc. This was detected after 7, 14, or 21 days of withdrawal, but not on the first day of withdrawal, indicating that AMPAR cell surface expression increases sometime during the first week of withdrawal (Boudreau and Wolf 2005; Boudreau et al. 2007, 2009). More recently, we have used a cocaine regimen that produces sensitization in all rats, and found a significant increase in AMPAR subunit surface/intracellular ratios in the total population of cocaine-treated rats on withdrawal day 14 (Ferrario et al. 2010).
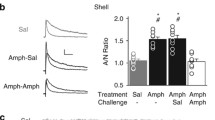
Evidence for AMPAR upregulation has also been obtained with other approaches. Electrophysiological studies found an increased AMPA/NMDA ratio in the NAc of cocaine-sensitized mice after 10–14 days of withdrawal (Kourrich et al. 2007). A more recent study found that synaptic strength was increased on withdrawal day 35 due to the addition of GluA2-lacking AMPAR (Mameli et al. 2009; see next paragraph for more discussion). Biochemical studies found increased GluA1 and GluA2 levels in synaptosomal membrane fractions prepared from the NAc of sensitized rats after 21 days but not 1 day of withdrawal (Ghasemzadeh et al. 2009a; Schumann and Yaka 2009), while increased GluA1 in tissue homogenates was reported after 1–3 weeks but not 1 day of withdrawal (Churchill et al. 1999; Scheggi et al. 2002). Finally, the locomotor response elicited by infusion of AMPA into the NAc is enhanced after withdrawal from a sensitizing cocaine regimen (Pierce et al. 1996; Bell and Kalivas 1996; Bachtell and Self 2008). The cellular mechanisms that trigger and maintain AMPAR upregulation in cocaine-sensitized rats remain unclear, but NMDAR stimulation and ERK activation during withdrawal are implicated (Boudreau et al. 2007, 2009; Schumann and Yaka 2009; Huang et al. 2009). CaMKII and PKA may also be involved (Boudreau et al. 2009). Interestingly, despite the fact that both cocaine and amphetamine elevate DA transmission in the NAc, amphetamine sensitization is not associated with increased AMPAR surface expression, arguing against a major role for increased DA transmission in triggering AMPAR upregulation in cocaine-sensitized rats (Nelson et al. 2009; Wolf and Ferrario 2010).
The NAc is divided into two subregions, core and shell, that can be distinguished based on morphology, connectivity, and functional role (Meredith et al. 2008). While our studies and others (Schumann and Yaka 2009) used a combined core/shell dissection, other studies examined these subregions separately and obtained results that, taken together, support the idea that AMPAR upregulation occurs in both subregions (Pierce et al. 1996; Bell and Kalivas 1996; Kourrich et al. 2007; Mameli et al. 2009; Ghasemzadeh et al. 2009a). Differences and similarities between core and shell subregions are beyond the scope of this review, although AMPAR transmission in both regions is implicated in drug seeking behavior in cocaine-experienced rats (Wolf and Ferrario 2010).
Most results suggest that GluA1A2-containing AMPAR, rather than GluA2-lacking AMPAR, increase after withdrawal from non-contingent cocaine treatment leading to sensitization. Thus, both GluA1 and GluA2 subunits exhibit increased surface expression (Boudreau and Wolf 2005; Boudreau et al. 2007) and enrichment in synaptosomal membrane fractions (Ghasemzadeh et al. 2009a) on withdrawal days 14–21, and electrophysiological studies found no evidence for GluA2-lacking AMPAR in the shell of cocaine-sensitized mice on withdrawal days 10–14 (Kourrich et al. 2007). In contrast, results in the incubation model (see “AMPAR Plasticity in the Incubation Model”) indicate that GluA2-lacking AMPAR are added to excitatory synapses in the NAc after prolonged withdrawal from extended access cocaine self-administration; furthermore, these receptors mediate the “incubated” cue-induced craving observed after prolonged withdrawal (Conrad et al. 2008; Mameli et al. 2009). Why do GluA1A2 receptors increase in the sensitization model while GluA2-lacking AMPAR increase in the incubation model? It is possible that this is due to different patterns of neuronal activity in pathways projecting to the NAc as a result of non-contingent (sensitization) versus contingent (incubation) cocaine exposure. However, Mameli et al. (2009) found evidence for GluA2-lacking AMPAR in the NAc of cocaine-treated mice after 35 days of withdrawal regardless of whether they received 10 daily i.p. cocaine injections or 8 days of cocaine self-administration (4 h/day). This suggests that a long withdrawal period leads to synaptic incorporation of GluA2-lacking AMPAR regardless of whether cocaine administration is contingent or non-contingent, and raises the possibility that GluA1A2-containing AMPAR increase during the first week of withdrawal and are then replaced by GluA2-lacking AMPAR. Alternatively, the results of Mameli et al. (2009) in the sensitization model may be related to the age of the mice when cocaine treatment began (P16-35). This is an important point, since there are developmental changes in the prevalence of GluA2-lacking AMPAR (e.g., Ho et al. 2007) and plasticity mechanisms (McCutcheon and Marinelli 2009).
