
Abstract
The influence of the atmosphere composition (CO, Ar, air), in which wet synthesis of Pt/C electrocatalyst was carried out, on the structural and morphological characteristics, and electrochemical behavior of electrocatalysts have been studied. For comparison, commercial Pt/C electrocatalysts with the same platinum loading were also studied. It has been shown that the adsorption of CO molecules on the surface of the growing platinum nuclei leads to the decrease in the average size of the nanoparticles and the narrowing of the size distribution in the Pt/C. Homemade electrocatalysts, with the values of electrochemically active surface area being from 94 to 139 m2 g−1 (Pt), prove to be in no way inferior to their commercial counterparts in oxygen reduction reaction mass activity. Durability of the homemade Pt/C samples in accelerated stress tests exceeds durability of the commercial ones.

ᅟ
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Over the past decades, platinum nanoparticles deposited on a carbon support have been the best electrocatalysts for the proton exchange membrane fuel cell (PEMFC) [1]. Platinum alloying by some d-metals improves activity and, in certain cases, durability of electrocatalysts [2–4]. Reduction in the mass fraction of the precious metal in the catalysts also results in the reduction of their cost [1]. Platinum replacement by the Pt–Ru alloy has significantly increased PEMFC tolerance of the hydrogen electrode to the CO impurity [4]. At the same time, replacement of platinum by its alloys on the oxygen electrode is not widely used in PEMFC manufacture. The point is that degradation of a catalyst, as well as corrosion of metal nanoparticles, takes place in the process of PEMFC operation [5]. The degradation phenomenon is more pronounced on the oxygen electrode whose potential can achieve high values, while intermediates of the oxygen electroreduction are highly reactive [6]. In the case of the two-component platinum alloys, an alloying component mostly undergoes selective dissolution. This component has a lower thermodynamic stability compared to platinum [6]. The formed metal cations replace hydrogen ions in the ionomer sulfo groups. As a result, proton conductivity of the membrane and the thickness of the catalyst layer are reduced. This effect leads to accelerated degradation of the membrane and damages the fuel cell system [6].
The activity and durability of Pt/C catalysts are connected with their structural and morphological characteristics. The point is that the physicochemical properties of the noble metal nanostructures strongly depend on their shape, size, and size distribution; the strength of anchoring; and the character of spatial distribution on the carbon support surface [4, 7]. By controlling these parameters, one can change the properties of the resulting electrocatalysts [4, 7]. The problem, however, consists in the following: (i) mass activity and stability of the catalysts generally antisymbatically depend on the size of nanoparticles [4] and (ii) optimization of one morphological characteristic during the synthesis of nanoparticles, e.g., the size, may be accompanied by undesirable changes in other characteristics, such as the shape [8]. The search for a compromise between the activity and stability of electrocatalyst, as well as the search for the ways of obtaining the Pt/C microstructure with the optimum combination of several characteristics, stipulates continuous efforts to improve the methods of Pt/C synthesis [7].
In the search of the optimal synthesis method, researchers usually stick to the following widely accepted characteristics of Pt/C electrocatalysts with the desired microstructure (morphology): the optimum value of platinum nanoparticles being 2–4 nm, a narrow size distribution, a uniform spatial distribution of nanoparticles (when no aggregation of nanoparticles is formed), and strong adhesion of the nanoparticles to the surface of the support. An important feature of the catalyst morphology, which directly depends on the previous characteristics, is an electrochemically active surface area (ESA) of platinum. In view of the negative correlation between the nanoparticle size and specific activity (A cm−2 (Pt)) of the catalyst [9], high mass activity is usually observed for Pt/C catalyst with the ESA values from 70 to 90 m2 g−1 (Pt) [10]. While discussing the stability of the oxygen Pt/C catalyst, many authors point out that a decrease of the nanoparticles less than 3 nm in the size leads to a dramatic reduction in stability [3]. However, this issue requires further study, because it is necessary to take into account different mechanisms of catalyst degradation and the change in the contribution of each mechanism to the degradation process depending on PEMFC operation conditions [3, 11].
