
Abstract
Leishmaniases are a complex of diseases that range from the deadly visceral disease and some self-curing lesions to gross disfigurations. About 12 million peoples from 88 different countries get infected by this protozoan parasite through the sand flies. Visceral leishmaniasis is a potentially fatal disease endemic to large parts of Asia and Africa, primarily caused by the protozoan parasite Leishmania donovani. L. donovani is a species of Leishmania, a hemoflagellate parasite and causative agent of visceral leishmaniasis. Leishmanolysin is the major surface protein of the parasitic Leishmania. Leishmanolysin has been described as a parasite virulence factor and is involved in the direct interaction of promastigotes and host macrophage receptors and interaction with the complement cascade. In the current study we predicted the 3D structure of leishmanolysin using homology modeling as 3D structure prediction approach. Leishmanolysin is a stable extracellular stable protein of 561 amino acid residues. 3D structure of the leishmanolysin was determined using Protein Structure Prediction Server (PS2 Server) selecting MODELLER as 3D structure prediction method. Quality analysis of the model through its Ramchandran Plot and ERRAT value (94.25) indicated that it is a reliable model. Functional annotation showed that this protein is a member of the superfamily cl18220. The information thus discussed provides insight to the molecular understanding of structure and function of leishmanolysin from L. donovani. The predicted 3-D model may be further used in characterizing the protein in wet laboratory.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the disease tropism the parasites of genus Leishmania are remarkably, clinically and epidemiologically diverse. Leishmania donovini species of parasite which most significantly causes visceral leishmaniasis, the complex disease that can spread to the internal organs and causes even death (World Health Organization 2010).
In many developing countries diseases caused by the protozoan parasite Leishmania are very common. Leishmania the kinetoplastid protozoan parasites cause diseases for which no effective vaccine exists and treatment is also very difficult (Desjeux 1992; WHO 1990).
The promastigotes injected during the feeding of infected sand flies parasitize macrophages. After infecting they show the various symptoms ranging in severity from self-healing sores, through severely disfiguring mucosal lesions, to a fatal systemic infection involving spleen and bone marrow (kalaazar).
Leishmanolysin is the abundant surface glycoprotein which secreted by Promastigotes of all Leishmania (Etges 1992). Leishmanolysin acts as a ligand involved in the interaction of the parasite with defensive systems of the host, along with the components of the complement system and the macrophage surface (Russell and Wilhelm 1986; Puentes et al. 1989; Connell et al. 1993). For the surface antigens of infectious protozoa despite their medical importance, there is very little structural information available, therefore leishmanolysin is an attractive vaccine candidate (Yu et al. 2006).
Methodology
Protein retrieval and sequence analysis
The protein sequence of leishmanolysin protein was retrieved from Uniprot Knowledgebase database using accession No. P23223. Physiochemical properties of the protein were computed by ProtParam tool (http://web.expasy.org/protparam/). The parameters computed by ProtParam included the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). Subcellular localization of any protein is important in understanding protein function. Prediction of subcellular localization of protein was carried out by CELLO v.2.5 (Yu et al. 2004; Rost et al. 2004).
Secondary structure prediction
PredictProtein (Yu et al. 2004) was employed for computing and analyzing the secondary structural features of leishmanolysin from L. donovani protein sequence.
3D structure prediction using homology approach
3D structure of protein was determined by homology modeling. BLASTP search with default parameters against the protein data bank (PDB) was used to find the best suitable templates for homology modeling. Based on maximum identity and lowest e-value, best suitable template with PDB ID 1LML_A having 80 % identity was selected. This template was used as a reference to determine the 3D structure of leishmanolysin protein from L. donovani. Protein structure prediction server (PS2) (Chen et al. 2006) predicted the homology model based on package MODELLER.
Quality and reliability assessments
Once the 3D model was generated, energy minimization was performed by GROMOS96 force field in a Swiss-PdbViewer. Structural evaluation and stereo-chemical analyses were performed using ProSA-web (Wiederstein and Sippl 2007; Sippl 1993), Z-scores and Procheck Ramachandran plot.
Function annotations of the protein
To functionally annotate leishmanolysin from L. donovani the Profunc was used and to find the conserved domains in protein to identify its family, it was searched against close orthologous family members. NCBI Conserved Domain Database (NCBI CDD) (Marchler-Bauer and Bryant 2004) was used to find the conserved domains or ancient domains in the protein sequence.
Results and discussion
The present study was to perform sequence and structure analysis of leishmanolysin from L. donovani. The protein sequence was retrieved using accession No. P23223 uniprot database.
Protein sequence analysis
ProtParam was used to find out the physiochemical properties from protein sequence. The protein was predicted to have 561 amino acids, with molecular weight of 60,471.4 Daltons and theoretical isoelectric point (PI) of 5.76. The instability index (II) is computed to be 39.28. This classifies the protein as stable. The N-terminal of the sequence considered is M (Met). The estimated half-life is: 30 h (mammalian reticulocytes, in vitro), >20 h (yeast, in vivo), and >0 h (Escherichia coli, in vivo). The negative GRAVY of 0.003 indicates that the protein is hydrophilic and soluble in nature. Total number of negatively charged residues (Asp + Glu) 55 and total number of positively charged residues (Arg + Lys) 44 are present.
Cellular functions are often localized in specific compartments. Therefore, predicting the subcellular localization of unknown proteins can give information about their functions and can also help in understanding disease mechanisms and developing drugs. The subcellular localization prediction using CELLO predicted that our protein is an extracellular.
PredictProtein was used to predict the secondary structure of the protein. Results showed that protein is a mixed protein having composition of Helix = 17.65 %, Strand = 13.19 %, Loop = 69.16 %. The solvent accessibility of leishmanolysin protein shows buried 55.97 %, intermediate 22.99 %, and exposed 21.03 %.
3D structure prediction using homology modeling approach
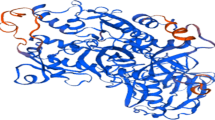
Proteins 3D structure is very important in understanding the protein interactions, functions and their localization. Homology modeling is the most common structure prediction method. To perform the homology modeling, the first and basic step is to find best matching template using similarity searching program like BLASTP against PDB database. Templates are selected on the basis of their sequence similarity with query sequence. PDB ID 1LML was selected for homology modeling which is an X-ray diffraction mode. The query sequence and template ID was then given as input to the (PS2 server) for homology modeling using MODELLER. 3D structure of protein showed that it has 544 hydrogen bonds (Fig. 1). Quality and reliability of structure was checked by several structure assessment methods including Z-score and Ramachandran plot. The Z-score is indicative of overall model quality and is used to check whether the input structure is within the range of scores typically found for native proteins of similar size (Fig. 2).

