
Abstract
Heat and free chlorine are among the most efficient and commonly used treatments to inactivate enteric viruses, but their global inactivation mechanisms have not been elucidated yet. These treatments have been shown to affect at least the capsid proteins of viruses and thus may affect the surface properties (i.e. electrostatic charge and hydrophobicity) of such particles. Our aim was to study the effects of heat and free chlorine on surface properties for a murine norovirus chosen as surrogate for human norovirus. No changes in the surface properties were observed with our methods for murine norovirus exposed to free chlorine. Only the heat treatment led to major changes in the surface properties of the virus with the expression of hydrophobic domains at the surface of the particles after exposure to a temperature of 55 °C. No modification of the expression of hydrophobic domains occurred after exposure to 60 °C, and the low hydrophobic state exhibited by infectious and inactivated particles after exposure to 60 °C appeared to be irreversible for inactivated particles only, which may provide a means to discriminate infectious from inactivated murine noroviruses. When exposed to a temperature of 72 °C or to free chlorine at a concentration of 50 mg/L, the genome became available for RNases.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pathogenic enteric viruses are commonly involved in waterborne and foodborne diseases in Europe and in the USA (EFSA 2016; Hall et al. 2014). Among them, human noroviruses (HuNoVs) are a leading cause of acute gastroenteritis worldwide (Verhoef et al. 2015) and two cell culture methods have recently been proposed for the detection of their infectious forms (Ettayebi et al. 2016; Jones et al. 2015). These methods are far from being immediately usable in routine analysis and standardized RT-PCR methods targeting viral genomes (ISO/TS 15216 standard 2013) are currently used by control laboratories (ISO 2013). However, the persistence of the genome is much greater than that of the corresponding infectious virus in most cases (de Roda Husman et al. 2009; Gassilloud et al. 2003; Prevost et al. 2016; Seitz et al. 2011). This makes questionable the use of viral genome data for risk assessment in food and environmental virology. Thus, it appears that knowledge about the differences between infectious and non-infectious viral particles, and more broadly about the mechanisms of inactivation, is crucial to improve the analysis of viruses in food and environmental matrices.
Numerous studies have been conducted on the mechanisms of virus inactivation which depend on the inactivation treatment. Moreover, the virus composition, structure and function surely affect the mechanisms of virus disinfection (Wigginton and Kohn 2012). The detection of a small fragment of genome by RT-PCR methods and the variation in susceptibility of some regions of the genome (Wigginton et al. 2012) have led to the development of methods focusing on the capsid integrity in terms of permeability and functionalities over the last ten years. Enzymatic treatments such as RNases limit the detection to genomes coming from intact particles under conditions leading to the disruption of the capsid (Brié et al. 2016) and have largely proven effective under very high temperature conditions (Nuanualsuwan and Cliver 2002; Pecson et al. 2009; Topping et al. 2009). Pre-treatment with the intercalating dyes ethidium monoazide or propidium monoazide coupled with molecular detection could allow better assessment of the viral risk in water when multiple disinfection treatments such as ozonation and chlorination are used (Prevost et al. 2016). The capacity of the virus to bind to its host cell, or to its receptor, reflects the preservation of this major function of the viral capsid. However, the loss of this function depends on the inactivation treatment. For example, the binding capacity of free chlorine-inactivated poliovirus and MS2 phage did not seem to be affected (O’Brien and Newman 1979; Wigginton et al. 2012), whereas a correlation between loss of infectivity and loss of this function was reported with heat treatment (Brié et al. 2016; Wigginton et al. 2012). Besides, permissive cell lines, or at least virus receptors, should be identified prior to applying such a method. New approaches evaluating the oxidation effect of free chlorine on viral capsid proteins with the use of a biotin-avidin system have concluded that the number of carbonyl groups on the viral capsid proteins of human and murine noroviruses increased with increasing free chlorine dose (Sano et al. 2010, 2015; Tojo et al. 2013). Beyond the permeability or the specific functionalities of the capsid, the surface properties of viral particles such as electrostatic charge and hydrophobicity seem to be affected during inactivation. Indeed, an increase in hydrophobicity was observed for poliovirus exposed to 50 °C, which was found to be linked to the externalization of the N-terminus part of the viral protein VP1 (Curry et al. 1996; Fricks and Hogle 1990). Recently, the expression of hydrophobic domains was also demonstrated for MS2 phage and this phenomenon was reported to be specific of particles remaining infectious after exposure to 60 °C (Brié et al. 2016). Concerning the electrostatic charge of viral particles, a decrease in the isoelectric point (IEP) was observed for poliovirus inactivated by heat (O’Brien and Newman 1979).
Thus, the aim of the present work was to evaluate the effect of heat and free chlorine on the surface properties of viral particles using a surrogate for HuNoVs. Heat and free chlorine are among the most cost-effective and easiest to apply processes and sanitizers in the food and water industries. Heat remains the most studied of the virucidal techniques for enteric viruses with studies focusing on poliovirus (Belnap et al. 2000; Breindl 1971; Curry et al. 1996; Fricks and Hogle 1990) and more recently on the F-specific RNA phage MS2 (Brié et al. 2016; Pecson et al. 2009; Wigginton et al. 2012). Less data are available about the mechanisms of inactivation induced by chlorine with few studies focusing on pathogenic enteric viruses but also on their surrogates such as MS2 phage or murine norovirus (MNV) (Li et al. 2002; Lim et al. 2010; O’Brien and Newman 1979; Sano et al. 2010, 2015; Sigstam et al. 2013; Tojo et al. 2013; Wigginton et al. 2012). Among norovirus surrogates, MNV and Tulane virus have recently been described as the best ones for the evaluation of inactivation and disinfection treatments (Cromeans et al. 2014). Although the application of HuNoV surrogates has been challenged because they may not mimic the viruses they represent (Knight et al. 2016; Richards 2012), MNV is widely used in areas of applied and fundamental virology such as the evaluation of food processing technologies (Hirneisen and Kniel 2013), agricultural practices (Wang and Kniel 2016), or the understanding of infection mechanisms (Karst and Wobus 2015). The first objective was to evaluate the effects of heat and free chlorine on infectivity, host cell binding capacity, genome stability and capsid permeability to RNase. RNase treatment was used to demonstrate the presence of non-infectious viruses still possessing a genome protected by the capsid after both heat and free chlorine treatments. The global electrostatic charge and hydrophobicity of MNV were then monitored for several temperatures and free chlorine concentrations.
