
Abstract
Platinum–cobalt (Pt–Co) alloy electrocatalysts were prepared on pretreated carbon cloth by electrodeposition from different plating baths with the aim of finding suitable plating bath conditions that could control the chemical composition of the Pt–Co alloy electrodeposits over a wide range (from 0 to 100 at% Pt) and then to study the relationship of the deposit composition on the oxygen reduction reaction (ORR) electroactivity. The type of supporting electrolyte, the solution pH, Pt and Co concentrations of the plating bath and the current density used for electrodeposition all play a crucial role in the composition of the electrodeposited Pt–Co alloy. Pt contents in all the Pt–Co alloys deposited in H2SO4 are at a relatively high and narrow level (83 at%–97 at% Pt), regardless of the electrodeposition conditions used. While the Pt contents in the Pt–Co alloys deposited in Na2SO4 could reach a very low and broad level (5 at%–97 at% Pt), evidence of Co oxide formation is observed. Of the three supporting electrolytes studied, only NaCl effectively produces Pt–Co deposits over a wide range of compositions (8–90 at% Pt) by controlling the Pt and Co concentrations of the plating bath and using the high enough current density. The results show a qualitative correlation between Pt–Co deposit composition and ORR activity, where Pt–Co alloy catalysts with ≥ 45 at% Pt tend to yield higher ORR activities than the pure Pt catalyst, with the best ORR performance obtained from the Pt–Co alloy catalyst with 78 at% Pt.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Proton exchange membrane fuel cells (PEMFCs) are attractive energy conversion devices for a wide range of applications ranging from small portable electronic devices to electric vehicles or even small stationary power generators [1,2,3]. However, due to their low operating temperature, the kinetics of the electrode reactions are very slow, resulting in a low current output and so a low fuel cell performance [4, 5]. As a consequence, expensive noble metals, such as platinum (Pt), are required for the electrocatalysis to enhance the power of the PEMFCs [6]. However, with a Pt catalyst, the oxygen reduction reaction (ORR) is several orders of magnitude slower than that of the hydrogen oxidation reaction, and so the whole electrochemical reaction process of the PEMFCs is controlled by the ORR kinetics [7]. One possible approach to overcome this problem is alloying Pt with other transition metals to enhance the electroactivity toward the ORR and/or to reduce the amount of Pt needed for fabricating the electrodes of the PEMFCs [8,9,10]. Among Pt-based alloys, Pt–Co is widely considered as one of the promising candidates as an ORR electrocatalyst due to its higher electrocatalytic activity toward the ORR compared to pure Pt or other Pt-based alloys [11,12,13,14] and its long-term stability under fuel cell operating conditions [15].
The Pt-based alloy catalysts for PEMFCs are normally prepared through a multi-step process. The typical approach is that the alloying metal is impregnated in the Pt, which is supported on a high surface area carbon, before undergoing the high-temperature alloying treatment [15,16,17,18,19,20]. The prepared Pt-based alloy catalyst is then applied on the electrode by painting or spraying. Alternatively, the preparation of Pt-based alloy catalysts can be achieved in a single step by electrodeposition, where the Pt-alloy catalyst formation and the process of applying the Pt-alloy catalyst onto the electrode take place simultaneously without the need for high-temperature treatment. Moreover, electrodeposition is relatively simple and easy to operate. It can be conducted in a galvanostatic or potentiostatic mode, where the amount of deposited metal, the metal particle size and structure and the alloy composition can easily be controlled by varying the electrodeposition parameters, such as the deposition time and magnitude of applied current density or potential [11, 21,22,23,24,25,26,27]. In the previous work, we tried to prepare Pt–Co alloys onto carbon cloths (CCs) by electrodeposition in a sulfuric acid (H2SO4) solution, aiming to broaden the range of the Pt–Co compositions deposited as much as possible (0–100 at% Pt) [28]. However, the Pt compositions of all the Pt–Co alloys obtained under the studied conditions were confined between 91 at% and 94 at% Pt, regardless of the deposition mode and the magnitude of the cathodic and anodic current densities applied.
In addition to the electrodeposition parameters, the plating bath composition and its interaction with the electrodeposition parameters can also influence the physical and chemical properties of the metal alloys deposited, which can then affect the catalytic activity when used as the electrocatalyst. A few works have reported on the manipulation of the electrodeposited Pt–Co alloy composition by controlling the concentrations of Pt and/or Co present in the plating bath [12], but the effects of other parameters, such as the solution pH and type of the supporting electrolyte, on the properties of the electrodeposited Pt–Co alloy have rarely been reported and are somewhat unclear. Normally, Pt electrodeposition has been conducted in a plating bath using H2SO4 as the supporting electrolyte [25, 27, 29,30,31,32,33,34,35,36,37,38], while H2SO4 [39,40,41,42,43,44], sodium sulfate (Na2SO4) [45] and sodium chloride (NaCl) [11, 12, 46,47,48] solutions were used for Pt-based alloy electrodeposition. To produce Pt–Co alloy catalysts by electrodeposition that have a good catalytic activity, or even better, than the pure Pt, the influences of the electrodeposition parameters and the plating bath composition on the properties of the deposited Pt–Co, especially on the alloy composition, should be understood.
In this present work, Pt–Co electrocatalysts were deposited by direction current (DC) electrodeposition onto carbon black (CB)-supported CC in different plating bath conditions. The influence of the type of supporting electrolytes, normally used for Pt and Pt-alloys electrodeposition (H2SO4, Na2SO4 and NaCl), solution pH and the concentrations of Pt and Co ions in the plating bath on the composition of the deposited Pt–Co alloy was investigated. The Pt–Co alloys obtained from the selected electrodeposition conditions were then used to study the ORR. The focus of the present work was to find a suitable plating bath that allowed the electrodeposition of Pt–Co alloys across a full range of Pt proportions (0–100 at% Pt), in order to then ascertain the electrocatalytic activities of the different Pt–Co alloys for the ORR.