Nowadays, different approaches to Pt/C catalyst synthesis coexist quite successfully [12]. The most commonly used methods are based on the chemical reduction of platinum from the precursor solution by ethylene glycol, borohydride, formaldehyde, and others [13–15]. These methods of synthesis are cost-effective and relatively simple for implementation. Unfortunately, some difficulties connected with the implementation of these methods arise, while materials with a narrow size distribution of the nanoparticles are obtained, especially in the synthesis of catalysts with the high platinum loading [16].
Control over the nucleation and growth of nanoparticles, their spatial distribution, and anchoring to the carbon surface during the chemical reduction plays the fundamental role for any process of the Pt/C catalyst synthesis [17]. It is of particular significance for both heterogeneous and homogeneous nucleation, when the metal nanoparticles are first formed in the solution, and only then deposited on the surface of the carbon support [18].
Researchers commonly use additive stabilizing agents to control the nanoparticle size and size distribution during the chemical synthesis of Pt/C catalysts. Molecules of stabilizers are adsorbed on the surface of the growing platinum nuclei and thus impede their growth and aggregation [19]. Unfortunately, this approach has several disadvantages: (i) pollution of catalysts by organic additives, which cannot be easily desorbed, causes reduction in catalytic activity [20] and (ii) mass fraction of a noble metal in the catalyst could be reduced due to additional difficulties in adsorption of nanoparticles on the carbon support surface. Some authors [20, 21] suggest removing stabilizing additives by thermal treatment at 150–200 °С. However, when heated, the coalescence of small-size nanoparticles is facilitated and this results in ESA reduction [21]. The problem of the adsorbent choice, which has a positive effect on the process of nucleation/growth of platinum nanoparticles on the surface of the carbon carrier, but which does not pollute the resulting catalyst, has not been resolved yet.
It is common knowledge that different media components can be easily and firmly adsorbed onto the surface of the platinum metal [22]. Adsorption of medium components also occurs on the highly developed surface of the carbon particles. However, the influence of the atmosphere on the microstructure of the platinum deposit, formed in the liquid phase synthesis of Pt/C catalysts, is poorly discussed in literature. We believe that the adsorption of gas molecules contained in the solution is one of the factors which affect the processes of nucleation, growth, and coalescence of the Pt nucleus. Of particular interest, in our opinion, is the study of the CO influence, which is a powerful adsorbent with regard to platinum [23–25]. It is important to note that the CO molecules, which are adsorbed on the surface of platinum and carbon, can be easily removed in the process of a successive drying of the synthesized catalysts.
The influence of the atmosphere (CO, air, Ar) on the microstructure and electrochemical behavior of Pt/C materials obtained by the chemical reduction of Pt(IV) in the liquid phase has been studied in the present paper. The research is based on the hypothesis that the adsorption of CO molecules on the surface of the growing platinum nuclei will slow down their growth and hamper the coalescence of the nanoparticles, resulting in the increase of the metal surface area in Pt/C. We took into account the possible effect of the CO, O2 gas molecules, adsorbed on the surface of platinum and carbon, on the conditions of platinum nanoparticle adhesion or the lack of such effect in the case of Ar. Thus, on the assumption that even in the case of a similar size and size distribution of the nanoparticles, we believe that the electrochemical performance of Pt/C catalysts could be different.
Experimental
The Pt/C materials were prepared by chemical reduction of the metal precursors (H2PtCl6·6H2O, Aurat, Moscow, Russia) from a carbon suspension based on water-organic solvents with formaldehyde (HCHO) (37 wt%) as a reducing agent. Vulcan XC-72 (Cabot Corp., S BET = 250–280 m2 g−1) was used as a carbon support. At first, 0.1 g of Vulcan XC-72 carbon powder was suspended in an aqueous solution of the appropriate amount of chloroplatinic acid. Then, 0.5 mL of 37% formaldehyde with a mole ratio of HCHO/PtCl6 2− 20:1 was added into the suspension. Thereafter, a 1M NaOH solution (H2O/EG 1:1) was gradually added to the suspended mixture until the pH of the mixed solution reached 11–12. Then, the suspension was maintained for 2 h at 90 °C heating (constant stirring). Finally, a strong electrolyte such as HCl solution was added as a sedimentation promoter. The catalysts were filtered, washed, and dried in air at 80 °C [26].
