
Abstract
The goal of this study was to determine if a cytopathogenic effects (CPE) cell culture assay and an integrated cell culture PCR (ICC-PCR) assay would yield similar or different results when used to assess virus survival in water. Poliovirus type 1 was added to dechlorinated tapwater and stored at room temperature (22.5–24°C) for a total of 50 days. Samples were assayed at defined time intervals by the most probable number (MPN) method on Buffalo green monkey kidney cells (BGM) by CPE and additionally by ICC-PCR. Monolayers that were CPE negative on first passage were passed onto fresh monolayers of cells for a second and third time if still negative. By CPE assay, second passage was observed to yield a greater titer (2,300 vs. 24,000 MPN/ml) and third passage also resulted in an increased titer. ICC-PCR proved to be a more rapid and sensitive method than conventional cell culture for determining virus inactivation rates in water. Poliovirus survived in tapwater for up to 32 days, as assessed by both third passage ICC-PCR and CPE. There was no statistical difference in the inactivation rates between the two methods. To determine the total number of infectious viruses, these findings indicate the need for performing three cell culture passages or, alternatively, ICC-PCR on first passage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Animal cell culture is still the major method used to determine virus infectivity. Assessment of viral infectivity is a critical tool in food and environmental virology to assess survival of viruses in the environment and removal by treatment processes. Detection and assay of enteroviruses can be done by observation for viral cytopathogenic effects (CPE) in animal cell culture, however, this method is time-consuming and suffers from the limitation that some enteric viruses do not always produce CPE, and toxicity factors can produce false positive results (Reynolds et al. 2001). Quantification by the TCID50 (tissue culture infectious dose) and most probable number (MPN) method is accomplished using a dilution series until CPE is no longer observed. Polymerase chain reaction (PCR), which involves amplification of the viral RNA or DNA, provides results in 2–4 h as opposed to cell culture which usually takes from 3 to 21 days for results. However, since it only detects nucleic acids, PCR cannot determine whether detected organisms are infectious (Rodriguez et al. 2009). Integrated cell culture (ICC) PCR combines conventional cell culture and PCR to provide rapid detection of infectious viruses, while reducing the disadvantages of each individual method (Chapron et al. 2000; Reynolds et al. 1996). The method has largely been used for the detection of naturally occurring enteric viruses in water and food (Reynolds 2004), and proved to be more effective than conventional methods for determining the efficacy of water disinfectants (Blackmer et al. 2000; Bigliardi and Sansebastiano 2006).
In a previous study (Blackmer et al. 2000) on the disinfection of poliovirus with free chlorine (6-min exposure with 0.5 mg/l free chlorine), cell culture lysate was found to be positive for infectious virus by first passage ICC-PCR and by CPE after second passage. However, first passage CPE results were negative suggesting that evaluation of the presence of viable poliovirus based solely on one conventional cell culture passage could be erroneous. The primary objective of the present study was to assess the survival of poliovirus in water by ICC-PCR and cell culture assay (CPE). Specifically, we were interested in determining whether multiple successive cell culture passages would increase the number of infectious viruses detected allowing for enhanced detection of viruses via CPE or ICC-PCR, and if it influenced the determination of the inactivation rate of the virus.
Materials and Methods
Virus and Cell Line
Poliovirus type 1 (strain LSc-2ab) was propagated in Buffalo green monkey kidney (BGM) cells as previously described (Blackmer et al. 2000). Briefly, cells were grown in cell culture flasks (Corning, Corning, NY) at 37°C until a confluent monolayer was formed using 5% minimum essential medium (MEM) (Irvine Sci., Santa Ana, CA) as the growth media, supplemented with 5% fetal bovine serum (FBS) (HyClone, Logan, UT) (Gibco BRL Life Technology, Grand Island, NY). Poliovirus was extracted by freeze-thawing flasks three times at room temperature, followed by centrifugation to remove cell debris at 1,000×g for 10 min at 4°C. The supernatant was stirred overnight at 4°C in 9% PEG (polyethylene glycol) and 5.8% sodium chloride (Sigma Chemical Co., St. Louis, MO), after which the virus preparation was centrifuged at 11,200×g for 30 min. The supernatant was discarded, and the pellet was resuspended in sterile Tris-buffered saline at pH 7.4 (Sigma Chemical Co., St. Louis, MO). The Tris virus suspension was Freon (1,1,2-2-trichlorotrifluorethane) (Sigma Chemical Co., St. Louis, MO) extracted in 20 ml amounts (20 ml virus suspension:20 ml Freon) by shaking for 5 min, followed by centrifuging at 4°C for 15 min at 1,000×g. The upper aqueous layer was collected and stored at −80°C.