What is the functional significance of AMPAR upregulation in the NAc of cocaine-sensitized rats? Some results indicate that enhanced AMPAR transmission in the NAc mediates the expression of locomotor sensitization (Pierce et al. 1996; Bell et al. 2000). On the other hand, several dissociations have been observed between the level of AMPAR transmission and the expression of sensitization (Bachtell and Self 2008; Bachtell et al. 2008; Ferrario et al. 2010). For example, locomotor sensitization is expressed on withdrawal day 1, yet AMPAR surface expression on withdrawal day 1 does not differ between cocaine-sensitized and saline-treated treated rats (Boudreau and Wolf 2005). Furthermore, if cocaine-sensitized rodents are administered a challenge injection of cocaine, AMPAR surface and synaptic expression are decreased 24 h later (Thomas et al. 2001; Boudreau et al. 2007; Kourrich et al. 2007). In other words, the AMPAR upregulation that occurs after withdrawal is reversed by cocaine re-exposure, although both behavioral and protein crosslinking results indicate that upregulation is restored within 7 days (Bachtell and Self 2008; Ferrario et al. 2010). However, if a second cocaine challenge is administered 24 h after the first, that is, when AMPAR surface and synaptic expression have been decreased by the first cocaine challenge, the magnitude of locomotor sensitization is not decreased (Ferrario et al. 2010). In fact, Bachtell and Self (2008) observed that it was enhanced. These and other results suggest that treatments which decrease AMPAR transmission in the NAc do not necessarily decrease the magnitude of the expression of locomotor sensitization to cocaine. Finally, results obtained after transient viral-mediated over-expression of wild-type GluA1 or a pore-dead GluA1 mutant suggest that the magnitude of sensitization varies inversely with the strength of AMPAR transmission (Bachtell et al. 2008). Related to this, one theory holds that rapid AMPAR internalization following drug challenge mediates the expression of locomotor sensitization (Brebner et al. 2005), although this is based on studies of amphetamine, which has very different effects on NAc AMPAR compared to cocaine (Nelson et al. 2009; Wolf and Ferrario 2010). Arguing against this theory as it applies to cocaine, we have not observed any change in AMPAR surface expression in the NAc 30 min after a cocaine challenge injection is administered to sensitized rats (Ferrario et al. 2010). In conclusion, some evidence suggests that enhanced AMPAR transmission in the NAc underlies locomotor sensitization, but other studies suggest a more complicated relationship.
In contrast, nearly all evidence supports the idea that cocaine seeking behavior, measured in a number of different animal models, requires activation of AMPAR on NAc neurons by glutamate afferents originating from PFC or limbic regions (Cornish and Kalivas 2000; Vorel et al. 2001; Di Ciano and Everitt 2001, 2004; Park et al. 2002; McFarland et al. 2003; Hayes et al. 2003; Ito et al. 2004; Di Ciano et al. 2007; Bäckstrom and Hyytiä 2007; Conrad et al. 2008; Famous et al. 2008; Ping et al. 2008; Suto et al. 2009; Sari et al. 2009). Furthermore, several studies have observed a positive correlation between the magnitude of drug seeking behavior and the level of AMPAR transmission in the NAc (Suto et al. 2004; Conrad et al. 2008; Anderson et al. 2008; see “AMPAR Plasticity in the Incubation Model” section for more discussion). As noted above, it is well established that rats administered non-contingent psychomotor stimulant treatment leading to locomotor sensitization will subsequently show enhanced acquisition of drug self-administration and will work harder to obtain the drug in progressive ratio experiments (Vezina 2004). Given that cocaine seeking seems to depend on the level of NAc AMPAR transmission, we propose that AMPAR upregulation in the NAc of cocaine-sensitized rats is responsible for this subsequent enhancement of drug taking and seeking. In other words, it is possible that AMPAR upregulation after non-contingent cocaine exposure is more directly related to incentive sensitization than to locomotor sensitization (see Wolf and Ferrario 2010 for more discussion).
There are studies that do not support a positive relationship between drug seeking and AMPAR transmission in the NAc. These involve experiments in which GluA1 has been over-expressed or deleted (Mead et al. 2007; Bachtell et al. 2008), situations in which compensatory changes may affect the outcome, or experiments in which extinction training followed cocaine self-administration (Sutton et al. 2003). Extinction training involves new learning (Self et al. 2004) and therefore, not surprisingly, different AMPAR changes are observed in the NAc if cocaine self-administration is followed by extinction training than if it is followed by withdrawal in home cages (Sutton et al. 2003; Ghasemzadeh et al. 2009b). Recognition of this difference may resolve at least some of the apparent discrepancies in this literature (Wolf and Ferrario 2010).