To study the influence of the atmosphere composition (air, CO, Ar) on the characteristics of Pt/C materials, the corresponding gas (CO or Ar) was bubbled through the reactionary mixture during 2 h in the course of the “formaldehyde” synthesis. Pt/C samples obtained in air, CO, or Ar atmospheres were referred to as Pt/C, Pt/CCO, and Pt/CAr, respectively [26].
The synthesis of the samples was repeated at least three times to assess the reproducibility of the obtained Pt/C catalyst characteristics.
Commercial Pt/C electrocatalysts with the same platinum content—E-TEK (E-TEK, 20 wt% Pt) and HiSPEC 3000 (Johnson Matthey, 20% Pt)—were also examined to compare their behavior with the behavior of the obtained materials.
The mass fraction of platinum (ω (Pt)) in the Pt/C was determined by thermogravimetric mass residue (Pt) after combustion (800 °C, 40 min). The measurement accuracy was ±3% (Table 1).
The XRD patterns of Pt/C materials were recorded in the range of 2 theta angles of 15–75° on a ARL X’TRA (Thermo Scientific, Switzerland) X-ray diffractometer with a Cu Kα radiation source (λ = 0.154056 nm) at room temperature. The fine structure was estimated from the broadening of the diffraction maxima using the procedure described in [29].
TEM study was performed using a JEM-2100 (JEOL, Japan) microscope operated at an accelerating voltage of 200 kV. Of electrocatalyst powders, 0.5 mg was placed in 1 mL of heptane to prepare the samples for TEM analysis. Then, the suspension was ultrasonically dispersed, and one drop of suspension was deposited onto a copper grid sputter-coated with carbon. The metal particle size distribution and the average size of the nanoparticles (D av) in the catalysts were obtained by direct measurement of 500 randomly selected particles by TEM images.
The active surface area was determined on a stationary electrode using the cycling voltammetry method. Measurements were performed in three-electrode cells at room temperature; a saturated silver chloride electrode was used as a reference electrode. All potentials in this work are referenced versus a reversible hydrogen electrode (RHE). A 0.1-M HClO4 solution saturated with Ar at atmospheric pressure was used as an electrolyte. The investigated thin layer of Pt/C, applied on glass carbon disc electrode (0.196-cm2 geometric surface area),was used as a working electrode.
To obtain “catalyst inks” containing a metal-carbon catalyst, 100 μL of 0.5% Nafion polymer aqueous emulsion and 900 μL of isopropanol were added to each sample (0.0060 g) and the suspension was further dispersed with ultrasonic waves for 10–15 min. A total of 6 μL of catalyst ink was selected with a microdozator upon continuous stirring and then applied as a drop on an electrode butt end checked the exact weight. To fix on the Pt/C electrode, 7 μL of 0.05% Nafion solution was applied over the catalyst layer so as to remain on the electrode after droplet has dried.
Initially, to standardize the electrode surface, 100 cycles were recorded in the potential sweep range of 0.03 to 1.26 V at 200 mV s−1. Then, two voltammograms were recorded in the same range of potentials, but the sweep rate was of 20 mV s−1. ESA of the catalyst was determined by the electricity amount spent on electrochemical adsorption/desorption of atomic hydrogen in the registration process when the second cyclic voltammogram (CV) scan rate was of 20 mV s−1, as described in [26]. The measurement accuracy was ±10% (Table 1). The correctness of determining ESA values by the amount of adsorbed/desorbed atomic hydrogen was also verified by the electrochemical CO desorption method. In this case, a constant electrode potential of 0.55 V was maintained, and the electrolyte was saturated with CO for 20 min and then with argon for 40 min. The CVs were measured in the 0.037–1.27-V range at a scan rate of 20 mV s−1 (3 cycles).
The electrochemical surface area of platinum was calculated from the expression ESA = Q/(R ∗ L). Here, Q is the charge consumed by CO oxidation or the half-sum of the charge consumed by hydrogen adsorption/desorption less the contribution from the double-layer region (μC); R is the charge consumed by the electrochemical conversion of the hydrogen or CO monolayer (210 or 420 μC cm−2 (Pt), respectively), L is the Pt loading on the electrode, and ESA is expressed in square centimeter per gram of Pt.