Materials and Methods
Cell Line, Virus and Purification
RAW 264.7 cells (ATCC TIB-71) were used in this study between passages 15 and 30. They were cultured in DMEM-10 which was Dulbecco’s modified Eagle medium (DMEM, ThermoFisher Scientific) supplemented with 10% foetal bovine serum (FBS, ThermoFisher Scientific), 1% non-essential amino acids (NEAA, ThermoFisher Scientific) and 1% HEPES solution (1 M, Dominique Dutscher). RAW 264.7 cells were grown at 37 °C in a 5% CO2 atmosphere.
MNV-1 strain S99 was kindly provided by the Friedrich Loeffler Institute (Germany). RAW 264.7 cells were infected with MNV-1 S99 at an MOI of 0.1 for 2 days. Three freeze/thaw cycles and centrifugation (400×g for 15 min) were then performed to remove cell debris from the viral lysate as previously described (Wobus et al. 2004). The supernatant was collected, aliquoted (1 mL) and stored at −80 °C.
Prior to each experiment, an MNV-1 aliquot was thawed, dialysed (Float-A-Lyzer G2 dialysis device 100 kDa 1 mL, Spectra/Por) overnight against phosphate-buffered saline (PBS) at a concentration of 1 mM (1 mM Na2HPO4 with 14 mM NaCl, Fisher Bioreagents) and kept at 4 °C before use.
Infectivity Assay
The TCID50 (tissue culture infectious dose50) assay was used to determine infectious virus titres (Hwang et al. 2015). RAW 264.7 cells were seeded into a 96-well plate at a density of 3 × 104 per well, the day before MNV titration. Serial tenfold dilutions of the working suspension of the virus were made in DMEM-2 which was DMEM supplemented with 2% FBS, 1% NEAA, 1% HEPES solution and 1% sodium pyruvate (ThermoFisher Scientific). Aliquots (100 µL) of each dilution were then inoculated into 16 wells of the 96-well plate with the RAW 264.7 cell monolayer. The plates were incubated for 3–5 days at 37 °C in a 5% CO2 atmosphere. The titre of the virus was then calculated using Spearman-Karber’s method (Karber 1931). The final concentration of infectious particles in dialysed MNV was around 107 TCID50/mL.
Genome Extraction and Quantification
Viral RNA was extracted from 50 µL samples using the QIAamp viral RNA kit (Qiagen) and eluted in 100 µL of the appropriate buffer, according to the manufacturer’s instructions. The extracts were immediately stored at −80 °C. According to the manufacturer’s recommendations, reverse transcription (RT) was performed from 7.5 µL of extracted RNA using a reverse primer (5′-CCATATCCAACTCGAGGTTGGT) at a final concentration of 0.4 µM, 100 U of SuperScript III (Life Technologies) and 20 U of RNaseOUT (Life Technologies) in a 20-µL reaction volume. The RT step was performed at 50 °C for 60 min and then at 70 °C for 15 min. Quantitative PCR (Life Technologies, StepOne Plus) was then carried out from 5 µL of cDNA with the TaqMan universal PCR master mix (Life Technologies) in a 25 µL reaction volume, with reverse and forward (5′-CCTGACATTGTGATGCAAGAATC) primers at a final concentration of 0.4 µM and the TaqMan MGB probe (5′-FAM-CGTCATCACCATAGAAG-MGBNFQ) at a concentration of 0.2 µM. PCR amplification was performed at 50 °C for 2 min and 95 °C for 10 min, followed by 45 cycles of 15 s at 95 °C and 60 s at 60 °C. The primers and probe used in the RT-qPCR (Life Technologies, StepOne Plus) were as previously defined (Belliot et al. 2008). Negative and positive controls (with known quantities of MNV) were included in each experiment to ensure no contamination and good reproducibility of the extractions and RT-qPCR steps. During the experiments, RT-qPCR efficiency ranged between 85 and 95%.
Determination of the number of MNV genome copies per mL (gc/mL) used for the standard curve was performed using an RNA transcript from a plasmid (Eurofins, pEX-A-A228235-MNV) quantified by a UV–Vis spectrophotometer (Thermo Scientific, NanoDrop 2000). No DNA contamination was detected in our RNA transcript solution. The calculation for the gc/mL was performed with a size of 286 nucleotides corresponding to the transcript size. The final number of genome copies in dialysed MNVs was around 5 × 107 gc/mL.
Inactivation of MNV by Heat and Free Chlorine
Dialysed viruses (≈250 µL) were used at a concentration of 1 × 107 TCID50/mL in 1 mM PBS (1 mM Na2HPO4 with 14 mM NaCl) for heat and free chlorine treatments.
MNV-1 was exposed to different temperatures—45, 50, 55, 60, 72, and 80 °C—for 10 min in prewarmed Protein LoBind tubes (Eppendorf) in a water bath. Heated samples were immediately placed on ice to avoid further inactivation.
To prepare our free chlorine stock solution, concentrated sodium hypochlorite solution (5%, Acros) was used and diluted using ultrapure water (>18 MΩ/cm). Determination of free chlorine concentration was performed according to the ISO 7393-2 (ISO 1985) procedure with the use of diethyl-p-phenylenediamine (DPD, Acros). The working concentrations of free chlorine were 20 and 50 mg/L with a contact time of 15 min at 4 °C. Residual free chlorine was immediately quenched by adding 2 mol of sodium thiosulfate equivalent to 1 mol of free chlorine.