Our discussion has not attempted to incorporate literature on the dorsal striatum. However, this region is also important for locomotor sensitization and shares some sensitization-related neuroadaptations with the NAc (Patrick et al. 1991; Robinson and Kolb 2004; Jedynak et al. 2007). Yet, cocaine sensitization is associated with different and in some respects opposite changes in AMPAR distribution in the dorsolateral striatum versus the NAc (Ferrario et al. 2010). This may argue further for dissociation between locomotor sensitization and the AMPAR redistribution observed in the NAc. However, AMPAR plasticity, albeit different from that induced by cocaine, may nevertheless contribute to amphetamine sensitization (e.g., Loweth et al. 2010).
AMPAR Plasticity in the Incubation Model
After extended access cocaine self-administration (e.g., 6 h/day × 10 days; Conrad et al. 2008), there is a progressive increase in cue-induced cocaine seeking over the first weeks to months of withdrawal that has been termed “incubation” (Neisewander et al. 2000; Grimm et al. 2001; Lu et al. 2004a, b; Sorge and Stewart 2005). We showed that GluA2-lacking AMPAR are added to NAc synapses in association with the incubation of cue-induced cocaine seeking (Conrad et al. 2008). Thus, they were not found in NAc synapses of drug-naïve rats or cocaine-experienced rats evaluated on withdrawal day 1, when cue-induced craving is low. However, on withdrawal days 42–47, when cue-induced craving is high, GluA2-lacking AMPAR were present. This was demonstrated using a variety of approaches, including protein crosslinking studies showing selective increases in surface GluA1, quantitative co-IP studies showing an increase in the portion of GluA1 not physically associated with GluA2, and electrophysiological studies showing inwardly rectifying and Naspm-sensitive evoked EPSCs (Conrad et al. 2008). Consistent with our results, inwardly rectifying AMPAR EPSC were demonstrated in the mouse NAc shell on withdrawal day 35 from cocaine self-administration (4 h/day for 8 days; Mameli et al. 2009). This represents a dramatic change in the synaptic function of MSN due to the Ca2+ permeability and resulting higher conductance of GluA2-lacking AMPAR (see “AMPAR Trafficking and Synaptic Plasticity”). Finally, we showed that blocking GluA2-lacking AMPAR, by injecting Naspm into the NAc core, prevented the expression of incubated cue-induced cocaine seeking on withdrawal day 45 (Conrad et al. 2008). Together, these results may suggest GluA2-lacking AMPAR as a target for the design of anti-craving medications.
Cocaine seeking requires activation of glutamate inputs to the NAc and a resultant increase in AMPAR transmission onto MSN (see “AMPAR Plasticity in Behavioral Sensitization”). Thus, the results described above (Conrad et al. 2008; Mameli et al. 2009) suggest that MSN become more responsive to these glutamate inputs after prolonged withdrawal from cocaine self-administration due to the synaptic incorporation of high conductance GluA2-lacking AMPAR. Thus, when a cue previously paired with cocaine is presented and glutamate is released in the NAc, MSN respond more strongly and cue-induced craving is enhanced. These ideas are consistent with electrophysiological data showing that incubation of cue-induced cocaine seeking is accompanied by an increase in the portion of NAc neurons exhibiting phasic activation in response to cocaine self-administration or cocaine-associated cues, as well as an increase in the strength of neuronal activation (Hollander and Carelli 2005, 2007). In addition to enabling stronger depolarization of NAc neurons due to their higher conductance, the synaptic incorporation of GluA2-lacking AMPAR after prolonged withdrawal from cocaine self-administration would be predicted to qualitatively alter synaptic transmission and subsequent plasticity at NAc synapses due to their ability to pass Ca2+ ions and thus activate Ca2+-dependent signaling pathways. It should be noted that the full circuitry responsible for the expression of incubated cocaine craving has not been worked out. We have focused in this review on NAc, but prior work has established a critical role for excitatory transmission in the central nucleus of the amygdala (Lu et al. 2005, 2007) and the ventral medial PFC (Koya et al. 2009a).
Results in the incubation model show that AMPAR transmission in the NAc is enhanced after withdrawal from extended access cocaine self-administration. What about limited access cocaine self-administration (e.g., 2 h/day)? Although AMPAR transmission in the NAc is required for cocaine seeking after such regimens (e.g., Cornish and Kalivas 2000), cell surface or synaptic AMPAR levels have not been measured. It will be important to determine if AMPAR upregulation occurs and, if so, what type of AMPAR is involved. Interestingly, withdrawal from limited access cocaine self-administration is associated with potentiation of extracellular field potentials in the NA core evoked by in vivo stimulation of the PFC, which could indicate AMPAR upregulation (Moussawi et al. 2009). Furthermore, there is evidence that a rapid increase in NAc cell surface levels of GluA1-containing AMPAR may contribute to cocaine-induced reinstatement of drug seeking following limited access cocaine self-administration and extinction training; D1 receptors, l-type Ca2+ channels, and CaMKII activation were implicated in this effect (Anderson et al. 2008). The latter results could be related to our demonstration that D1 receptor stimulation facilitates AMPAR synaptic insertion in cultured NAc neurons (Sun et al. 2008; see “D1-Class DA Receptors Prime AMPAR for Synaptic Insertion”).