2 Experimental
2.1 Electrodeposition of Pt–Co alloy
Electrodeposition of the Pt–Co alloy on the CC was conducted in a single-compartment electrochemical cell containing 80 cm3 of 0.004–0.010 mol·L−1 H2PtCl6·6H2O (Aldrich), 0.1–0.2 mol·L−1 CoSO4·7H2O (Aldrich) and 0.5 mol·L−1 of the supporting electrolyte (H2SO4 (Lab-scan), Na2SO4 (Fluka) or NaCl (Fluka)). To study the effect of the solution pH, H2SO4 or hydrochloric acid (HCl) was added into the plating solution containing Na2SO4 or NaCl supporting electrolyte, respectively, to the desired pH. Prior to electrodeposition, 2.3 cm × 3.0 cm CC (ElectroChem) was pretreated by applying two sub-layers, a hydrophobic and a hydrophilic layer, onto the CC as previously reported [49], with total hydrophobic and hydrophobic loadings of 1.9 and 0.8 mg·cm−2, respectively. The titanium gauze was used as the counter electrode.
The DC electrodeposition was performed at a current density of 10 or 20 mA·cm−2 using a potentiostat (Autolab PG STATO 30 potentiostat, Metrohm Autolab) until achieving a total charge density of 2 C·cm−2. The plating solution was stirred with a magnetic stirrer at 600 r·min−1 during the electrodeposition process. After electrodeposition, the electrode was dried at 110°C for 2 h. For comparison, the electrodeposition of pure Pt and Co was also performed in the NaCl plating solution containing 0.010 mol·L−1 H2PtCl6·6H2O or 0.2 mol·L−1 CoSO4·7H2O, respectively. The loading level of each Pt–Co alloy catalyst electrodeposited on the electrode was estimated from the weight difference before and after electrodeposition. The morphology and composition of the electrodeposited Pt–Co catalysts were studied by scanning electron microscope (SEM, JEOL: JSM 6400) and the energy-dispersive spectroscopy (EDS) attached to the SEM, respectively. The crystalline characteristics of the electrodeposited Pt–Co catalysts were investigated by X-ray diffractometer (XRD, Bruker AXS: D8 Discover).
2.2 Electrocatalytic activity toward ORR of Pt–Co alloys
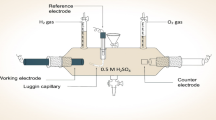
The electrocatalytic activity toward the ORR of the selected Pt–Co electrodeposits with different alloy compositions was investigated using a rotating disk electrode (RDE; Metrohm) in an O2-saturated 0.5 mol·L−1 H2SO4 solution. The working electrode was made of Pt–Co-electrodeposited CC, which was cut to a circular shape with a diameter of 1 cm (0.785 cm2 per area) and placed in a Teflon holder. A Pt rod and Ag/AgCl electrode (3 mol·L−1 KCl, Metrohm) were used as the counter and reference electrodes, respectively. During the electrochemical study, the potential was scanned from 0.90 to − 0.11 V (vs. Ag/AgCl) at a scan rate of 20 mV·s−1 using a potentiostat (PG STATO 30), while the RDE rotational speed was controlled at 500, 1000, 1500 and 2000 r·min−1. The electrode potentials were converted and reported in the RHE scale.
3 Results and discussion
3.1 Effect of supporting electrolytes and solution pH
The Pt–Co electrodeposition was performed at a current density of 10 mA·cm−2 in plating solutions containing 0.010 mol·L−1 H2PtCl6·6H2O, 0.1 mol·L−1 CoSO4·7H2O and 0.5 mol·L−1 of the selected supporting electrolyte (H2SO4, Na2SO4 or NaCl) at their natural pH and at pH 2.6. A very small change in the pH is obtained when the solution pH reaches 2.6. The amount and composition of the Pt–Co deposits prepared in different plating solutions are shown in Table 1. (Their deposited morphologies analyzed from SEM images are shown in Fig. 1.) Overall, the type of supporting electrolyte and the solution pH does not have a significant effect on the amount of Pt–Co that was electrodeposited on the CC, but does have a noticeable effect on the Pt–Co composition and deposited morphology. The Pt–Co deposited from the sulfate plating baths has a higher Pt content (94 at% Pt for H2SO4 and 90 and 97 at% Pt for Na2SO4 at the natural pH and pH 2.6, respectively) than those deposited in NaCl (81 at% and 87 at% Pt at the natural pH and pH 2.6, respectively). These results suggest that the presence of the chloride ion in the plating bath may influence the electrodeposition of Pt and/or Co, leading to the increased Co content in the Pt–Co alloy deposit.

SEM images of Pt–Co electrodeposits obtained from 0.5 mol·L−1a H2SO4 at natural pH, b Na2SO4 at natural pH, c Na2SO4 at pH 2.6, d NaCl at natural pH and e NaCl at pH 2.6
By adjusting the solution pH to the lower level (pH 2.6), the Pt contents in the Pt–Co deposits increase slightly (6 at%–7 at%) in both the Na2SO4 and NaCl plating solutions. Based on the equilibrium potentials, the hydrogen evolution reaction is more favorable to proceed than the Co reduction reaction. With proton concentration (lower pH) increasing in the plating bath, a greater amount of the hydrogen evolution reactions takes place at the expense of the Co reduction reaction. In addition, since Co, which is a non-noble metal, is considerably less stable than Pt, Co can preferentially dissolve during the electrodeposition process under the high acidic (pH 2.6) conditions. Thus, the amount of Co in the electrodeposited Pt–Co alloy decreases when deposited from the low pH (2.6) solutions. In addition, the solution pH has a substantial effect on the deposited morphology of the Pt–Co deposits, where the Pt–Co electrodeposited in a low pH (2.6) consists of bigger grains compared to those electrodeposited in their respective solutions at the natural pH (Fig. 1).