For the following experiments, MNV was tenfold diluted in the appropriate buffer corresponding to the assay.
RNase Assay
Suspensions of MNV-1 (final concentration of 107 gc/mL) were treated with RNase A (final concentration of 1 µg/mL, AppliChem) at 37 °C for 60 min as previously described for MS2 phage (Brié et al. 2016). MNV-1 suspensions without RNase treatment were used as negative controls for both TCID50 and RT-qPCR assays. The efficiency of the treatment was estimated using RNA extracted from MNV-1 (107 gc/mL) as positive control. The removal of RNase during the genome extraction procedure was assessed by a tenfold dilution of the samples which gave similar results (i.e. between 75 and 110%) to those of the undiluted samples.
Binding of MNV to RAW 264.7 Cells
Binding assays of MNV to its host cells, RAW 264.7, were performed in 24-well plates (5 × 105 cells/well in 1 mL DMEM-10). The medium was removed prior to adding 500 µL of viral suspension in DMEM-2. To allow viral particles to adsorb, the plate was incubated for 2 h at 37 °C in a 5% CO2 atmosphere. Next, each well was washed twice with 1 mL of DMEM-2. Lysis was carried out twice for 10 min with 250 µL of lysis buffer. The viral genome was then extracted and quantified by RT-qPCR. The modification of the binding capacity of MNV to RAW 264.7 cells was modelled as \({\text{log }}\frac{Q}{{Q_{ 0} }}\), where Q is the viral RNA of MNV found on the cells after treatment and Q 0 before treatment. Thus, the log reduction of the binding capacity to RAW 264.7 cells can also be expressed as a percentage of MNV particles which have lost this functionality.
Binding of MNV to Hydrophobic or Positively Charged Beads
Two types of magnetic beads were used to evaluate the hydrophobicity and the negative global charge of MNV (Brié et al. 2016). Hydrophobic beads and positively charged beads were coated with polystyrene (PolySciences) and primary secondary amine (PSA, Bioclone), respectively.
Briefly, prior to the experiments, the beads were washed twice in the corresponding buffer, which was either 10 mM PBS (10 mM Na2HPO4 with 150 mM NaCl) for hydrophobic beads or 50 mM PBS (50 mM Na2HPO4 with 20 mM NaCl) at different pH levels (7.0, 4.0, 3.0 and 2.5) for ion exchange beads. The binding assays were performed in Protein LoBind tubes with a viral concentration of 106 TCID50/mL and around 108 beads/mL, in a final volume of 2 mL. The binding assay using hydrophobic beads was carried out twice for 2 h at room temperature using Dynal MX1 Mixer (Life Technologies). New washed beads were used for the second run. With ion exchange beads, only one run was done for 10 min at room temperature using Dynal MX1 Mixer too. Then, the beads were separated from the supernatant with a magnet for 2–3 min. Supernatants were analysed by both TCID50 (infectious MNV) and RT-qPCR (total MNV) methods, and beads were analysed by RT-qPCR method only. The modification of the binding capacity of MNV to beads was estimated by \({ \log } \frac{{C_{\text{X}} }}{{C_{0} }}\), where C X is the concentration of either infectious MNV or MNV RNA after treatment and C 0 before treatment in suspension.
Statistical Analysis
Data were entered into Microsoft Excel 2010 (Microsoft Corporation) and analysed in StatEL 2.7 (Ad Science). Significant differences between groups were analysed by Kruskal–Wallis test. A p value of <0.05 was considered as significant.
Results
MNV production resulted in a viral stock of 107 TCID50/mL, with no free genome since no changes in the RT-qPCR signal were detected after RNase treatment. Different levels of heat and free chlorine were applied on MNV to monitor their effects on infectivity, genome persistence, capsid integrity and binding capacity to host cells. The conditions allowing preservation of the capsid integrity of inactivated MNVs were then selected to evaluate their impact on both global charge and hydrophobicity.
Global Effects of Heat and Free Chlorine on MNV Particles
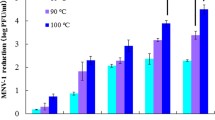
MNV was exposed to various temperature conditions, from 45 to 80 °C, for 10 min (Fig. 1). As expected, infectivity decreased as temperature increased: −0.4 ± 0.1 log10 at 45 °C, −1.1 ± 0.2 log10 at 50 °C, −2.6 ± 0.6 log10 at 55 °C and −5.6 ± 0.3 log10 at 60 °C. Complete inactivation, above 6 log10, was observed at 72 and 80 °C. The RT-qPCR signal slightly decreased for temperatures above 55 °C: −0.5 ± 0.3 log10 at 60 °C, −0.6 ± 0.2 log10 at 72 °C and −0.4 ± 0.2 log10 at 80 °C. Therefore, heat exposure did not involve an effective loss of the RT-qPCR signal since the decrease was not temperature dependent. Next, MNV capsid integrity was evaluated by performing RNase treatment after heat exposure. The efficiency of the RNase treatment was checked on extracted MNV RNA, and a decrease of over 3 log10 was obtained. An increase in RNase sensitivity was then noticed only for MNV exposed to 72 or 80 °C with an additional genome decay of −1.5 log10 for 72 °C and −2.3 log10 for 80 °C, suggesting impairment of the capsid integrity. Only the three temperatures at which the capsid integrity was conserved (i.e. 50, 55, and 60 °C) were kept for the binding assays to RAW 264.7 host cells. The binding capacity to RAW 264.7 cells decreased very slowly when temperature increased: −0.6 ± 0.1 log10 at 50 °C, −0.8 ± 0.3 log10 at 55 °C, and −1.0 ± 0.1 log10 at 60 °C. Nevertheless, these results showed that the loss of infectivity was related at least to the loss of binding capacity to the host cells for 75–90% of the MNV particles.