Another important challenge is to understand the relationship between AMPAR plasticity and observations of altered LTP or LTD at NAc synapses after cocaine withdrawal (Thomas et al. 2001; Yao et al. 2004; Goto and Grace 2005; Martin et al. 2006; Mameli et al. 2009; Moussawi et al. 2009). Overall, these studies indicate that repeated cocaine exposure impairs normal plasticity mechanisms in the NAc. However, some divergent results have been reported, probably because these studies differ with respect to whether drug exposure is contingent or non-contingent, whether extinction training has occurred, and the duration of withdrawal (see Wolf and Ferrario 2010, for more discussion).
Global Versus Ensemble-Specific Neuroadaptations
The upregulation of NAc AMPAR after sensitization and incubation is likely to occur in many or all NAc neurons, based on the ability to detect changes in tissue homogenates (e.g., Boudreau and Wolf 2005; Conrad et al. 2008). Furthermore, in our electrophysiological studies in the incubation model, we detected inwardly rectifying AMPAR EPSC in all MSN recorded from cocaine-experienced rats after 30–47 days of withdrawal (Conrad et al. 2008 and unpublished findings of Kuei-Yuan Tseng, Marina Wolf, and Carrie Ferrario). Some of the other cocaine-induced neuroadaptations that influence MSN output are also likely to affect a large number of MSN, such as decreased extracellular glutamate levels (Kalivas 2009) and decreased intrinsic excitability of MSN (Zhang et al. 1998, 2002; Hu et al. 2004, 2005; Dong et al. 2006; Ishikawa et al. 2009; Kourrich and Thomas 2009; Mu et al. 2010). In contrast, many results demonstrate the importance of functional ensembles of NAc neurons in mediating responses to natural rewards and drugs (e.g., Peoples et al. 2007; Koya et al. 2009b; Wheeler and Carelli 2009). This is probably due in large part to selective activation or modulation of MSN by various presynaptic inputs. For example, DA is known to gate the activation of MSN by excitatory inputs (Nicola et al. 2000; O’Donnell 2003; Wheeler and Carelli 2009). Perhaps DA and other inputs select the population of MSN that is activated in a particular behavioral context (e.g., Owesson-White et al. 2009), and then global adaptations such as AMPAR upregulation influence the strength of activation. A challenge for future studies is to understand the relationship between presynaptic and postsynaptic changes over the course of cocaine withdrawal.
Conclusions
It is now well established that drugs of abuse produce both transient and long-lasting changes in excitatory transmission in brain circuits that control motivation and reward. This review focused on a few examples involving cocaine and AMPAR upregulation, but the number of mechanistically distinct forms of drug-induced plasticity suggested by recent studies is impressive. The challenge of relating drug-induced synaptic changes to addiction-related behavioral changes is daunting, but progress is being made. Indeed, due in part to its strong animal models and relatively well characterized underlying circuitry, addiction research is emerging as a major front in ongoing efforts to characterize the cellular mechanisms that link synaptic and behavioral plasticity.
References
Abraham WC, Williams JM (2008) LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem 89:260–268
Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC (2008) CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci 11:344–353
Argilli E, Sibley DR, Malenka RC, England PM, Bonci A (2008) Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci 28:9092–9100
Bachtell RK, Self DW (2008) Renewed cocaine exposure produces transient alterations in nucleus accumbens AMPA receptor-mediated behavior. J Neurosci 28:12808–12814
Bachtell RK, Choi KH, Simmons DL, Falcon E, Monteggia LM, Neve RL, Self DW (2008) Role of GluR1 expression in nucleus accumbens neurons in cocaine sensitization and cocaine-seeking behavior. Eur J Neurosci 27:2229–2240
Bäckstrom P, Hyytiä P (2007) Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 192:571–580
Bell K, Kalivas PW (1996) Context-specific cross-sensitization between systemic cocaine and intra-accumbens AMPA infusion in the rat. Psychopharmacology (Berl) 127:377–383
Bell K, Duffy P, Kalivas PW (2000) Context-specific enhancement of glutamate transmission by cocaine. Neuropsychopharmacology 23:335–344
Bellone C, Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641
Berke JD, Hyman SE (2000) Addiction, dopamine and the molecular mechanisms of memory. Neuron 26:515–532
Bernard V, Somogyi P, Bolam JP (1997) Cellular, subcellular, and subsynaptic distribution of AMPA-type glutamate receptor subunits in the neostriatum of the rat. J Neurosci 17:819–833
Boudreau AC, Wolf ME (2005) Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25:9144–9151
Boudreau AC, Reimers JM, Milovanovic M, Wolf ME (2007) Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci 27:10621–10635
Boudreau AC, Ferrario CR, Glucksman MJ, Wolf ME (2009) Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J Neurochem 110:363–377
Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT (2005) Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310:1340–1343
Campioni MR, Xu M, McGehee DS (2009) Stress-induced changes in nucleus accumbens glutamate synaptic plasticity. J Neurophysiol 101:3192–3198
Carroll RC, Beattie EC, von Zastrow M, Malenka RC (2001) Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci 2:315–324