A series of linear voltammograms with the potential scan rate of 20 mV s−1 was measured in the range of 0.12 to 1.20 V at the speed of disc electrode rotation 1600 rpm to compare the activity of catalysts in the reaction of oxygen electroreduction. These curves were normalized as follows. The cell voltage, E, is defined as E = E reg − J i *R, where E reg is the registered value of potential, J i * R is the ion conduction resistance. Сompensation for the resistance of the electrolyte has been applied for R = 26 Ω. This value well corresponded to [30]. Correction of rotating disk electrode (RDE) linear sweep voltammogram (LSV) curves depicting the contribution of the processes in oxygen-free (Ar) solution (J i (O2) − J i (Ar)), as it was written in [31], has been made. The electrocatalytic activities of Pt/C catalysts in the oxygen reduction reaction (ORR) (kinetic currents) were extracted from the normalized RDE LSVs using a well-known mass transport correction for RDE measurements [32, 33], resulting from Koutecky-Levich equation:
where J is the experimentally obtained current, J d refers to the measured diffusion-limited current, and J k is the mass transport free kinetic current.
To assess electrocatalyst stability, a long-time (5000 cycles) voltammetric cycling method in a three-electrode cell with the potential sweep in the range of 0.6–1.1 V with a scan rate of 100 mV s−1 was selected [34]. After every 500 cycles, two CVs were recorded to calculate ESA (potential sweep rate of 20 mV s−1, the potential range of 0.03 to 1.26 V). Stability was evaluated by the value of the ESA j /ESA0 ratio, where j is the number of cycles.
To calculate the values of the specific currents and stability of the catalysts, the ESA values determined from the desorption/adsorption of atomic hydrogen were used.
Results and discussion
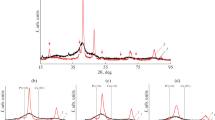
The obtained Pt/C materials contain from 13 to 20 wt% of platinum (Table 1). Only one, the most intensive Pt <111> reflection, characterized by 2 theta maxima of about 39.9°, is distinctly seen on the diffraction patterns of the samples (Fig. 1). This is the consequence of a small average crystallite size (D (111)) in these materials (Table 1). The broadening of the reflection peak for platinum <111> is also caused by the small size of the crystallites. The calculation, conducted by the Scherrer equation, shows that D (111) value increases in the line: Pt/CCO < Pt/CAr ≤ Pt/C from 1.5 to 2.2 nm.

XRD patterns of the samples. (1) Pt/C. (2) Pt/CCO. (3) Pt/CAr
The results of XRD treatment are well corroborated by electron microscopy studies (Fig. 2). The platinum nanoparticles are rounded in shape, and on the photos, they are seen as dark dotted areas on the light-colored surface of the carbon support. The coalescence of the nanoparticles is expressed very weakly, and large platinum agglomerates are missing on the carbon surfaces. The average size of nanoparticles in each material is the same as the calculated crystallite size (Table 1). The narrow size distribution and a more uniform spatial distribution of platinum nanoparticles are mostly characteristic for Pt/CCO electrocatalyst (Fig. 2b).

Transmission electron microscopy photographs and diagrams of the nanoparticle size distribution for Pt/C (a), Pt/CCO (b), and Pt/CAr (c)
The results of XRD and TEM study confirm our hypothesis: adsorption of CO molecules on the platinum surface impedes the process of Pt nuclei growth during the synthesis. Molecules of carbon oxide (II) act as a stabilizing agent. They prevent the aggregation of Pt nanoparticles and hamper the anchoring of Pt particles formed in solution on the surface of carbon support. This causes the decrease of platinum content in the Pt/CCO compared to Pt/C and Pt/CAr (Table 1).
Molecular oxygen chemisorption on platinum takes place at very positive potentials. In the solution with the excess of reducing agent, platinum potential should take a highly negative value. Therefore, the dissolved O2 molecules are weakly adsorbed on the surface of platinum nanoparticles during their synthesis in the air atmosphere. Argon atoms cannot be adsorbed on the surface of the platinum nuclei. Apparently, adsorption of the formaldehyde molecules and intermediates, formed during the synthesis, prevails in these conditions. A less uniform spatial distribution of platinum nanoparticles on the carbon support surface in the case of synthesis in air atmosphere can be due to the influence of O2 molecules on the number of active centers on the carbon surface (Fig. 2a). Blocking a part of such centers may cause a less-uniform distribution of the nanoparticles on the surface of the support, which is characteristic for this sample.