Decay of MNV concentration [log10 (C x /C 0)] after exposure to different temperatures for 10 min as determined by RT-qPCR (circle forms) and TCID50 (square forms) assays without (empty forms) or with (full forms) RNase treatment. The dotted lines are only guides to the eye. In the molecular assay, decay was calculated using the threshold cycle (C T) values and the slope of the standard curve: log10 (C x /C 0) = (C T0−C TX)/slope. Three independent experiments were performed for each point (n = 3). Error bars indicate standard deviations. Downward arrow below the detection limit; C X, concentration after inactivation; C 0, initial concentration
For MNV exposed to free chlorine, inactivation rates of −1.6 ± 0.4 log10 for 20 mg/L and −4.2 ± 0.8 log10 for 50 mg/L were observed (Fig. 2); the residual chlorine level reached 2 and 9 mg/L, respectively. A decrease in the RT-qPCR signal was noted only for 50 mg/L, reaching −1.3 ± 0.6 log10. An increase in RNase sensitivity (−1.1 log10) was observed only with 50 mg/L. This suggested impairment of the capsid integrity by the highest concentration of free chlorine. Decreases in the binding capacity of −0.4 ± 0.1 log10 for 20 mg/L and −1.4 ± 0.1 log10 for 50 mg/L were observed, meaning that the loss of infectivity by chlorine was related at least to the loss of binding capacity to the host cells for 60% of the MNV particles.

Decay of MNV concentration [log10 (C x /C 0)] after exposure to 20 and 50 mg free chlorine/L for 15 min at 4 °C as determined by RT-qPCR (circle forms) and TCID50 (square forms) assays without (empty forms) or with (full forms) RNase treatment. The dotted lines are only guides to the eye. In the molecular assay, decay was calculated using the threshold cycle (C T) values and the slope of the standard curve: log10 (C x /C 0) = (C T0−C TX)/slope. Three independent experiments were performed for each point (n = 3). Error bars indicate standard deviations. C X, concentration after inactivation; C 0, initial concentration
Effect of Heat and Free Chlorine on the Charge of MNV Particles
Based on the results of a previous study (Brié et al. 2016) and on the concentration of our MNV suspension, the effect of heat and free chorine on the global charge of MNV was estimated by adhesion to positively charged beads only. These beads allowed the global charge of infectious viruses (determined by TCID50 assay) and that of total viruses (determined by RT-qPCR) to be evaluated separately. Experiments were done for one temperature condition (55 °C for 10 min) and one concentration of free chlorine (20 mg/L) at which sufficient inactivation of around 1 log was reached, while keeping enough infectious particles to compare their surface properties to those of inactivated ones. Adhesion experiments were performed in triplicate in 50 mM PBS for different pH levels (2.5, 3.0, 4.0 and 7.0) for native and treated MNVs. The profile of adhesion to the positively charged beads was compared between native viruses and heated (Fig. 3a) or chlorinated (Fig. 3b) viruses. For native viruses, slight adhesion was observed at pH 7.0, which increased at pH 4.0. No adhesion was observed at pH 3.0 and 2.5. For heated MNVs, greater adhesion was observed at pH 7.0 when compared to native MNVs (p < 0.05, Kruskal–Walis test). No difference in the adhesion profile was observed between native MNVs and those exposed to free chlorine (p < 0.05, Kruskal–Walis test). Overall, the IEP of native MNVs and MNVs exposed to heat or free chlorine was estimated to be between 3.0 and 4.0. Moreover, heat and free chlorine did not seem to affect the global charge of MNV.

Binding assay to positively charged beads of MNV [log10 (C x /C 0)] in 50 mM PBS in the case of heated viruses (empty forms) at 55 °C for 10 min (a), and chlorinated viruses (empty forms) at 20 mg/L for 15 min at 4 °C (b). Native viruses (full forms) were used as reference in both cases. The circle forms and the square forms represent the total viruses detected by RT-qPCR and the infectious viruses detected by TCID50 assay, respectively. The dotted lines are only guides to the eye. In the molecular assay, binding was calculated using the threshold cycle (C T) values and the slope of the standard curve: log10 (C x /C 0) = (C T0−C TX)/slope. Three independent experiments were done for each point (n = 3). Error bars indicate standard deviations. C X, concentration after the binding assay; C 0, initial concentration
Effect of Heat and Free Chlorine on the Hydrophobicity of MNV Particles
The effect of heat and free chlorine on the hydrophobicity of MNV was studied by binding assays to hydrophobic beads. The experiments were performed at pH 7 and higher ionic strength than that used for positively charged beads (10 mM Na2HPO4 and 150 mM NaCl) in order to maximize hydrophobic interactions. For native MNVs, no adhesion was observed on these beads for both total (−0.1 ± 0.1 log10) and infectious (−0.2 ± 0.2 log10) viruses. The hydrophobicity of MNV particles was not affected by both concentrations of free chlorine, 20 and 50 mg/L (data not shown). The binding capacity to hydrophobic beads was then evaluated for MNV exposed to 45, 50, 55, and 60 °C for 10 min (Fig. 4). Interestingly, subjecting MNV to 55 °C highly increased the binding capacity to hydrophobic beads (p < 0.05, Kruskal–Walis test). A temperature of 60 °C for 10 min inactivated almost all viruses as described above, which exhibited a low level of hydrophobicity similar to that of native viruses. These inactivated viruses were then exposed again to 55 °C for 10 min. Contrary to the results obtained with native particles, no increase in hydrophobicity was then observed suggesting that the capsid proteins of inactivated particles had reached an irreversible conformation when exposed to 60 °C. We then investigated whether this low hydrophobic stable state was also reached by MNV remaining infectious after a 60 °C heat treatment. In order to obtain a sufficient concentration of infectious MNV after such a heat treatment, it was performed for only 1 min instead of 10 min. A concentration of infectious MNVs of 4.5 × 105 ± 2.7 × 105 TCID50/mL (n = 3) was obtained. The adhesion test performed immediately after this heat treatment resulted in no adhesion of infectious MNV particles onto hydrophobic beads showing that infectious as well as inactivated MNVs exhibited a low level of hydrophobicity after exposure to 60 °C. These MNVs were then exposed to 55 °C for 10 min and a second adhesion test on hydrophobic beads was done. An increase in adhesion corresponding to 6 × 105 genome copies was noted; this value was equivalent to that of infectious particles. Moreover, a very low concentration of infectious MNVs (4.3 ± 0.6 TCID50/mL) was detected in suspension at the end of the entire experiment confirming that all infectious MNV particles had adsorbed onto hydrophobic beads. In conclusion, after 60 °C heat treatment, infectious and inactivated MNVs exhibited a low hydrophobic state which was irreversible for inactivated particles only.