3.2 Effect of current density
The effect of the current density on the composition of the Pt–Co deposits was studied at 10 and 20 mA·cm−2 in the plating solutions containing different supporting electrolytes (0.5 mol·L−1 H2SO4, Na2SO4 or NaCl) at their natural pH. Two levels of metal ion concentrations in the plating bath were investigated, using (1) 0.010 mol·L−1 H2PtCl6·6H2O and 0.1 mol·L−1 CoSO4·7H2O (Co-to-Pt atomic ratio of 10) and (2) 0.004 mol·L−1 H2PtCl6·6H2O and 0.2 mol·L−1 CoSO4·7H2O (Co-to-Pt atomic ratio of 50). The amount and composition of the Pt–Co deposits prepared under different conditions are shown in Table 2. Their morphologies are shown in Fig. 2.

SEM images of Pt–Co electrodeposits obtained from 0.5 mol·L−1a–d H2SO4, e–h Na2SO4 and i–l NaCl supporting electrolytes at different current densities and Co-to-Pt atomic ratios
In Pt–Co electrodeposition at a low applied current density level (10 mA·cm−2) in all supporting electrolytes, decreasing PtCl62− concentration (increasing Co-to-Pt atomic ratio from 10 to 50) in a plating solution does not substantially alter the deposited alloy content, where the Pt contents in all deposited Pt–Co alloys are still at a relatively high level (~ 80 at% Pt), but the amount of Pt–Co deposited is noticeably decreased (from 0.2 to 0.1 mg·cm−2 level). These results indicate that Pt–Co electrodeposition approaches the mass transport limitation when decreasing PtCl62− concentration from 0.010 to 0.004 mol·L−1. As Pt ion at the electrode surface is depleted, the hydrogen evolution reaction starts to take place at a higher rate despite a large amount of Co ion present in the plating solution since hydrogen reduction reaction is more thermodynamically favorable to proceed than the Co deposition reaction. Thus, the amount of Pt–Co deposited is decreased with a minimal change in the Pt content. However, when the applied current density increases from 10 to 20 mA·cm−2, the results are different depending on the type of supporting electrolyte.
In the H2SO4 supporting electrolyte, increasing the applied current density from 10 to 20 mA·cm−2 does not substantially alter the deposited alloy content, where the Pt contents in all the deposited Pt–Co alloys in H2SO4 are at a relatively high and narrow level of 83 at%–97 at% Pt. Only a slight decrease in Pt content is observed, from 94 to 90 at% and from 87 at% to 83 at% Pt for a plating solution with a Co-to-Pt atomic ratio of 10 and 50, respectively, when the applied current density increases from 10 to 20 mA·cm−2, respectively. These results suggest that as the applied current density increases, the surface concentration of Pt ion is strongly depleted and the electrodeposition proceeds toward the mass transfer limitation of the Pt ion. Despite the abundance of Co ion in the plating solution and higher energy available (via higher applied current) to force the less thermodynamically favorable reaction of Co reduction to proceed, most of the extra applied current was used for hydrogen evolution rather than Co electrodeposition, leading to a lower amount of Pt–Co being deposited with only a slight increase in the Co content.
While the applied current density has only a marginal influence on the composition of the Pt–Co deposited in H2SO4 solution, it has a significant effect on the composition of the Pt–Co deposited in Na2SO4 and NaCl solutions. In Na2SO4, increasing applied current density from 10 and 20 mA·cm−2 significantly reduces Pt content in the Pt–Co deposit from 90 at% to 40 at% Pt at a Co-to-Pt atomic ratio of 10, and to as low as 5 at% Pt at a Co-to-Pt atomic ratio of 50. When a current density of 10 mA·cm−2 was used, the Pt content of the Pt–Co deposits remains in 90 at% even when the Co-to-Pt atomic ratio increases from 10 to 50. However, when a current density of 20 mA·cm−2 was used, the Pt content of the Pt–Co deposits significantly decreases from 40 at% to 5 at% Pt when the Co-to-Pt atomic ratio in the solution increases from 10 to 50. However, the Pt–Co deposited in Na2SO4 at 20 mA·cm−2 at both Co-to-Pt atomic ratios appears to develop a dark green deposit (Fig. 3a) in form of Co oxide, as shown in SEM images (Figs. 2g, h, 3b) and EDS (Fig. 3c). Thus, even though a wide range of Pt–Co deposit compositions (5 at%–90 at% Pt) could be obtained in the Na2SO4 plating solution, it is not suitable as a supporting electrolyte for Pt–Co electrodeposition.

a Macro-image, b SEM image and c EDS spectrum of Pt–Co electrodeposited on CC at 20 mA·cm−2 in Na2SO4 containing 0.004 mol·L−1 PtCl62− and 0.20 mol·L−1 Co2+
In NaCl, while increasing the applied current density from 10 to 20 mA·cm−2 does not influence the Pt content level in the Pt–Co deposits (~ 80 at% Pt) in a plating solution containing a Co-to-Pt atomic ratio of 10, it dramatically reduces the Pt contents of the Pt–Co deposits (from 78 at% to 8 at% Pt) for the plating solution containing a Co-to-Pt atomic ratio of 50. These results indicate that at the Co-to-Pt atomic ratio of 10 (0.01 mol·L−1 PtCl62− in the plating solution) despite increasing the applied current density to 20 mA·cm−2, there might be enough Pt ion at the electrode surface for Pt electrodeposition to proceed so that the Pt content of the Pt–Co deposit remains relatively unchanged (78 at%−81 at% Pt), while the amount of Pt–Co deposited increases (from 0.25 to 0.32 mg·cm−2). On the other hand, at the Co-to-Pt atomic ratio of 50 when increasing the applied current density to 20 mA·cm−2, due to significantly low amount of Pt ion in the plating bath (0.004 mol·L−1 PtCl62−), there might not be enough Pt ion at the electrode surface for Pt electrodeposition to proceed. When the applied current density is high enough (20 mA·cm−2), there is enough energy to force the less favorable Co deposition reaction to proceed, and so the amount of Pt–Co deposited in the plating bath with a Co-to-Pt atomic ratio of 50 is at a comparable level (~ 0.3 mg·cm−2) and has a lower Pt content (8 at% Pt compared to 78 at% Pt) than those deposited in the plating bath with a Co-to-Pt atomic ratio of 10.