Survival of Poliovirus in Tapwater
City of Tucson tapwater was collected directly from a tap at the University of Arizona and dechlorinated by passage through an activated carbon filter. The tapwater had a turbidity of less than 0.1 NTU, pH of 7.8, and a total organic concentration (TOC) of less than 1 mg/ml. Two separate 25-ml tapwater samples were collected and designated as tapwater replicate samples A and B. Poliovirus was then added to the tapwater samples. Samples A and B had a starting concentration of 2,000 and 2,200 MPN/ml poliovirus, respectively. Tapwater samples were stored at room temperature (22.5–24°C) in 50 ml polypropylene tubes (Falcon, Becton–Dickinson Labware, Franklin Lakes, NJ) wrapped in aluminum foil to prevent exposure to light. Subsamples for virus assay were collected at the start of the experiment and every 2–4 days afterward for up to 50 days.
CPE Cell Culture Assay
BGM cells in 6-well trays were inoculated with samples from tapwater A and B. The following dilutions of tapwater samples were assayed: undiluted sample, 10−1, 10−2, 10−3, and 10−4. Dilutions were prepared using Eagle’s Minimal Essential Media (MEM). For tapwater A and B replicate samples, each dilution was assayed in duplicate. Cell monolayers were inoculated with 0.1 ml of sample and allowed to incubate for 1 h at 37°C and 5% CO2. During this incubation period, samples were gently rocked every 15 min. The monolayer was then covered with MEM maintenance media containing 2% fetal bovine serum and checked daily for cytopathic effect (CPE). After CPE was observed or after 14 days for CPE negative wells, the wells were frozen at −20°C. Cell lysate was produced for the next passage by thawing the wells at room temperature and using cell scrapers (blade length 1.3 cm) (Phoenix Research Products, Hayward, CA) to physically disrupt the cell monolayer. For second passage, 0.1 ml of these lysates were placed onto a fresh BGM cell monolayer in the 6-well plates and incubated for 7 days. If negative, a third passage was performed in the same manner. Poliovirus concentrations were determined by a MPN program (Hurley and Roscoe 1983).
Integrated Cell Culture-PCR
BGM cells in 6-well plates were inoculated with samples from tapwater A only. Dilutions were prepared using MEM, and each dilution was assayed in duplicate. The cell assay procedure for ICC-PCR was identical to that of the conventional cell culture assay except cells were only incubated for 2 days at 37°C and 5% CO2. After incubation, the BGM cells were frozen at −20°C and thawed once at room temperature. The cells were detached using cell scrapers as previously described, placed into sterile 50 ml centrifuge tubes (Nalgene, Rochester, NY), and centrifuged at 4°C for 20 min at 1,000×g to remove cellular debris. Aliquots of the supernatant were stored at −20°C until analyzed by PCR or the next cell culture passage.
PCR was performed using 50 µl reactions. A master mix containing 1.5 µl of 10× buffer II, 3.5 µl of 25 mM MgCl2, and 4 µl of 10 mM deoxynucleotide triphosphate (dNTP’s) was made for each reaction. Template (9 µl), in the form of cell culture lysate was added to each reaction, and the sample was heat extracted for 5 min at 99°C. A reverse transcription master mix consisting of 0.5 µl of 20 U/µl ribonuclease inhibitor, 0.5 µl of 50 µM random hexamers, and 0.5 µl of 50 U/µl MulV reverse transcriptase was added for each reaction. Samples were then reverse transcribed via a single cycle of 24°C for 10 min, 44°C for 60 min, 99°C for 5 min, and 5°C for 5 min. A third master mix was prepared for PCR consisting of 1.5 µl of 25 mM MgCl2, 3.5 µl of 10× buffer II, 0.25 µl of upstream and downstream primer each (3), 28.75 µl nuclease free water, and 0.25 µl AmpliTaq Gold Polymerase (Perkin-Elmer, Cetus Corp., Norwalk, CT) per reaction. Samples were amplified with a 95°C 10 min hot start, 25 cycles of 95°C for 20 s, 60°C for 25 s, and 72°C for 20 s, followed by a 10 min extension at 72°C.