Binding of heated MNV (from 45 to 60 °C for 10 min) [log10 (C x /C 0)] to hydrophobic beads at pH 7 in 10 mM PBS (10 mM Na2HPO4 with 150 mM NaCl). The circle forms and the square forms represent the total phages detected by RT-qPCR and the infectious phages detected by plaque assay, respectively. At 60 °C, no infectious viruses were present. The dotted lines are only guides to the eye. In the molecular assay, binding was calculated using the threshold cycle (C T) values and the slope of the standard curve: log10 (C x /C 0) = (C T0−C TX)/slope. Three independent experiments were done for each point (n = 3). Error bars indicate standard deviations. C X, concentration after the binding assay; C 0, initial concentration
Discussion
MNV is increasingly used as a cultivable surrogate for human noroviruses in numerous areas such as evaluation of food processing technologies (Girard et al. 2016; Hirneisen and Kniel 2013; Predmore et al. 2015) and sanitization of contact surfaces (Park et al. 2015; Yeap et al. 2016). Heat and free chlorine are quite easy to apply in those contexts, but the distinction between inactivated viruses and those remaining infectious after such a kind of treatment is still challenging.
In that context, we aimed to determine if heat and free chlorine affect the surface properties of MNV particles and thus could allow methods based on surface properties to be developed for the specific detection of infectious MNV. Prior to any inactivation experiments, the surface properties of native MNV should be defined. The adhesion experiments performed with native MNV using positively charged beads at different pH levels allowed approaching its IEP which may be estimated between 3.0 and 4.0. To our knowledge, only a theoretical IEP value of around 4.8 has previously been described for MNV considering the major capsid protein (Bolton et al. 2013). IEP values of around four have recently been described for VLPs (Virus-like particles) of human norovirus GI.1 and GII.4 and feline calicivirus (Samandoulgou et al. 2015). Few hydrophobic domains appeared to be expressed at the surface of native MNV as previously described for native poliovirus and MS2 phage through adhesion to liposomes or hydrophobic supports, respectively (Curry et al. 1996; Dika et al. 2013).
The first issue discussed here is the preservation of the virus structure under our conditions of inactivation by heat or free chlorine. Indeed, heat has previously been shown to disrupt viral particles for temperatures ranging from 60 to 72 °C (Ausar et al. 2006; Brié et al. 2016; Ruokola et al. 2014). RNase and transmission electronic microscopy were in agreement in determining the critical temperature leading to the disruption of the capsid of MS2 phage (Brié et al. 2016). In the present study, RNase was the only relevant method, due the concentration of MNV in our working suspension. From our results, it appeared that the critical temperature for MNV leading, at least, to its sensitivity to RNase is strictly above 60 °C and below or equal to 72 °C. Exposure to 72 °C led to genome accessibility to RNase for poliovirus and hepatitis A virus (Nuanualsuwan and Cliver 2002; Pecson et al. 2009), whereas 60 °C was sufficient for feline calicivirus (Topping et al. 2009). Free chlorine has been shown to target both the viral capsid and the genome (Nuanualsuwan and Cliver 2003; Wigginton et al. 2012). Between the two concentrations of free chlorine, only the highest (50 mg/L) led to a decrease in the RT-qPCR signal of around 1 log10 and to an additional 1−log10 reduction by RNase; these conditions resulted in a 4 log10 reduction in the infectivity of MNV. Similarly, for MNV suspended in natural seawater, a more than 4 log10 reduction in infectivity was obtained with 2.5 mg of free chlorine/L over 60 min; the subsequent use of RNase yielded genome copy numbers closer to those of infectious particles than to those of the genome copies obtained without enzymatic treatment (de Abreu Corrêa et al. 2012).
Thus, RNase could be used to reduce false positives of up to 2 log10 of genome copies after heat exposure for temperatures strictly above 60 °C. This enzyme may also be useful after free chlorine exposure, but its conditions of use need to be more clearly defined.
Under inactivation conditions that preserve the impermeability of the viral capsid to RNase, a decrease in the host cell binding capacity was observed when temperature or free chlorine concentration increased. However, no explicit correlation between inactivation and decrease in the host cell binding capacity was found for these two inactivation parameters. Previous studies have shown that heat-inactivated poliovirus, hepatitis A virus and MS2 phage lost their ability to attach to host cells after exposure to heat (Brié et al. 2016; Nuanualsuwan and Cliver 2003; Wigginton et al. 2012). Some studies have concluded that free chlorine did not alter the host cell binding capacity of poliovirus, adenovirus and MS2 phage (O’Brien and Newman 1979; Page et al. 2010; Wigginton et al. 2012), whereas another study has shown that all inactivated poliovirus and hepatitis A virus particles failed to attach to their host cells (Nuanualsuwan and Cliver 2003). For MS2 phage in particular, the inactivation by free chlorine has been related to a loss in injection and replication of its genome (Wigginton et al. 2012). The binding capacity to host cells did not appear to be relevant for the detection of infectious MNVs, especially for those exposed to free chlorine. Nevertheless, this functionality of the capsid was affected by heat and free chlorine, suggesting that changes in global charge or hydrophobicity could also occur.