It is important to make sure that the observed differences in the morphology of the synthesized electrocatalysts are reflected in their ESA values and in the electrochemical behavior. It is interesting to compare the activity and stability of the obtained materials with the commercial analogues, while considering the small size of the platinum nanoparticles, their uniform distribution on the carbon surface, and the narrow size distribution. Finally, based on the total estimation of the ESA, electrocatalyst activity, and stability, one can make a conclusion of their quality.
During the preliminary standardization of the electrode surface, stationary current values are set (the shape of voltammograms is stabilized) after 10–15 CV cycles (Fig. 3). In fact, standardization ends as quickly as in the case of commercial Pt/C materials. That confirms the purity of the obtained catalysts and demonstrates relative stability of the microstructure. CVs of the standardized electrodes have the shape, which is characteristic for the Pt/C electrocatalysts (Fig. 4).

a–e One hundred CV curves of pretreatment for Pt/C materials in 0.1 M HClO4 saturated by Ar at room temperature. The scan rate is 200 mV s−1. f CV curves for standardized Pt/C and carbon support (Vulcan XC-72) materials in 0.1 M HClO4 saturated by Ar at room temperature. The scan rate is 20 mV s−1

CV curves for different Pt/C catalysts in 0.1 M HClO4 saturated by Ar at room temperature. Scan rate 20 mV s−1
Commercial E-TEK and Pt/CCO electrocatalysts, which contain the platinum nanoparticles of the smallest sizes, demonstrate the highest specific currents (A g−1 (Pt)) in the hydrogen areas of the voltammograms. CVs of E-TEK are characterized by a wider area of a double-layer part compared to other catalysts. Apparently, this is due to the higher surface area of the carbon support used in the production of this catalyst.
The ESA values of the standardized Pt/C catalysts, calculated by the results of the cyclic voltammetry (Fig. 4) (“Experimental” section), generally correlate with the size of the nanoparticles (crystallites) (Table 1). For Pt/C, Pt/CAr, and HiSPEC 3000 catalysts, ESA values are about 94–100 m2 g−1 (Pt), for Pt/CCO about 140 m2 g−1 (Pt), and for E-TEK about 85 m2 g−1 (Pt) (Fig. 4; Table 2). Note that the calculated values of ESA do not take into account a possible spillover of hydrogen atoms, but nevertheless, they correlate well with the published data for the commercial catalysts [31, 32].
To evaluate the activity of materials in the ORR, LSV of the obtained Pt/C material and their commercial analogues were compared (Fig. 5). All LSVs took into account an ohmic resistance of electrolyte and background correction of RDE LSVs (J i (O2) − J i (Ar)) (see “Experimental” section). The values of kinetic currents (Am−2 (Pt) and Ag−1 (Pt)) corresponding to the potential value of 0.9 V and half-wave potentials, used as indicators of activity for all catalysts [33, 35], are given in Table 2.

Normalized LSV curves for different Pt/C catalysts in 0.1 M HClO4 saturated by O2 at room temperature. Scan rate 20 mV s−1 and rotating speed 1600 rpm. Absolute (a) and specific (b) values of electric currents
The values of half-wave potentials are almost identical to all electrocatalysts studied (Table 2). Commercial sample HiSPEC 3000 shows the highest value of kinetic current and therefore specific activity (A cm−2 (Pt)). Kinetic current and specific activity of Pt/CCO at E = 0.9 V are a bit lower compared to other catalysts. Mass activities (A g−1 (Pt)) of HiSPEC 3000 and Pt/CCO at E = 0.9 V are a bit higher compared to other catalysts. Since J mas = J spec • ESA, one would expect a much greater mass activity for the Pt/CCO catalyst, which demonstrated a significantly higher ESA in comparison with other samples (Table 2). However, the decrease in specific activity (J spec) of platinum due to the size effect (see “Introduction” section) partly compensates the ESA effect. In conclusion, we would like to note that the observed minor difference in the values of the limiting currents in diffusion regions of the voltammograms (Fig. 5) can be caused by the structural features of the porous catalyst layer for each material.