This study shows that the combination of the applied current density and the amount of metal ions present in the plating solution have a significant influence on the composition of the Pt–Co deposits, which could be varied over the wide range of Pt–Co deposit compositions when Na2SO4 or NaCl was used as the supporting electrolyte. However, unlike the Pt–Co deposited in the Na2SO4 plating solution, those deposited in the NaCl do not have a dark green deposit of Co oxide and normal Pt–Co-deposited morphologies are obtained (Fig. 2k, l). The incapability of the H2SO4 electrolyte to produce the low and broad range of Pt–Co deposit compositions should be due to the instability of Co in a strong acid like H2SO4. Co deposit might not form strong bonds or could preferentially dissolve during the electrodeposition process in H2SO4 supporting electrolyte, and Pt–Co electrodeposits with low Pt contents are difficult to produce.
3.3 Varying electrodeposited Pt–Co compositions
From the above results, a broad range of the Pt–Co deposits (78 at%–8 at% Pt) could be obtained from the NaCl plating solution without Co oxide formation. Accordingly, the NaCl plating solution containing Pt and Co concentrations of 0.004–0.010 mol·L−1 and 0.1–0.2 mol·L−1, respectively, at the natural pH was used to fabricate the Pt–Co catalysts with different compositions for evaluation of their effect on the ORR. The electrodeposition was performed at a current density of 20 mA·cm−2. The morphologies of the Pt–Co deposits obtained in different plating solutions are shown in Fig. 4, where Pt composition electrodeposited in Pt–Co deposit is present in SEM image. The electrodeposited pure Pt and Co are also included for comparison.

SEM images of Pt–Co electrodeposits obtained from NaCl supporting electrolyte containing different concentrations of PtCl62− and Co2+ at a fixed current density of 20 mA·cm−2 with different Pt-to-Co deposited atomic ratios: a Pt78Co22, b Pt55Co45, c Pt45Co55, d Pt24Co76, e Pt21Co79, f Pt10Co80, g Pt13Co87, h Pt11Co89, i Pt8Co92, j pure Pt, and k pure Co
By varying Pt and Co compositions in plating solutions, Pt–Co deposits with different compositions, ranging from Pt78Co22 to Pt8Co92, are obtained (Fig. 4). When the PtCl62− concentration is at a high level, increasing Co2+ concentration substantially increases the amount of electrodeposited Co in Pt–Co alloy (Pt content decreases from 78 at% to 45 at%), although less pronounced changes in the chemical composition are obtained at lower PtCl62− concentrations and minimal changes in the Pt–Co deposit composition are observed when Pt content is lowered to 13 at%. Like the pure Pt deposit (Fig. 4j), all Pt–Co deposits consist of granular-shaped particles. The pure Co deposit, on the other hand, has a large leaf-like structure (Fig. 4k). The grained particles of the Pt–Co deposits with high Pt contents (≥ 45 at%) consist of smaller particles than those of the pure Pt. The particle sizes are smallest at the highest Pt content (Pt78Co22) and gradually increase as Pt content decreases. Interestingly, Pt–Co deposits with a low Pt content (≤ 13 at%) still maintain the granular-shaped structure of the pure Pt rather than change to the large leaf-like structure of the pure Co.
XRD results of the selected Pt–Co deposits, along with that of the pure Pt deposit, are shown in Fig. 5. Only the Pt–Co deposits with high Pt contents (Pt78Co22 and Pt55Co45) show distinct XRD patterns, where it is observed that Pt(111), Pt(200) and Pt(220) peaks slightly shift to higher 2θ values compared to those of the Pt deposit, indicating the alloy formation between Pt and Co [50]. The lattice constants of these Pt–Co deposits (0.3856 and 0.3665 nm for Pt78Co22 and Pt55Co45, respectively) become smaller than that of the Pt deposit (0.3924 nm) and decreases with Co content in the Pt–Co alloys increasing. The contraction of the Pt lattices, due to the incorporation of the smaller Co atoms into the larger Pt structure, also indicates the Pt–Co alloy formation [16, 50,51,52] during the Pt–Co electrodeposition. When the Pt content in the Pt–Co deposits decreases, XRD peaks are depleted and so a very small peak of Pt(111) is observed for Pt45Co55. This suggests that as Co becomes the base metal, XRD peaks representing the Pt host tend to disappear.

XRD patterns of Pt–Co electrodeposits with different compositions
3.4 Electrocatalytic activity toward ORR of electrodeposited Pt–Co alloys
The electrocatalytic activity toward the ORR of selected Pt–Co-electrodeposited CC with different Pt–Co compositions was performed by a linear sweep voltammetry using a RDE in an O2-saturated 0.5 mol·L−1 H2SO4 solution. In fact, it was also tried to investigate the electrochemical surface area of the electrodeposited Pt–Co electrodes using the hydrogen underpotential deposition (HUPD) technique but were not able to obtain the desirable results due to the nature of electrode (i.e., CC) studied. The HUPD technique is based on the hydrogen adsorption/desorption process on the electrode surface using the cyclic voltammetry (CV) technique in the N2-purged H2SO4 or HClO4 solution. With the suitable cathodic limiting potential used, the peaks corresponding to adsorption and desorption of hydrogen on the electroactive surface should appear. Various cathodic limiting potentials were used (in the range of − 0.1 to 0.1 V vs. RHE), but none give the desirable hydrogen underpotential adsorption peak on the CV. If the potential was scanned to the more negative potentials, the tail of cathodic current due to the occurrence of hydrogen evolution is observed, but without a sign of the hydrogen underpotential adsorption peak. On the other hand, if the scanned potential is limited to the less negative potentials, no peaks appear and a flat CV is obtained. The obtained results should be due to the nature of the CC electrodes investigated herein. CC was made from bundles of carbon yarns and then has a porous and rough surface. It is possible that the desirable hydrogen adsorption/desorption peaks corresponding to the Pt deposited on the CC surface might be concealed by the capacitive response due to a very high surface area of the CC. It is worth noting that a number of studies have investigated the electrochemical surface area of the Pt or Pt-based alloy catalysts by the HUPD technique, but most were done on a glassy carbon [19, 24, 31, 32, 53,54,55,56,57,58,59,60,61,62,63,64,65,66,67], graphite [32, 34, 68] or metal [61, 69, 70] electrode, which has a very smooth surface. Since the desirable HUPD response is not obtained in this study, the results are not shown and discussed in detail. Only the electrocatalytic activity toward the ORR was investigated, and the linear sweep voltammograms are presented in Fig. 6.