Free chlorine seemed to have no effect on the global charge and hydrophobicity of MNV. The impact of free chlorine on the surface properties of viral particles remains poorly described. A previous study concluded only that free chlorine did not affect the IEP of poliovirus (O’Brien and Newman 1979). Heat seemed to impact the surface properties of MNV more deeply than free chlorine. Contradictory effects of heat on the global charge have been described for enteric viruses. Indeed, no change in the IEP has been observed for MS2 phage exposed to heat (Brié et al. 2016), whereas a drastic decrease in the IEP from 7.5 to 4.5 has been noted for poliovirus and related to a loss of the capsid protein VP4 (Breindl 1971; O’Brien and Newman 1979). The slight increase in adhesion capacity to positively charged beads at pH 7.0 observed for MNV heated at 55 °C may be explained by the ionic strength applied during adhesion experiments which could favour the detection of hydrophobic domains expressed after this heat treatment. Indeed, subjecting MNV to 55 °C led to an increase in hydrophobicity for both infectious and inactivated viruses. Interestingly, MNVs exposed to 60 °C showed a very low level of hydrophobicity, similar to that of native particles. For MS2 phage exposed to 60 °C for 10 min, an increase in hydrophobicity was observed for remaining infectious phages only (Brié et al. 2016). A transient hydrophobic state has also been observed for poliovirus 1 exposed to heat (Fricks and Hogle 1990; Tuthill et al. 2010); nevertheless, these hydrophobic particles remained infectious only for Chinese Hamster Ovary and murine L cells which are not susceptible to infection by native poliovirus (Curry et al. 1996). More interestingly, the low hydrophobic state reached by MNVs after exposure to 60 °C seemed to be irreversible for inactivated viruses only and may provide prospects for discriminating between infectious and inactivated viruses.
In light of our results, it appeared that above the critical temperature, which is probably around 72 °C for MNV, treatment with RNase improved the specific detection of RNA from infectious MNV. For heat treatments performed below 60 °C, the main effect on the surface properties of MNV was shown for hydrophobicity. Exposure to 55 °C led to the expression of hydrophobic domains at the surface of MNV particles for both infectious and inactivated particles. No expression of hydrophobic domains was observed after exposure to 60 °C, but this low hydrophobic state appeared to be irreversible for inactivated particles only. Indeed, we succeeded in increasing the hydrophobicity of MNV remaining infectious after a first exposure to 60 °C by a second exposure to 55 °C. Critical temperatures, permeability to RNase and expression of hydrophobic domains should now be explored for pathogenic enteric viruses prior to their detection by molecular methods. Free chlorine did not seem to affect the surface properties of MNV, but the permeability to RNase and the decrease in host cell binding capacity suggested at least the weakening of the capsid structure. Thus, this oxidant may increase the efficiency of other inactivating treatments such as temperature.
References
Ausar, S. F., Foubert, T. R., Hudson, M. H., Vedvick, T. S., & Middaugh, C. R. (2006). Conformational stability and disassembly of Norwalk virus-like particles. Effect of pH and temperature. The Journal of Biological Chemistry, 281(28), 19478–19488. doi:10.1074/jbc.M603313200.
Belliot, G., Lavaux, A., Souihel, D., Agnello, D., & Pothier, P. (2008). Use of murine norovirus as a surrogate to evaluate resistance of human norovirus to disinfectants. Applied and Environmental Microbiology, 74(10), 3315–3318. doi:10.1128/AEM.02148-07.
Belnap, D. M., Filman, D. J., Trus, B. L., Cheng, N., Booy, F. P., Conway, J. F., et al. (2000). Molecular tectonic model of virus structural transitions: The putative cell entry states of poliovirus. Journal of virology, 74(3), 1342–54. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=111469&tool=pmcentrez&rendertype=abstract
Bolton, S. L., Kotwal, G., Harrison, M. A., Law, S. E., Harrison, J. A., & Cannon, J. L. (2013). Sanitizer efficacy against murine norovirus, a surrogate for human norovirus, on stainless steel surfaces when using three application methods. Applied and Environmental Microbiology, 79(4), 1368–1377. doi:10.1128/AEM.02843-12.
Breindl, M. (1971). The structure of heated poliovirus particles. The Journal of General Virology, 11(3), 147–156. doi:10.1099/0022-1317-11-3-147.
Brié, A., Bertrand, I., Meo, M., Boudaud, N., & Gantzer, C. (2016). The Effect of Heat on the Physicochemical Properties of Bacteriophage MS2. Food and Environmental Virology. doi:10.1007/s12560-016-9248-2.
Cromeans, T., Park, G. W., Costantini, V., Lee, D., Wang, Q., Farkas, T., et al. (2014). Comprehensive comparison of cultivable norovirus surrogates in response to different inactivation and disinfection treatments. Applied and Environmental Microbiology, 80(18), 5743–5751. doi:10.1128/AEM.01532-14.
Curry, S., Chow, M., & Hogle, J. M. (1996). The poliovirus 135S particle is infectious. Journal of virology, 70(10), 7125–7131. Accessed Decembe 5, 2014. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=190765&tool=pmcentrez&rendertype=abstract.
de Abreu Corrêa, A., Carratala, A., Barardi, C. R. M., Calvo, M., Girones, R., & Bofill-Mas, S. (2012). Comparative inactivation of murine norovirus, human adenovirus, and human JC polyomavirus by chlorine in seawater. Applied and Environmental Microbiology, 78(18), 6450–6457. doi:10.1128/AEM.01059-12.
de Roda Husman, A. M., Lodder, W. J., Rutjes, S. A., Schijven, J. F., & Teunis, P. F. M. (2009). Long-term inactivation study of three enteroviruses in artificial surface and groundwaters, using PCR and cell culture. Applied and Environmental Microbiology, 75(4), 1050–1057. doi:10.1128/AEM.01750-08.