Cepeda C, Levine MS (2006) Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE 333:p320
Chao SZ, Ariano MA, Peterson DA, Wolf ME (2002a) D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem 83:704–712
Chao SZ, Lu W, Lee HK, Huganir RL, Wolf ME (2002b) D1 dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J Neurochem 81:984–992
Churchill L, Swanson CJ, Urbina M, Kalivas PW (1999) Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem 72:2397–2403
Collingridge GL, Olsen RW, Peters J, Spedding M (2009) A nomenclature for ligand-gated ion channels. Neuropharmacology 56:2–5
Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454:118–121
Cornish JL, Kalivas PW (2000) Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci 20:RC89
Cornish JL, Duffy P, Kalivas PW (1999) A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience 93:1359–1367
Cull-Candy S, Kelly L, Farrant M (2006) Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol 16:288–297
Derkach VA, Oh MC, Guire ES, Soderling TR (2007) Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci 8:101–113
Di Chiara G (1998) A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol 12:54–67
Di Ciano P, Everitt BJ (2001) Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology 25:341–360
Di Ciano P, Everitt BJ (2004) Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J Neurosci 24:7167–7173
Di Ciano P, Benham-Hermetz J, Fogg AP, Osborne GE (2007) Role of the prelimbic cortex in the acquisition, re-acquisition or persistence of responding for a drug-paired conditioned reinforcer. Neuroscience 150:291–298
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61
Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC (2006) CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci 9:475–477
Duffy SN, Nguyen PV (2003) Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci 23:1142–1150
Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6:136–143
Everitt BJ, Robbins TW (2005) Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8:1481–1489
Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC (2008) Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J Neurosci 28:11061–11070
Ferrario CR, Li X, Wang X, Reimers JM, Uejima JL, Wolf ME (2010) The role of glutamate receptor redistribution in locomotor sensitization to cocaine. Neuropsychopharmacology 35:818–833
Frey U, Schroeder H, Matthies H (1990) Dopaminergic antagonists prevent long-term maintenance of posttetanic LTP in the CA1 region of rat hippocampal slices. Brain Res 522:69–75
Frey U, Matthies H, Reymann KG (1991) The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro. Neurosci Lett 129:111–114
Frey U, Huang YY, Kandel ER (1993) Effects of cAMP stimulate a late stage of LTP in hippocampal CA1 neurons. Science 260:1661–1664
Gao C, Wolf ME (2007) Dopamine alters AMPA receptor synaptic expression and subunit composition in dopamine neurons of the ventral tegmental area cultured with prefrontal cortex neurons. J Neurosci 27:14275–14285
Gao C, Sun X, Wolf ME (2006) Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem 98:1664–1677
Ghasemzadeh MB, Mueller C, Vasudevan P (2009a) Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience 159:414–426
Ghasemzadeh MB, Vasudevan P, Mueller C, Seubert C, Mantsch JR (2009b) Region specific alterations in glutamate receptor expression and subcellular distribution following extinction of cocaine self-administration. Brain Res. Feb 4 Epub
Gold SJ, Ambros-Ingerson J, Horowitz JR, Lynch G, Gall CM (1997) Stoichiometries of AMPA receptor subunit mRNAs in rat brain fall into discrete categories. J Comp Neurol 385:491–502
Goldstein RZ, Volkow ND (2002) Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159:1642–1652
Goto Y, Grace AA (2005) Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron 47:255–266
Goto Y, Grace AA (2008) Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci 31:552–558
Greger IH, Esteban JA (2007) AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol 17:289–297
Greger IH, Khatri L, Kong X, Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40:763–774
Grimm JW, Hope BT, Wise RA, Shaham Y (2001) Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature 412:141–142
Groc L, Choquet D (2006) AMPA and NMDA glutamate receptor trafficking: multiple roads for reaching and leaving the synapse. Cell Tissue Res 326:423–438
Groenewegen HJ, Wright CI, Beijer AV, Voorn P (1999) Convergence and segregation of ventral striatal inputs and outputs. Ann NY Acad Sci 877:49–63
Guire ES, Oh MC, Soderling TR, Derkach VA (2008) Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci 28:6000–6009
Harvey J, Lacey MG (1996) Endogenous and exogenous dopamine depress EPSCs in rat nucleus accumbens in vitro via D1 receptor activation. J Physiol 492(1):143–154
Hayes RJ, Vorel SR, Spector J, Liu X, Gardner EL (2003) Electrical and chemical stimulation of the basolateral complex of the amygdala reinstates cocaine-seeking behavior in the rat. Psychopharmacology (Berl) 168:75–83
Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ (2007) Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci 27:11651–11662
Hollander JA, Carelli RM (2005) Abstinence from cocaine self-administration heightens neural encoding of goal-directed behaviors in the accumbens. Neuropsychopharmacology 30:1464–1474
Hollander JA, Carelli RM (2007) Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci 27:3535–3539
Hu XT, White FJ (1996) Glutamate receptor regulation of rat nucleus accumbens neurons in vivo. Synapse 23:208–218