RDE voltammograms for ORR on a Pt78Co22 catalyst at different rotational speeds and b different Pt–Co catalysts at 1000 r·min−1
Figure 6a shows the linear sweep voltammograms of the ORR on Pt78Co22 deposits at different rotational speeds. Note that the voltammograms of the Pt–Co deposits with other compositions follow a similar trend and so are not shown here for brevity. After the onset of the ORR initiates, the voltammograms can be divided into three regions: kinetic-limited, mixed kinetic–diffusion and diffusion-limited regions. In the kinetic-limited region, the rotational speed does not influence the voltammogram. As the potential is scanned to the more cathodic direction, the voltammogram starts to be influenced by the rotational speed and the ORR is controlled by both kinetic and diffusion. In the diffusion-limited region, the magnitude of the current density is solely governed by the rotational speed (i.e., the limiting current density, iL), and the voltammogram reaches a higher current density with rotational speed increasing.
The linear sweep voltammograms obtained from various catalysts coated on CC at a rotational speed of 1000 r·min−1 are shown in Fig. 6b. The voltammograms of the Co-deposited and Co-uncoated CC appear flat, indicating that the ORR does not proceed on these surfaces since Co and carbon are not good for the ORR. The surfaces that have Pt-deposited (Pt and Pt–Co alloys) exhibit an electrocatalytic activity for the ORR. The onset of the ORR for all Pt-deposited CC appears to take place at relatively similar potentials, but the current density in the kinetic region rises at different rates depending on the Pt content of the catalysts. The voltammograms of the Pt–Co alloy deposits with ≥ 45 at% Pt have steeper slopes (i.e., more positive half-wave potentials) than that of the pure Pt deposit, while those with Pt content of < 45 at% rise slower (i.e., lower positive half-wave potentials) than that of the pure Pt deposit. Thus, the ORR activity of the Pt–Co alloys is dependent on their composition. As reported in the studies, the relationships between the composition of the bi-metallic Pt-based alloys and the ORR activity normally follow the volcano-type behavior, which relates surface electronic structure (the d-band center) to the activity for the ORR [14, 20, 71, 72]. The d-band center determines the strength of the metal–adsorbate bonding. Thus, alloy composition influences adsorption energies of reactive intermediates and blocking species on the surface, which affect the ORR kinetics [13]. When Pt deposits are alloyed by appropriate amounts of Co, their ORR electroactivity is improved. Of the Pt–Co compositions investigated in this study, the Pt78Co22 yields the highest electroactivity, which is consistent with other reports [12,13,14, 17, 73,74,75,76], where the Pt3Co alloy has the best ORR performance, a composition of which the adsorption strength of oxygen and intermediates is optimal [13, 14, 76] for the ORR.
Quantitatively, the ORR kinetics can be investigated using the Koutecky–Levich (K–L) equation, where the K–L plot (i.e., 1/i vs. rotational speed−1/2) can be used to estimate the kinetic-controlled current density (ik) and the number of electrons involved in the reaction (n) of the ORR on the catalysts [77]. However, they were not investigated in this study due to differences in the nature of the CC from the smooth glassy carbon. The K–L is normally used to estimate the ik and n of the ORR based on a very flat and smooth surface, i.e., glassy carbon [13, 54, 55, 58, 62, 65,66,67, 78] or graphite [34] or metal [11, 69, 70] electrode. As mentioned earlier, the CC is made from bundles of carbon yarns leading to a very rough surface and porous structure, which leads to a higher actual surface area and a different fluid flow pattern or stagnant film in the region adjacent to the catalyst sites. Thus, the voltammograms of the ORR on the RDE with CC surface obtained in this study have significantly higher current densities (range between ~ 18 and 55 mA·cm−2 for the rotational speeds of 500 and 2000 r·min−1, respectively) than those on the RDE made of the very flat and smooth surface, which have seldom been reported to exceed 7 mA·cm−2 even at a rotational speed as high as 2500 r·min−1 (references listed above). To our knowledge, few investigations of the ORR on the RDE made of CC have been reported. Without proper corrections for the relevant factors due to the structure of the CC surface, the estimated values of the kinetic parameters of the catalysts deposited on the CC using the K–L equation will be very inaccurate and misleading. Nevertheless, without the quantitative analysis, the voltammograms still reveal that the alloy composition of the Pt–Co catalyst influences the ORR activity and that the Pt–Co alloy with ~ 75 at% Pt (78 at% Pt in this study) has the best ORR activity.