Dika, C., Ly-Chatain, M. H., Francius, G., Duval, J. F. L., & Gantzer, C. (2013). Non-DLVO adhesion of F-specific RNA bacteriophages to abiotic surfaces: Importance of surface roughness, hydrophobic and electrostatic interactions. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 435, 178–187. doi:10.1016/j.colsurfa.2013.02.045.
EFSA. (2016). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2014. 191p. doi:10.2903/j.efsa.2015.4329
Ettayebi, K., Crawford, S. E., Murakami, K., Broughman, J. R., Karandikar, U., Tenge, V. R., et al. (2016). Replication of human noroviruses in stem cell-derived human enteroids. Science. doi:10.1126/science.aaf5211.
Fricks, C. E., & Hogle, J. M. (1990). Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. Journal of virology, 64(5), 1934–45. Accessed 5, 2014. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=249347&tool=pmcentrez&rendertype=abstract.
Gassilloud, B., Schwartzbrod, L., & Gantzer, C. (2003). Presence of viral genomes in mineral water: a sufficient condition to assume infectious risk? Applied and Environmental Microbiology, 69(7), 3965–3969. doi:10.1128/AEM.69.7.3965.
Girard, M., Mattison, K., Fliss, I., & Jean, J. (2016). Efficacy of oxidizing disinfectants at inactivating murine norovirus on ready-to-eat foods. International Journal of Food Microbiology, 219, 7–11. doi:10.1016/j.ijfoodmicro.2015.11.015.
Hall, A. J., Wikswo, M. E., Pringle, K., Gould, L. H., & Parashar, U. D. (2014). Vital signs: foodborne norovirus outbreaks - United States, 2009–2012. MMWR. Morbidity and mortality weekly report, 63(22), 491–5. Accessed September 12, 2014, http://www.ncbi.nlm.nih.gov/pubmed/24898166.
Hirneisen, K. A., & Kniel, K. E. (2013). Norovirus surrogate survival on spinach during preharvest growth. Phytopathology, 103(4), 389–394. doi:10.1094/PHYTO-09-12-0231-FI.
Hwang, S., Alhatlani, B., Arias, A., Caddy, S. L., Christodoulou, C., Cunha, J. B., et al. (2015). Murine norovirus: propagation, quantification, and genetic manipulation. Current protocols in microbiology, 33(2), 15K.2.1–61. doi:10.1002/9780471729259.mc15k02s33
ISO (1985). ISO 7393-2. Water quality: Determination of free chlorine and total chlorine. Part 2: Colorimetric method using N,N-diethyl-1,4-phenylenediamine, for routine control purposes.
ISO (2013). ISO/TS 15216-1: Microbiology of food and animal feed—Horizontal method for determination of hepatitis A virus and norovirus in food using real-time RT-PCR—Part 1: Method for quantification.
Jones, M. K., Grau, K. R., Costantini, V., Kolawole, A. O., de Graaf, M., Freiden, P., et al. (2015). Human norovirus culture in B cells. Nature Protocols, 10(12), 1939–1947. doi:10.1038/nprot.2015.121.
Karber, G. (1931). Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Archiv für Experimentelle Pathologie und Pharmakologie, 162(4), 480–483. doi:10.1007/BF01863914.
Karst, S. M., & Wobus, C. E. (2015). A working model of how noroviruses infect the intestine. PLoS Pathogens, 11, e1004626. doi:10.1371/journal.ppat.1004626.
Knight, A., Haines, J., Stals, A., Li, D., Uyttendaele, M., Knight, A., et al. (2016). A systematic review of human norovirus survival reveals a greater persistence of human norovirus RT-qPCR signals compared to those of cultivable surrogate viruses. International Journal of Food Microbiology, 216, 40–49. doi:10.1016/j.ijfoodmicro.2015.08.015.
Li, J. W., Xin, Z. T., Wang, X. W., Zheng, J. L., & Chao, F. H. (2002). Mechanisms of inactivation of hepatitis a virus by chlorine. Applied and Environmental Microbiology, 68(10), 4951–4955. doi:10.1128/AEM.68.10.4951.
Lim, M. Y., Kim, J.-M., & Ko, G. (2010). Disinfection kinetics of murine norovirus using chlorine and chlorine dioxide. Water Research, 44(10), 3243–3251. doi:10.1016/j.watres.2010.03.003.
Nuanualsuwan, S., & Cliver, D. O. (2002). Pretreatment to avoid positive RT-PCR results with inactivated viruses. Journal of virological methods, 104(2), 217–25. http://www.ncbi.nlm.nih.gov/pubmed/12088831
Nuanualsuwan, S., & Cliver, D. O. (2003). Capsid functions of inactivated human picornaviruses and feline calicivirus. Applied and Environmental Microbiology, 69(1), 350–357. doi:10.1128/AEM.69.1.350.
O’Brien, R. T., & Newman, J. (1979). Structural and compositional changes associated with chlorine inactivation of polioviruses. Applied and Environmental Microbiology, 38(6), 1034–1039. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=291240&tool=pmcentrez&rendertype=abstract
Page, M. A., Shisler, J. L., & Mariñas, B. J. (2010). Mechanistic aspects of adenovirus serotype 2 inactivation with free chlorine. Applied and Environmental Microbiology, 76(9), 2946–2954. doi:10.1128/AEM.02267-09.
Park, S. Y., Kim, A.-N., Lee, K.-H., & Ha, S.-D. (2015). Ultraviolet-C efficacy against a norovirus surrogate and hepatitis A virus on a stainless steel surface. International Journal of Food Microbiology, 211, 73–78. doi:10.1016/j.ijfoodmicro.2015.07.006.
Pecson, B. M., Martin, L. V., & Kohn, T. (2009). Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation, and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Applied and Environmental Microbiology, 75(17), 5544–5554. doi:10.1128/AEM.00425-09.
Predmore, A., Sanglay, G., Li, J., & Lee, K. (2015). Control of human norovirus surrogates in fresh foods by gaseous ozone and a proposed mechanism of inactivation. Food Microbiology, 50, 118–125. doi:10.1016/j.fm.2015.04.004.