Hu XT, Basu S, White FJ (2004) Repeated cocaine administration suppresses HVA-Ca2+ potentials and enhances activity of K+ channels in rat nucleus accumbens neurons. J Neurophysiol 92:1597–1607
Hu XT, Ford K, White FJ (2005) Repeated cocaine administration decreases calcineurin (PP2B) but enhances DARPP-32 modulation of sodium currents in rat nucleus accumbens neurons. Neuropsychopharmacology 30:916–926
Hu H, Real E, Takamiya K, Kang M-G, Ledoux J, Huganir RL, Malinow R (2007) Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131:160–173
Huang YY, Kandel ER (1995) D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci USA 92:2446–2450
Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schlüter OM, Zukin RS, Dong Y (2009) In vivo cocaine experience generates silent synapses. Neuron 63:40–47
Isaac JT, Ashby M, McBain CJ (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54:859–871
Ishikawa M, Mu P, Moyer JT, Wolf JA, Quock RM, Davies NM, Hu XT, Schlüter OM, Dong Y (2009) Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci 29:5820–5831
Ito R, Robbins TW, Everitt BJ (2004) Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nat Neurosci 7:389–397
Jay TM (2003) Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol 69:375–390
Jedynak JP, Uslaner JM, Esteban JA, Robinson TE (2007) Methamphetamine-induced structural plasticity in the dorsal striatum. Eur J Neurosci 25:847–853
Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572
Kalivas PW, Volkow ND (2005) The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162:1403–1413
Kauer JA, Malenka RC (2007) Synaptic plasticity and addiction. Nat Rev Neurosci 8:844–858
Kelley AE (1999) Functional specificity of ventral striatal compartments in appetitive behaviors. Ann NY Acad Sci 877:71–90
Kelley AE (2004) Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev 27:765–776
Kelley AE, Berridge KC (2002) The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci 22:3306–3311
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61:340–350
Kourrich S, Thomas MJ (2009) Similar neurons, opposite adaptations: psychostimulant experience differentially alters firing properties in accumbens core versus shell. J Neurosci 29:12275–12283
Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007) Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27:7921–7928
Koya E, Uejima JL, Wihbey KA, Bossert JM, Hope BT, Shaham Y (2009a) Role of ventral medial prefrontal cortex in incubation of cocaine craving. Neuropharmacology 56(Suppl. 1):177–185
Koya E, Golden SA, Harvey BK, Guez-Barber DH, Berkow A, Simmons DE, Bossert JM, Nair SG, Uejima JL, Marin MT, Mitchell TB, Farquhar D, Ghosh SC, Mattson BJ, Hope BT (2009b) Targeted disruption of cocaine-activated nucleus accumbens neurons prevents context-specific sensitization. Nat Neurosci 12:1069–1073
Krugers HJ, Hoogenraad CC (2009) Hormonal regulation of AMPA receptor trafficking and memory formation. Front Synap Neurosci 1:Article 2
Kruzich PJ, Xi J (2006) Different patterns of pharmacological reinstatement of cocaine-seeking behavior between Fischer 344 and Lewis rats. Psychopharmacology (Berl) 187:22–29
Li S, Cullen WK, Anwyl R, Rowan MJ (2003) Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci 6:526–531
Lin D-T, Makino Y, Sharma K, Kayashi T, Neve R, Takamiya K, Huganir RL (2009) Regulation of AMPA receptor extrasynaptic insertion by 4.1 N, phosphorylation and palmitoylation. Nat Neurosci 12:879–887
Lisman JE, Grace AA (2006) The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron 46:703–713
Lisman JE, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190
Liu SQ, Cull-Candy SG (2000) Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405:454–458
Liu SJ, Zukin RS (2007) Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci 30:126–134
Loweth JA, Singer BF, Baker LK, Wilke G, Inamine H, Bubula N, Alexander JK, Carlezon WA Jr, Neve RL, Vezina P (2010) Transient overexpression of α-CaMKII in the nucleus accumbens shell enhances behavioral responding to amphetamine. J Neurosci 30:939–949
Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254
Lu L, Grimm JW, Dempsey J, Shaham Y (2004a) Cocaine seeking over extended withdrawal periods in rats: different time courses of responding induced by cocaine cues versus cocaine priming over the first 6 months. Psychopharmacology (Berl) 176:101–108
Lu L, Grimm JW, Hope BT, Shaham Y (2004b) Incubation of cocaine craving after withdrawal: a review of preclinical data. Neuropharmacology 47(Suppl 1):214–226
Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y (2005) Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci 8:212–219
Lu L, Uejima JL, Gray SM, Bossert JM, Shaham Y (2007) Systemic and central amygdala injections of the mGluR(2/3) agonist LY379268 attenuate the expression of incubation of cocaine craving. Biol Psychiatry 61:591–598
Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62:254–268
Malinow R (2003) AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc B 358:707–714
Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126
Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C (2009) Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci 12:1036–1041
Man HY, Sekine-Aizawa Y, Huganir RL (2007) Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci USA 104:3579–3584
Mangiavacchi S, Wolf ME (2004a) D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J Neurochem 88:1261–1271