4 Conclusion
In this work, Pt–Co alloys on a carbon-supported CC gas diffusion layer were synthesized by DC electrodeposition in an attempt to find the suitable plating bath conditions that can produce a wide range of Pt–Co compositions and then to investigate the relationship between the composition of Pt–Co catalyst and ORR activity. It is found that using the suitable supporting electrolyte allows the electrodeposition process to effectively fine-tune the chemical composition of the Pt–Co alloys by adjusting the other electrodeposition parameters such as the applied current density, solution pH and the concentrations of the Pt and Co ions in the plating solution. The Pt–Co electrodeposition in H2SO4 produces Pt–Co alloys with very high Pt contents that could not be reduced to a level lower than 80 at% Pt even though the applied current density and the Co-to-Pt ratio are substantially increased. Even if the Pt–Co electrodeposition in Na2SO4 is able to yield a very low and wide range of chemical compositions (5 at%–97 at% Pt), cobalt oxide is formed on the Pt–Co deposit surfaces. Only the Pt–Co electrodeposition in NaCl is found to effectively produce Pt–Co deposits over a wide range of compositions (8 at%–90 at% Pt). The qualitative study reveals that the Pt–Co composition has a substantial impact on ORR activity, where alloying Pt with a Co-to-Pt content of ≥ 45 at% improves the ORR activity, and the Pt–Co alloy catalyst with 78 at% Pt content yields the best ORR performance.
References
de Bruijn F. The current status of fuel cell technology for mobile and stationary applications. Green Chem. 2005;7(3):132.
Wee JH. Applications of proton exchange membrane fuel cell systems. Renew Sustain Energy Rev. 2007;11(8):1720.
Wang Y, Chen KS, Mishler J, Cho SC, Adroher XC. A review of polymer electrolyte membrane fuel cells: technology, applications, and needs on fundamental research. Appl Energy. 2011;88(4):981.
Shao Y, Yin G, Wang Z, Gao Y. Proton exchange membrane fuel cell from low temperature to high temperature: material challenges. J Power Sour. 2007;167(2):235.
Murthi VS, Urian RC, Mukerjee S. Oxygen reduction kinetics in low and medium temperature acid environment: correlation of water activation and surface properties in supported Pt and Pt alloy electrocatalysts. J Phys Chem B. 2004;108(30):11011.
Holton OT, Stevenson JW. The role of platinum in proton exchange membrane fuel cells. Platin Met Rev. 2013;57(4):259.
Larminie J, Dicks A. Fuel Cell Systems Explained. 2nd ed. West Sussex: Wiley; 2003. 48.
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B. 2005;56(1–2):9.
Sharma S. Recent developments in electrocatalysts and hybrid electrocatalyst support systems for polymer electrolyte fuel cells. In: Maiyalagan T, Saji VS, editors. Electrocatalysts for Low Temperature Fuel Cells: Fundamentals and Recent Trends. Weinheim: Wiley-VCH; 2017. 197.
Stacy J, Regmi N, Leonard B, Fan M. The recent progress and future of oxygen reduction reaction catalysis: a review. Renew Sustain Energy Rev. 2017;69:401.
Jayasayee K, Dam V-AT, Verhoeven T, Celebi S, de Bruijn FA. Oxygen reduction kinetics on electrodeposited PtCo as a model catalyst for proton exchange membrane fuel cell cathodes: stability as a function of PtCo composition. J Phys Chem C. 2009;113(47):20371.
Woo S, Kim I, Lee JK, Bong S, Lee J, Kim H. Preparation of cost-effective Pt–Co electrodes by pulse electrodeposition for PEMFC electrocatalysts. Electrochim Acta. 2011;56(8):3036.
Choi S-I, Lee S-U, Kim WY, Choi R, Hong K, Nam KM, Han SW, Park JT. Composition-controlled PtCo alloy nanocubes with tuned electrocatalytic activity for oxygen reduction. ACS Appl Mater Interfaces. 2012;4(11):6228.
Zhao Y, Liu J, Zhao Y, Wang F. Composition-controlled synthesis of carbonsupported Pt–Co alloy nanoparticles and the origin of their ORR activity enhancement. Phys Chem Chem Phys. 2014;16(36):19298.
Yu P, Pemberton M, Plasse P. PtCo/C cathode catalyst for improved durability in PEMFCs. J Power Sour. 2005;144(1):11.
Neergat M, Shukla AK, Gandhi KS. Platinum-based alloys as oxygen-reduction catalysts for solid-polymer-electrolyte direct methanol fuel cells. J Appl Electrochem. 2001;31(4):373.
M-k Min, Cho J, Cho K, Kim H. Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications. Electrochim Acta. 2000;45(25–26):4211.
Salgado JRC, Antolini E, Gonzalez ER. Carbon supported Pt70Co30 electrocatalyst prepared by the formic acid method for the oxygen reduction reaction in polymer electrolyte fuel cells. J Power Sour. 2005;141(1):13.
Lu Y, Reddy RG. The electrochemical behavior of cobalt phthalocyanine/platinum as methanol-resistant oxygen-reduction electrocatalysts for DMFC. Electrochim Acta. 2007;52(7):2562.
Aricò AS, Stassi A, Gatto I, Monforte G, Passalacqua E, Antonucci V. Surface properties of Pt and PtCo electrocatalysts and their influence on the performance and degradation of high-temperature polymer electrolyte fuel cells. J Phys Chem C. 2010;114(37):15823.
Paunovic M, Schlesinger M. Fundamentals of Electrochemical Deposition. New York: Wiley; 1998. 6.
Choi KH, Kim HS, Lee TH. Electrode fabrication for proton exchange membrane fuel cells by pulse electrodeposition. J Power Sour. 1998;75(2):230.
Kim H, Subramanian NP, Popov BN. Preparation of PEM fuel cell electrodes using pulse electrodeposition. J Power Sour. 2004;138(1–2):14.
Lee J, Seo J, Han K, Kim H. Preparation of low Pt loading electrodes on Nafion (Na+)-bonded carbon layer with galvanostatic pulses for PEMFC application. J Power Sour. 2006;163(1):349.
Duarte MEE, Pilla AS, Sieben JM, Mayer CE. Platinum particles electrodeposition on carbon substrates. Electrochem Commun. 2006;8(1):159.
Rajalakshmi N, Dhathathreyan KS. Nanostructured platinum catalyst layer prepared by pulsed electrodeposition for use in PEM fuel cells. Int J Hydrog Energy. 2008;33(20):5672.