Predicted 3D structure of leishmanolysin from Leishmania donovani

Z-score of query protein using PROSA web
PROSAweb was used to find the Z-score of template and query. Z-score of query protein was—8.64. Procheck checks the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and overall structure geometry. This tool was used to determine the Ramachandran plot to assure the quality of the model. The result of the Ramachandran plot showed 89.9 % of residues in favorable region, 9.8 % in additional allowed regions [a, b, l, p], 0.2 % in generously allowed regions [~a, ~b,~l, ~p] and only 0.2 % in disallowed regions. 89.9 % of residues in favorable region representing that it is a reliable and good quality model. A model having 90 % residues in favorable region is considered as good quality model (Fig. 3).

Ramachandran plot of leishmanolysin from Leishmania donovani using Procheck
Reliability of the model was further checked by ERRAT that analyzes the statistics of non-bonded interactions between different atom types and plots the value of the error function versus position of a 9-residue sliding window, calculated by a comparison with statistics from highly refined structures. Results from ERRAT showed overall model quality of 94.25 value (Fig. 4). The Z-scores confirm the quality of the homology model of leishmanolysin protein.

Overall quality factor of leishmanolysin from Leishmania donovani checked by ERRAT
Function annotation of the protein
ProFunc tool was used to hypothetically annotate the function of the leishmanolysin from L. donovani,. The structure reveals three domains, two of which have novel folds. The N-terminal domain has a similar structure to the catalytic modules of zinc proteinases. The structure clearly shows that leishmanolysin is a member of the metzincin class of zinc proteinases.
To further investigate about the function of protein by finding its family; it was searched in the NCBI Conserved Domain Database (NCBI CDD) to find conserved domains so that its family can be identified. The results showed that leishmanolysin from L. donovani is a member of the superfamily cl18220.
Conclusion
Our main objective of this study was to perform sequence analysis, structure analysis and homology modeling on leishmanolysin from L. donovani.
We have used various sequence and structure analysis tools that helped in understanding of the protein sequence and its structure. Furthermore, protein was functionally annotated by using ProFunc and by searching conserved domains of the protein. As a part of present study, we used homology modeling approach to propose the first 3D structure of the leishmanolysin from L. donovani.
The predicted 3D structure will provide more insight in understanding the structure and function of the protein. The results from the present study can be used as basis for drug designing as well as for understanding the protein–protein interactions and interactions with the prospective therapeutic agents.
References
Chen CC, Hwang JK, Yang JM (2006) (PS)2: protein structure prediction server. Nucleic Acids Res 34:W152–W157
Connell ND, Medina-Acosta E, McMaster WR, Bloom BR, Russell DG (1993) Effective immunization against cutaneous leishmaniasis with recombinant bacille Calmette–Guerin expressing the Leishmania surface proteinase gp63. Proc Natl Acad Sci USA 90:11473–11477
Desjeux P (1992) Human leishmaniases: epidemiology and publichealth aspects. World Health Stat Q 45:267–275
Etges R (1992) Identification of a surface metalloproteinase on 13 species of Leishmania isolated from humans, Crithidia fasciculata, and Herpetomonas samuelpessoai. Acta Tropica 50:205–217
Marchler-Bauer A, Bryant SH (2004) CD-search: protein domain annotations on the fly. Nucleic Acids Res 32:327–331 (Web Server issue)
Puentes SM, Dwyer DM, Bates PA, Joiner KA (1989) Bindingand release of C3 from Leishmania donovani promastigotes during incubation in normal human serum. J Immunol 143:3743–3749
Rost B, Yachdav G, Liu J (2004) The predictprotein server. Nucleic Acids Res 32:W321–W326 (Web Server issue)
Russell DG, Wilhelm H (1986) The involvement of the majorsurface glycoprotein (gp63) of Leishmania promastigotes in attachment to macrophages. J Immunol 136:2613–2621
Sippl MJ (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17:355–362
WHO (1990) Control of the leishmaniases: report of a WHO expert committee. WHO Technical Report Series 793:1–158
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35:W407–W410
World Health Organization (2010) Control of the leishmaniases: report of a meeting of the WHO expert committee on the control of leishmaniasis, Geneva, 22–26 March 2010., WHO Technical Report Series, no. 949WHO, Geneva
Yu CS, Chen YC, Lu CH, Hwang JK (2004) Prediction of protein subcellular localization. Proteins: Struct Funct Bioinf 64:643–651
Yu CS, Lin CJ, Hwang JK (2006) Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci 13:1402–1406
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Waghmare, S., Buxi, A., Nandurkar, Y. et al. In silico sequence analysis, homology modeling and function annotation of leishmanolysin from Leishmania donovani . J Parasit Dis 40, 1266–1269 (2016). https://doi.org/10.1007/s12639-015-0665-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12639-015-0665-1