Mangiavacchi S, Wolf ME (2004b) Stimulation of N-methyl-D-aspartate receptors, AMPA receptors or metabotropic glutamate receptors leads to rapid internalization of AMPA receptors in cultured nucleus accumbens neurons. Eur J Neurosci 20:649–657
Mano I, Teichberg VI (1998) A tetrameric subunit stoichiometry for a glutamate receptor-channel complex. NeuroReport 9:327–331
Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A (2006) Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci 9:868–869
Matthies H, Becker A, Schroeder H, Kraus J, Höllt V, Krug M (1997) Dopamine D1-deficient mutant mice do not express the late phase of hippocampal long-term potentiation. Neuroreport 8:3533–3535
McCutcheon JE, Marinelli M (2009) Age matters. Eur J Neurosci 29:997–1014
McFarland K, Lapish CC, Kalivas PW (2003) Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23:3531–3537
Mead AN, Zamanillo D, Becker N, Stephens DN (2007) AMPA-receptor GluR1 subunits are involved in the control over behavior by cocaine-paired cues. Neuropsychopharmacology 32:343–353
Meredith GE, Totterdell S (1999) Microcircuits in nucleus accumbens’ shell and core involved in cognition and reward. Psychobiology 27:165–186
Meredith GE, Baldo BA, Andrezjewski ME, Kelley AE (2008) The structural basis for mapping behavior onto the striatum and its subdivisions. Brain Struct Funct 213:17–27
Milstein AD, Nicoll RA (2008) Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci 29:333–339
Mogenson GJ (1987) Limbic-motor integration. Prog Psychobiol Physiol Psychol 12:117–170
Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O’Carroll C (2003) Elements of a neurobiological theory of the hippocampus; the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc B 358:773–786
Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW (2009) N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci 12:182–189
Mu P, Moyer J, Ishikawa M, Zhang Y, Panksepp J, Sorg BA, Schlüter OM, Dong Y (2010) Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J Neurosci 30:3689–3699
Neisewander JL, Baker DA, Fuchs RA, Tran-Nguyen LT, Palmer A, Marshall JF (2000) Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J Neurosci 20:798–805
Nelson CL, Milovanovic M, Wetter JB, Ford KA, Wolf ME (2009) Behavioral sensitization to amphetamine is not accompanied by changes in glutamate receptor surface expression in the rat nucleus accumbens. J Neurochem 109:35–51
Nicola SM, Kombian SB, Malenka RC (1996) Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci 16:1591–1604
Nicola SM, Surmeier DJ, Malenka RC (2000) Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci 23:185–215
O’Donnell P (2003) Dopamine gating of forebrain neural ensembles. Eur J Neurosci 17:429–435
Oh MC, Derkach VA, Guire ES, Soderling TR (2006) Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281:752–758
Otmakhova NA, Lisman JE (1996) D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci 16:7478–7486
Owesson-White CA, Ariansen J, Stuber GD, Cleaveland NA, Cheer JF, Wightman RM, Carelli RM (2009) Neural encoding of cocaine-seeking behavior is coincident with phasic dopamine release in the accumbens core and shell. Eur J Neurosci 30:1117–1127
Palmer CL, Cotton L, Henley JM (2005) The molecular pharmacology and cell biology of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Pharmacol Rev 57:253–277
Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, Pierce RC (2002) Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci 22:2916–2925
Passafaro M, Piech V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4:917–926
Patrick SL, Thompson TL, Walker JM, Patrick RL (1991) Concomitant sensitization of amphetamine-induced behavioral stimulation and in vivo dopamine release from rat caudate nucleus. Brain Res 538:343–346
Pennartz CM, Dolleman-Van der Weel MJ, Kitai ST, Lopes da Silva FH (1992) Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studied in vitro. J Neurophysiol 67:1325–1334
Pennartz CM, Groenewegen HJ, Lopes da Silva FH (1994) The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol 42:719–761
Peoples LL, Kravitz AV, Guillem K (2007) The role of accumbal hypoactivity in cocaine addiction. ScientificWorldJournal 7:22–45
Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D (2009) Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63:92–105
Pi HJ, Lisman JE (2008) Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J Neurosci 28:13132–13138
Pierce RC, Bell K, Duffy P, Kalivas PW (1996) Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16:1550–1560
Ping A, Xi J, Prasad BM, Wang MH, Kruzich PJ (2008) Contributions of nucleus accumbens core and shell GluR1 containing AMPA receptors in AMPA- and cocaine-primed reinstatement of cocaine-seeking behavior. Brain Res 1215:173–182
Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JR (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci 9:602–604
Porrino LJ, Smith HR, Nader MA, Beveridge TJ (2007) The effects of cocaine: a shifting target over the course of addiction. Prog Neuropsychopharmacol Biol Psychiatry 31:1593–1600
Reimers JM, Milovanovic M, Wolf ME (2007) Quantitative analysis of AMPA receptor subunit composition in addiction-related brain regions. Soc Neurosci Abstr 33:815.19
Robinson TE, Berridge KC (2008) Review. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc Lond B 363:3137–3146
Robinson TE, Kolb B (2004) Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 47(Suppl 1):33–46
Rosenmund C, Stern-Bach Y, Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280:1596–1599
Sari Y, Smith KD, Ali PK, Rebec GV (2009) Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci 29:9239–9243