To the best of our knowledge as well as the information published in [31], data accuracy in determining the kinetic currents does not exceed 10% of their absolute values. Thus, in our opinion, the established difference in LSV parameters does not give grounds for selecting the most active catalyst. It is clear, however, that the homemade Pt/C samples are no worse than the commercial HiSPEC 3000 and E-TEK samples by the ORR mass activity.
Another important functional characteristic of Pt/C catalysts, along with the ORR activity, is their durability, i.e., stability of characteristics during a long-time operation. Rapid assessment of this indicator can be carried out in various modes of voltammetric cycling. Each of them corresponds to one or another mode of the operation in a fuel cell [11, 36, 37]. Selected cycling conditions (see “Experimental” section) correspond to the load cycle conditions of PEMFC exploitation [36, 38].
During the stress test, all the studied electrocatalysts degraded but as a result showed significantly different stability (Table 2): a relative change of ESA ranged from 17% (Pt/C sample) to 39% (HiSPEC 3000 sample). Thus, with cycling, ESA decreases evenly only for the commercial E-TEK catalyst (Fig. 6a, b). Other catalysts degrade at a faster rate during the first 1500–2000 cycles (Fig. 6a, b; Table 2). For example, HiSPEC 3000 ESA decreases by 39% from the initial value after 5000 cycles, while 28% of them occur in the first 2000 cycles (Table 2). Similar degrees of degradation by 17 and 11%, respectively, are determined for Pt/C; 32 and 23%, respectively, for Pt/CCO; and 30 and 23%, respectively, for Pt/CAr (Table 2). Сomparison of values for relative stability shows that (i) the samples that we have synthesized are more stable compared to the commercial electrocatalysts and (ii) Pt/C electrocatalyst is the most durable under conditions of the stress test (Fig. 6a; Table 2). The Pt/CCO electrode retains the highest ESA values during the stress test. This becomes evident when we compared the absolute values of ESA (Table 2) and analyzed the nature of their changes (Fig. 6b).

Change a of the relative stability (ESAj/ESA0) and b of ESA absolute values for Pt/C electrocatalysts during the stress test (5000 cycles)
Interpretation of the accelerated stress testing results for electrocatalysts is hampered by the lack of knowledge about the contribution of various mechanisms (see “Introduction” section) to the degradation of these materials [3]. According to [39, 40], the higher Pt/C electrocatalyst stability based on the same support is, the more Pt loading, size, and average distance values between nanoparticles are. The authors [39] have shown that the broader the size distribution of the nanoparticle is, the more probable the location of large and small nanoparticles adjacent to each other becomes. Consequently, the contribution of platinum re-precipitation processes (from small nanoparticles to the big ones) to the total ESA decline is higher in such material.
The previous considerations explain only in part the results obtained during the accelerated test. Among the homemade catalysts, the Pt/C material is characterized by the greatest D av values of the nanoparticles (Table 1). This may be the main reason which causes its high stability compared to Pt/CCO and Pt/CAr. At the same time, the sample synthesized in the CO atmosphere, which contained the smallest nanoparticles (Table 1), in its stability is in no way inferior to stability of the Pt/CAr catalyst ( Fig. 6a; Table 2). The smallest size distribution of nanoparticles (Fig. 2) and a relatively large average distance between the nanoparticles [3] are the features of the Pt/CCO positive morphology, which may increase resistance to degradation. It is caused by the lowest Pt loading in all samples of the studied materials. The lack of large nanoparticles slows down “Ostwald ripening” for deposited platinum, and a large distance between the nanoparticles reduces the probability of their coalescence. We cannot also exclude a stronger anchoring of nanoparticles to the carbon surface, caused by specific state of the platinum and carbon surfaces during the synthesis in the CO atmosphere.
The process of clarifying the reasons for a comparatively low stability of commercial electrocatalysts HiSPEC 3000 and E-TEK, which contained the largest nanoparticles (Table 1), as well as the reasons which cause E-TEK uniform degradation during the cycling (Fig. 6), was hampered by the lack of precise knowledge about the methods for their preparation and conditions for pretreatment of carbon supports.