Prevost, B., Goulet, M., Lucas, F. S., Joyeux, M., Moulin, L., & Wurtzer, S. (2016). Viral persistence in surface and drinking water: Suitability of PCR pre-treatment with intercalating dyes. Water Research, 91, 68–76. doi:10.1016/j.watres.2015.12.049.
Richards, G. P. (2012). critical review of norovirus surrogates in food safety research: Rationale for considering volunteer studies, 6–13. doi:10.1007/s12560-011-9072-7
Ruokola, P., Dadu, E., Kazmertsuk, A., Häkkänen, H., Marjomäki, V., & Ihalainen, J. A. (2014). Raman spectroscopic signatures of echovirus 1 uncoating. Journal of Virology, 88(15), 8504–8513. doi:10.1128/JVI.03398-13.
Samandoulgou, I., Fliss, I., & Jean, J. (2015). Zeta potential and aggregation of virus-like particle of human norovirus and feline calicivirus under different physicochemical conditions. Food and Environmental Virology, 7(3), 249–260. doi:10.1007/s12560-015-9198-0.
Sano, D., Ohta, T., Nakamura, A., Nakagomi, T., Nakagomi, O., & Okabe, S. (2015). Culture-independent evaluation of non-enveloped virus infectivity reduced by free chlorine disinfection. Applied and Environmental Microbiology, 81(8), AEM.03802–AEM.03814. doi:10.1128/AEM.03802-14.
Sano, D., Pintó, R. M., Omura, T., & Bosch, A. (2010). Detection of oxidative damages on viral capsid protein for evaluating structural integrity and infectivity of human norovirus. Environmental Science and Technology, 44(2), 808–812. doi:10.1021/es9018964.
Seitz, S. R., Leon, J. S., Schwab, K. J., Lyon, G. M., Dowd, M., McDaniels, M., et al. (2011). Norovirus infectivity in humans and persistence in water. Applied and Environmental Microbiology, 77(19), 6884–6888. doi:10.1128/AEM.05806-11.
Sigstam, T., Gannon, G., Cascella, M., Pecson, B. M., Wigginton, K. R., & Kohn, T. (2013). Subtle differences in virus composition affect disinfection kinetics and mechanisms. Applied and Environmental Microbiology, 79(11), 3455–3467. doi:10.1128/AEM.00663-13.
Tojo, K., Sano, D., Miura, T., Nakagomi, T., Nakagomi, O., & Okabe, S. (2013). A new approach for evaluating the infectivity of noncultivatable enteric viruses without cell culture. Water science and technology: A Journal of the International Association on Water Pollution Research, 67(10), 2236–2240. doi:10.2166/wst.2013.114.
Topping, J. R., Schnerr, H., Haines, J., Scott, M., Carter, M. J., Willcocks, M. M., et al. (2009). Temperature inactivation of Feline calicivirus vaccine strain FCV F-9 in comparison with human noroviruses using an RNA exposure assay and reverse transcribed quantitative real-time polymerase chain reaction-A novel method for predicting virus infectivity. Journal of Virological Methods, 156(1–2), 89–95. doi:10.1016/j.jviromet.2008.10.024.
Tuthill, T. J., Groppelli, E., Hogle, J. M., & Rowlands, D. J. (2010). Picornaviruses. Current Topics in Microbiology and Immunology, 343, 43–89. doi:10.1007/82_2010_37.
Verhoef, L., Hewitt, J., Barclay, L., Ahmed, S. M., Lake, R., Hall, A. J., et al. (2015). Norovirus genotype profiles associated with foodborne transmission, 1999–2012. Emerging Infectious Diseases, 21(4), 592–599. doi:10.3201/eid2104.141073.
Wang, Q., & Kniel, K. E. (2016). Survival and transfer of murine norovirus within a hydroponic system during kale and mustard microgreen harvesting. Applied and Environmental Microbiology, 82(2), 705–713. doi:10.1128/AEM.02990-15.
Wigginton, K. R., & Kohn, T. (2012). Virus disinfection mechanisms: the role of virus composition, structure, and function. Current Opinion in Virology, 2(1), 84–89. doi:10.1016/j.coviro.2011.11.003.
Wigginton, K. R., Pecson, B. M., Bosshard, F., Kohn, T., & Sigstam, T. (2012). Virus inactivation mechanisms: impact of disinfectants on virus function and structural integrity. Environmental Science and Technology, 46(21), 12069–12078. doi:10.1021/es3029473.
Wobus, C. E., Karst, S. M., Thackray, L. B., Chang, K.-O., Sosnovtsev, S. V., Belliot, G., et al. (2004). Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biology, 2(12), e432. doi:10.1371/journal.pbio.0020432.
Yeap, J. W., Kaur, S., Lou, F., Dicaprio, E., Morgan, M., & Linton, R. (2016). Inactivation kinetics and mechanism of a human norovirus surrogate on stainless steel coupons via chlorine dioxide gas. Applied and Environmental Microbiology, 82(1), 116–123. doi:10.1128/AEM.02489-15.Editor.
Acknowledgements
The results of this study were obtained within the scope of CapsiVir, a project coordinated by ACTALIA and funded by the “Conseil Régional de Basse Normandie”. This study, labelled by the competitiveness cluster VALORIAL, was also supported by the Joint Technical Unit ACTIA VIROcontrol and the “Syndicat des Fabricants des Produits Frais Prêts à l’Emploi” (cluster of ready-to-eat food industries: Florette, Bonduelle, Crudettes, and Rosée des Champs).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brié, A., Razafimahefa, R., Loutreul, J. et al. The Effect of Heat and Free Chlorine Treatments on the Surface Properties of Murine Norovirus. Food Environ Virol 9, 149–158 (2017). https://doi.org/10.1007/s12560-016-9271-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-016-9271-3