Zhang H, Zhou W, Du Y, Yang P, Wang C. One-step electrodeposition of platinum nanoflowers and their high efficient catalytic activity for methanol electro-oxidation. Electrochem Commun. 2010;12(7):882.
Chaisubanan N, Tantavichet N. Pulse reverse electrodeposition of Pt–Co alloys onto carbon cloth electrodes. J Alloys Compd. 2013;559:69.
Mikhaylova AA, Khazova OA, Bagotzky VS. Electrocatalytic and adsorption properties of platinum microparticles electrodeposited onto glassy carbon and into Nafion® films. J Electroanal Chem. 2000;480(1–2):225.
Thompson SD, Jordan LR, Forsyth M. Platinum electrodeposition for polymer electrolyte membrane fuel cells. Electrochim Acta. 2001;46(10–11):1657.
Selvaraju T, Ramaraj R. Electrochemically deposited nanostructured platinum on Nafion coated electrode for sensor applications. J Electroanal Chem. 2005;585(2):290.
Domínguez-Domínguez S, Arias-Pardilla J, Berenguer-Murcia Á, Morallón E, Cazorla-Amorós D. Electrochemical deposition of platinum nanoparticles on different carbon supports and conducting polymers. J Appl Electrochem. 2008;38(2):259.
Martín AJ, Chaparro AM, Gallardo B, Folgado MA, Daza L. Characterization and single cell testing of Pt/C electrodes prepared by electrodeposition. J Power Sour. 2009;192(1):14.
Li J, Ye F, Chen L, Wang T, Li J, Wang X. A study on novel pulse preparation and electrocatalytic activities of Pt/C-Nafion electrodes for proton exchange membrane fuel cell. J Power Sour. 2009;186(2):320.
Alcaide F, Álvarez G, Blázquez JA, Cabot PL, Miguel O. Development of a novel portable-size PEMFC short stack with electrodeposited Pt hydrogen diffusion anodes. Int J Hydrog Energy. 2010;35(11):5521.
Liu J, Zhong C, Du X, Wu Y, Xu P, Liu J, Hu W. Pulsed electrodeposition of Pt particles on indium tin oxide substrates and their electrocatalytic properties for methanol oxidation. Electrochim Acta. 2013;100:164.
Le Vot S, Roué L, Bélanger D. Study of the electrochemical oxidation of ammonia on platinum in alkaline solution: effect of electrodeposition potential on the activity of platinum. J Electroanal Chem. 2013;691:18.
Fouda-Onana F, Guillet N, AlMayouf AM. Modified pulse electrodeposition of Pt nanocatalyst as high-performance electrode for PEMFC. J Power Sour. 2014;271:401.
Jyoko Y, Schwarzacher W. Characterization of electrodeposited magnetic Co/Pt multilayered nanostructures. Electrochim Acta. 2001;47(1–2):371.
Coutanceau C, Rakotondrainibé AF, Lima A, Garnier E, Pronier S, Léger J-M, Lamy C. Preparation of Pt–Ru bimetallic anodes by galvanostatic pulse electrodeposition: characterization and application to the direct methanol fuel cell. J Appl Electrochem. 2004;34(1):61.
Martín AJ, Chaparro AM, Daza L. Electrochemical quartz crystal microbalance study of the electrodeposition of Co, Pt and Pt–Co alloy. J Power Sour. 2007;169(1):65.
Liu J, Cao L, Huang W, Li Z. Direct electrodeposition of PtPd alloy foams comprised of nanodendrites with high electrocatalytic activity for the oxidation of methanol and ethanol. J Electroanal Chem. 2012;686(1):38.
Zhao W, Yang Y, Zhang H. Electrodeposition preparation of highly dispersed Pt/HxWO3 composite catalysts for PEMFCs. Electrochim Acta. 2013;99:273.
Zhang J, Li D, Zhu Y, Chen M, An M, Yang P, Wang P. Properties and electrochemical behaviors of AuPt alloys prepared by direct-current electrodeposition for lithium air batteries. Electrochim Acta. 2013;151:415.
Leistner K, Oswald S, Thomas J, Fähler S, Schlörb H, Schultz L. Potential dependence of composition and structure of electrodeposited Fe–Pt films. Electrochim Acta. 2006;52(1):194.
Hwang SM, Lee CH, Kim JJ, Moffat TP. Oxygen reduction reaction on electrodeposited Pt100−x−yNixPdy thin films. Electrochim Acta. 2010;55(28):8938.
Ra Y, Lee J, Kim I, Bong S, Kim H. Preparation of Pt–Ru catalysts on Nafion(Na+)-bonded carbon layer using galvanostatic pulse electrodeposition for proton-exchange membrane fuel cell. J Power Sour. 2009;187(2):363.
Jayasayee K, Van Veen JAR, Hensen EJM, de Bruijn FA. Influence of chloride ions on the stability of PtNi alloys for PEMFC cathode. Electrochim Acta. 2011;56(20):7235.
Lertviriyapaisan S, Tantavichet N. Sublayers for Pt catalyst electrodeposition electrodes in PEMFC. Int J Hydrog Energy. 2010;35(19):10464.
Lopes T, Antolini E, Colmati F, Gonzalez ER. Carbon supported Pt–Co (3:1) alloy as improved cathode electrocatalyst for direct ethanol fuel cells. J Power Sour. 2007;164(1):111.
Zhang X, Chan KY. Microemulsion synthesis and electrocatalytic properties of platinum–cobalt nanoparticles. J Mater Chem. 2002;100(4):1203.
Travitsky N, Ripenbein T, Golodnitsky D, Rosenberg Y, Burshtein L, Peled E. Pt-, PtNi- and PtCo-supported catalysts for oxygen reduction in PEM fuel cells. J Power Sour. 2006;161(2):782.
Xiong L, Manthiram A. Nanostructured Pt–M/C (M = Fe and Co) catalysts prepared by a microemulsion method for oxygen reduction in proton exchange membrane fuel cells. Electrochim Acta. 2005;50(11):2323.