Scheggi S, Leggio B, Masi F, Grappi S, Gambarana C, Nanni G, Rauggi R, De Montis MG (2002) Selective modifications in the nucleus accumbens of dopamine synaptic transmission in rats exposed to chronic stress. J Neurochem 83:895–903
Schultz W (2000) Multiple reward signals in the brain. Nat Rev Neurosci 1:199–207
Schumann J, Yaka R (2009) Prolonged withdrawal from repeated noncontingent cocaine exposure increases NMDA receptor expression and ERK activity in the nucleus accumbens. J Neurosci 29:6955–6963
Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klöcker N (2009) Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 323:1313–1319
Seamans JK, Yang CR (2004) The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol 74:1–58
Self DW, Choi KH, Simmons D, Walker JR, Smagula CS (2004) Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn Mem 11:648–657
Sesack SR, Carr DB, Omelchenko N, Pinto A (2003) Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann NY Acad Sci 1003:36–52
Shepherd JD, Huganir RL (2007) The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol 23:613–643
Smith WB, Starck SR, Roberts RW, Schuman EM (2005) Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron 45:765–769
Soel GH, Ziburkus J, Huang SY, Song L, Kim IT, Takamiya K, Huganir RL, Lee H-K, Kirkwood A (2009) Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron 55:919–929
Song I, Huganir RL (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25:578–588
Sorge RE, Stewart J (2005) The contribution of drug history and time since termination of drug taking to footshock stress-induced cocaine seeking in rats. Psychopharmacology (Berl) 183:210–217
Stefani A, Chen Q, Flores-Hernandez J, Jiao Y, Reiner A, Surmeier DJ (1998) Physiological and molecular properties of AMPA/Kainate receptors expressed by striatal medium spiny neurons. Dev Neurosci 20:242–252
Sun X, Wolf ME (2009) Nucleus accumbens neurons exhibit synaptic scaling that is occluded by repeated dopamine pre-exposure. Eur J Neurosci 30:539–550
Sun X, Zhao Y, Wolf ME (2005) Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25:7342–7351
Sun X, Milovanovic M, Zhao Y, Wolf ME (2008) Acute and chronic dopamine receptor stimulation modulates AMPA receptor trafficking in nucleus accumbens neurons cocultured with prefrontal cortex neurons. J Neurosci 28:4216–4230
Suto N, Tanabe LM, Austin JD, Creekmore E, Pham CT, Vezina P (2004) Previous exposure to psychostimulants enhances the reinstatement of cocaine seeking by nucleus accumbens AMPA. Neuropsychopharmacology 29:2149–2159
Suto N, Ecke LE, Wise RA (2009) Control of within-binge cocaine-seeking by dopamine and glutamate in the core of nucleus accumbens. Psychopharmacology (Berl) 205:431–439
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D, Karanian DA, Monteggia LM, Neve RL, Self DW (2003) Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421:70–75
Swanson-Park JL, Coussens CM, Mason-Parker SE, Raymond CR, Hargreaves EL, Dragunow M, Cohen AS, Abraham WC (1999) A double dissociation within the hippocampus of dopamine D1/D5 receptor and β-adrenergic receptor contributions to the persistence of long-term potentiation. Neuroscience 92:485–497
Thiagarajan TC, Lindskog M, Malgaroli A, Tsien RW (2007) LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology 52:156–175
Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4:1217–1223
Thomas MJ, Kalivas PW, Shaham Y (2008) Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol 154:327–342
Tigaret C, Choquet D (2009) More AMPAR garnish. Science 323:1295–1296
Turrigiano GG (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435
Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5:97–107
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411:583–587
Vezina P (2004) Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci Biobehav Rev 27:827–839
Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL (2001) Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292:1175–1178
Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16:1982–1989
Wheeler RA, Carelli RM (2009) Dissecting motivational circuitry to understand substance abuse. Neuropharmacology 56(Suppl 1):149–159
Wolf ME, Ferrario CR (2010) AMPA receptor plasticity after repeated cocaine exposure. Neurosci Biobehav Rev. Epub Jan 28
Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004) Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47(S1):61–79
Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG (2004) Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41:625–638
Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, von Zastrow M (2007) Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J Neurosci 27:11112–111121
Zhang XF, Hu XT, White FJ (1998) Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18:488–498
Zhang XF, Cooper DC, White FJ (2002) Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J Pharmacol Exp Ther 301:1119–1125
Acknowledgments
I thank members of my laboratory who contributed to the ideas presented in this article, including Drs. Amy Boudreau, Kelly Conrad, Carrie Ferrario, Can Gao, Christopher Nelson, and Xiu Sun. Our research is supported by USPHS grants DA009621, DA015835, and DA000453.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wolf, M.E. Regulation of AMPA Receptor Trafficking in the Nucleus Accumbens by Dopamine and Cocaine. Neurotox Res 18, 393–409 (2010). https://doi.org/10.1007/s12640-010-9176-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-010-9176-0