Conclusions
Pt/CCO, Pt/CAr, and Pt/C electrocatalysts with platinum loading from 13 to 20 wt% have been obtained by the method of chemical reduction of platinum(IV) by formaldehyde in the suspension of carbon black Vulcan XC-72 in CO, Ar, and air atmospheres. The assumption about the impact of the gas atmosphere on the Pt/C morphology during the synthesis was confirmed by the results of the study that we have performed. The size distribution of nanoparticles broadens, and their average size increases in the line Pt/CCO < Pt/CAr < Pt/C from 1.5 to 2.1 nm. The presence of the CO molecules adsorbed on the surface of the platinum nuclei makes more complicated not only the growth of the nanoparticles, but also their adhesion to the carbon support.
ESA of the synthesized Pt/C catalysts changes from 94 to 97 m2 g−1 (Pt) (Pt/C and Pt/CAr) to 139 m2 g−1 (Pt) (Pt/CCO). Values of the ORR mass activity of the homemade catalysts are not inferior to the mass activity of commercial counterparts HiSPEC 3000 (Johnson Matthey) and E-TEK, which contain 20 wt% Pt. At the same time, they are superior to the commercial catalysts by their durability under the accelerated CV stress tests with the potential range of 0.6–1.1 V.
The rate of degradation during the cycling is maximal in the first 1500–2000 cycles and is markedly reduced upon further cycling for the majority of the catalysts that have been studied. Degradation with almost the constant rate is fixed only for the commercial E-TEK catalyst. The Pt/C sample, synthesized in the air atmosphere, demonstrated abnormally high stability. Its ESA decreased by only 17% after 5000 cycles. The Pt/CCO catalyst retained the highest values of the ESA throughout the testing process. It makes these catalysts promising candidates for further study.
The established features in the electrochemical behavior of the synthesized Pt/C catalysts are caused by the peculiarities in their morphology, which are, in turn, caused by the adsorption of gases on the surfaces of the carbon support and on the growing platinum nuclei during the synthesis. Probably, the change in the state of the surfaces affects the strength of the platinum nanoparticle adhesion to the carbon support.
Alterations in the composition of the atmosphere during wet synthesis of nanoparticles (particularly, the synthesis in the CO atmosphere) can be used as one of parameters to control morphology and electrochemical behavior of Pt/C materials.
References
Holton OT, Stevenson JW (2013) Platin Met Rev 57:259–271
Singh RN, Awasthi R, Sharma CS (2014) Int J Electrochem Sci 9:5607–5639
Meier JC, Galeano C, Katsounaros I, Witte J, Bongard HJ, Topalov AA, Baldizzone C, Mezzavilla S, Schüth F, Mayrhofer KJJ (2014) Beilstein J Nanotechnol 5:44–67
Thompsett D (2003) Handbook of fuel cells. In: Fundamentals technology and applications, 3. Wiley Ltd, New York pp. 6–1–6-23
Shao Y, Yin G, Gao Y (2007) J of Power Sources 171:558–566
Wu J, Yuan XZ, Martin JJ, Wang H, Zhang J, Shen J, Wu S, Merida W (2008) J Energy Power Sources 184:104–119
Yang Z, Hafez IH, Berber MR, Nakashima N (2015) ChemCatChem 7:808–813
Leontyev IN, Belenov SV, Guterman VE, Haghi-Ashtiani P, Shaganov AP, Dkhil B (2011) J Phys Chem C 115:5429–5434
Li D, Wang C, Strmcnik DS, Tripkovic DV, Sun X, Kang Y, Chi M, Snyder DJ, Van Der Vliet D, Tsai Y, Stamenkovic VR, Sun S, Markovic MN (2014) Energy Environ 7:4061–4069
Kinoshita K (1990) J Electrochem Soc 137:845–848
Zhang S, Yuan X, Hin JNC, Wang H, Friedrich KA, Schulze M (2009) J Power Sources 194:588–600
Saha MS, Neburchilov V, Ghosh D, Zhang J (2013) Wiley Interdiscip. Rev. Energy Environ 2:31–51
You DJ, Kwon K, Joo SH, Kim JH, Kim JM, Pak C, Chang H (2012) Int J Hydrog Energy 37:6880–6885
Alegre C, Gálvez ME, Moliner R, Baglio V, Aricò AS, Lázaro MJ (2014) Appl Catal B 147:947–957
Jiang Q, Peng Z, Xie X, Du K, Hu G, Liu Y (2011) Trans Nonferrous Met Soc China 21:127–132
Chan K, Ding J, Ren J, Cheng S, Tsang KY (2004) J Mater Chem 14:505–516
Ye F, Liu H, Feng Y, Li J, Wang X, Yang J (2014) Electrochim Acta 117:480–485
Latsuzbaia R, Negro E, Koper G (2015) Fuel Cells 15:628–638
Coutanceau C, Baranton S, e-Lyse TWN (2012) The delivery of nanoparticles (Eds.: Dr. Abbass A. Hashim) pp. 403–430.