Luo J, Njoki PN, Lin Y, Wang L, Zhong CJ. Activity-composition correlation of AuPt alloy nanoparticle catalysts in electrocatalytic reduction of oxygen. Electrochem Commun. 2006;8(4):581.
Jeon MK, Zhang Y, McGinn PJ. A comparative study of PtCo, PtCr, and PtCoCr catalysts for oxygen electro-reduction reaction. Electrochim Acta. 2010;55(19):5318.
Ye JH, Fedkiw PS. Electrodeposition of high-surface area platinum in a well adherent nafion film on glassy carbon. Electrochim Acta. 1996;41(2):221.
Paulus UA, Schmidt TJ, Gasteiger HA, Behm RJ. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: a thin-film rotating ring-disk electrode study. J Electroanal Chem. 2001;495(2):134.
Paulus UA, Wokaun A, Scherer GG, Schmidt TJ, Stamenkovic V, Markovic NM, Ross PN. Oxygen reduction on high surface area Pt-based alloy catalysts in comparison to well defined smooth bulk alloy electrodes. Electrochim Acta. 2002;47(22):3787.
Mo Y, Sarangapani S, Le A, Scherson DA. Electrochemical characterization of unsupported high area platinum dispersed on the surface of a glassy carbon rotating disk electrode in the absence of Nafion® or other additives. J Electroanal Chem. 2002;538–539:35.
Liu Z, Gan LM, Hong L, Chen W, Lee JY. Carbon-supported Pt nanoparticles as catalysts for proton exchange membrane fuel cells. J Power Sour. 2005;139(1–2):73.
Plyasova LM, Molina IYU, Gavrilov AN, Cherepanova SV, Cherstiouk OV, Rudina NA, Savinova ER, Tsirlina GA. Electrodeposited platinum revisited: tuning nanostructure via the deposition potential. Electrochim Acta. 2006;51(21):4477.
Wang R, Li H, Ji S, Wang H, Lei Z. Pt decorating of PdNi/C as electrocatalysts for oxygen reduction. Electrochim Acta. 2010;55(5):1519.
Punyawudho K, Blom DA, Van Zee JW, Monnier JR. Comparison of different methods for determination of Pt surface site concentrations for supported Pt electrocatalysts. Electrochim Acta. 2010;55(19):5349.
Ohashi M, Beard KD, Ma S, Blom DA, St-Pierre J, Van Zee JW, Monnier JR. Electrochemical and structural characterization of carbon-supported Pt–Pd bimetallic electrocatalysts prepared by electroless deposition. Electrochim Acta. 2010;55(24):7376.
Chen D, Tao Q, Liao LW, Liu SX, Chen YX, Ye S. Determining the active surface area for various platinum electrodes. Electrocatalysis. 2011;2(3):207.
Zhang S, Chen S. Enhanced-electrocatalytic activity of Pt nanoparticles supported on nitrogen-doped carbon for the oxygen reduction reaction. J Power Sour. 2013;240:60.
Geboes B, Ustarroz J, Sentosun K, Vanrompay H, Hubin A, Balsand S, Breugelmans T. Electrochemical behavior of electrodeposited nanoporous Pt catalysts for the oxygen reduction reaction. ACS Catal. 2016;6(9):5856.
Perez J, Gonzalez ER, Ticianelli EA. Oxygen electrocatalysis on thin porous coating rotating platinum electrodes. Electrochim Acta. 1998;44(8–9):1329.
Van Brussel M, Kokkinidis G, Hubin A, Buess-Herman C. Oxygen reduction at platinum modified gold electrodes. Electrochim Acta. 2003;48(25–26):3909.
Benítez R, Chaparro AM, Daza L. Electrochemical characterisation of Pt/C suspensions for the reduction of oxygen. J Power Sour. 2005;151:2.
Stamenkovic VR, Mun BS, Arenz M, Mayrhofer KJJ, Lucas CA, Wang G, Ross PN, Markovic NM. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat Mater. 2007;6(3):241.
Viswanathan V, Hansen HA, Rossmeisl J, Nørskov JK. Universality in oxygen reduction electrocatalysis on metal surfaces. ACS Catal. 2012;2(8):1654.
Mukerjee S, Srinivasan S. Enhanced electrocatalysis of oxygen reduction on platinum alloys in proton exchange membrane fuel cells. J Electroanal Chem. 1993;357(1–2):201.
Salgado RC, Antolini E, Gonzalez ER. Structure and activity of carbon-supported Pt–Co electrocatalysts for oxygen reduction. J Phys Chem B. 2004;108(46):17767.
Koh S, Toney MF, Strasser P. Activity–stability relationships of ordered and disordered alloy phases of Pt3Co electrocatalysts for the oxygen reduction reaction (ORR). Electrochim Acta. 2007;52(8):2765.
Stamenkovic VR, Mun BS, Arenz M, Mayrhofer KJJ, Lucas CA, Wang GF, Ross PN, Markovic NM. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat Mater. 2007;6(3):241.
Bard AJ, Faulkner LR. Electrochemical Methods—Fundamentals and Applications. 2nd ed. New York: Wiley; 2001. 341.
Park JH, Sohn Y, Jung DH, Kim P, Joo JB. Pt deposited Pt–Pd/C electrocatalysts with the enhanced oxygen reduction activity. J Ind Eng Chem. 2016;36:109.
Acknowledgements
This project was financially supported by the Ratchadapisek Sompoch Endowment Fund, Chulalongkorn University (Sci-Super 2014-026), the Thailand Research Fund (TRF IRG5780001) and the Faculty of Science, Chulalongkorn University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Puangsombut, P., Tantavichet, N. Effect of plating bath composition on chemical composition and oxygen reduction reaction activity of electrodeposited Pt–Co catalysts. Rare Met. 38, 95–106 (2019). https://doi.org/10.1007/s12598-018-1082-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12598-018-1082-2