Chandravathanam S, Viswanathan B (2011) Varadarajan. Sci Adv Mater 3:1031–1037
Ch W, Chi M, Li D, Strmcnik D, Vliet D, Komanicky WV G, Chang K, Paulikas AP, Tripkovic D, Pearson J, More KL, Markovic NM, Stamenkovic VR (2011) J Am Chem Soc 133:14396–14403
Zhou Z, Wang S, Zhou W, Jiang L, Wang G, Sun G, Zhou B, Xi Q (2003) Phys Chem Chem Phys 5:5485–5488
Corradini P, Antolini E, Perez J (2013) Phys Chem Chem Phys 15:11730–11739
Spendelow JS, Lu GQ, Kenis PJA, Wieckowski A (2004) J Electroanal Chem 568:215–224
Torigoe K, Nakajima Y, Esumi K (1993) J Phys Chem 97:8304–8309
Guterman VE, Alekseenko AA, Volochaev VA, Tabachkova NY (2016) Inorg Mater 52:23–28
Chiang Y-C, Liang C-C, Chung C-P (2015) Materials 8:6484–6497
Perez J, Paganin VA, Antolini E (2011) J Electroanal Chem 654:108–115
Shelekhov EV, Sviridova TA (2000) Metal Science and Heat Treatment 42:309–313
Vliet D, Strmcnik DS, Wang C, Stamenkovic VR, Markovic NM, Kope MTM (2010) J Electroanal Chem 647:29–34
Shinozaki K, Zack JW, Pylypenko S, Pivovar BS, Kocha SS (2015) J Electrochem Soc 162:F1384–F1396
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT (2005) Appl Catal B 56:9–35
Khudhayer WJ, Kariuki NN, Wang X, Myers DJ, Shaikh AU, Karabacak T (2011) J Electrochem Soc 158:B1029–B1041
Weimin C, Xin Q, Sun G, Wang Q, Mao Q, Su H (2008) J Power Sources 180:199–204
Lin R, Zhao T, Shang M, Wang J, Tang W, Guterman VE, Ma J (2015) J Power Sources 293:274–282
Ohma A, Shinohara K, Iiyama A, Yoshida T, Daimaru A (2011) ECS Trans 41:775–784
Cherevko S, Kulyk N, Mayrhofer KJJ (2016) Nano Energy. Articles in Press. doi:10.1016/j.nanoen.2016.03.005
Neyerlin KC, Srivastava R, Yu C, Strasser P (2009) J Power Sources 186:261–267
Holby EF, Sheng W, Shao-Horn Y, Morgan D (2009) Energy Environ Sci 2:865–871
Rinaldo SG, Stumper JR, Eikerling M (2011) J Phys Chem C 114:5773–5785
Acknowledgements
The authors express their gratitude to the Russian Science Foundation (grant 16-19-10115), which has supported this work. We are grateful to Dr. Vadim Volochaev for the assistance in registering X-ray spectra of the catalysts as well as to Dr. Sergey Belenov for the fruitful discussion of the results obtained.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Alekseenko, A.A., Guterman, V.E., Tabachkova, N.Y. et al. Impact of the atmosphere composition in the process of synthesis on the morphology and electrochemical performance of Pt/C electrocatalysts. J Solid State Electrochem 21, 2899–2907 (2017). https://doi.org/10.1007/s10008-017-3581-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-017-3581-8