Seminested reactions were performed to confirm RT-PCR reaction results. Five microliter of PCR product served as template. The seminested master mix consisted of 5 µl of 25 mM MgCl2, 5 µl of 10× buffer II, 4 µl of 10 mM dNTP’s, 0.25 µl of upstream and internal primer each (3), 30.25 µl nuclease free water, and 0.25 µl AmpliTaq Gold Polymerase (Perkin-Elmer, Cetus Corp., Norwalk, CT) per reaction. Samples were amplified for 20 cycles using the conditions described for PCR.
The primers used were: P1 upstream 5′-CCT CCG GCC CCT GAA TG-3′ and p2 downstream 5′-ACC GGA TGG CCA ATC CAA-3′ and Ent 33 downstream 5′-CCC AAA GTA GTC GGT TCC GC-3′ (Schwab et al. 1996). The first round of PCR used the set of primers P1 and P2, with an amplified fragment of 195 bp. The second round of PCR used the set of primers P1 and P33, with an amplified fragment of 105 bp.
Amplified PCR products were visualized by 1.5% low electroendosmosis (EEO) agarose gel electrophoresis. Ten microliters of product was combined with 3 µl of Ficoll loading buffer (20% Ficoll, 1% sodium dodecyl sulfate, 0.25% bromophenol blue, and 0.1 M EDTA, pH 8.0) and subjected to agarose gel electrophoresis at 120 V for 1.25 h. The gel was then stained with ethidium bromide solution for 15 min and destained in double distilled water for 20 min. RT-PCR and semi-nested bands representative of 195 and 106 bp products, respectively, were recorded as a positive result and photographed with an AlphaImager 2000 (Alpha Innotech Corp., San Leandro, CA). Poliovirus concentrations were then determined by a MPN program (Hurley and Roscoe 1983).
Statistical Analysis
Survival of poliovirus over time was examined for all three cell culture passages and ICC-PCR passages using the equation
where N t is the number of surviving microorganisms (MPN/ml) at time t, N 0 is the initial number of microorganisms (MPN/ml), t is time (days), and k d is inactivation rate (1/day). Linear regressions were generated using Microsoft Excel 97 (Microsoft Corp., Bellevue, WA) for statistical analysis of poliovirus inactivation rates. The f test was used to test for equality of the slopes of the regression lines. For each regression line, an f value and the corresponding P value were generated using the SAS System for Windows version 6.12 (SAS Institute Inc., Cary, NC). Differences between inactivation rates were considered statistically significant if P < 0.05.
Results
Viral Assay by CPE
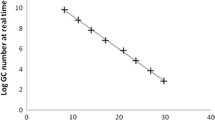
The titer of the virus stock was determined to be 1.1 × 108 MPN/ml. The titer of poliovirus as measured by CPE was observed to decrease over time in the tapwater samples (Figs. 1, 2). However, the titer of poliovirus was always greater after a second passage. For example, on day 1, first passage yielded a titer of 2,300 MPN/ml while the second passage titer was 24,000 MPN/ml. A second passage always yielded a 1–2 log10 greater concentration of virus. Although no viruses were detected after 23 days in tapwater A (Fig. 1) or in tapwater B after 21 days (Fig. 2) by first passage, they could still be detected on second and third passages. Titers on third passage were either equal to or greater than those by second passage. Furthermore, with each successive cell culture passage, the length of time that infectious poliovirus could be detected increased. For example, in tapwater sample A, the day at which poliovirus was last detected by first, second, and third passage was day 17, 28, and 32, respectively (Fig. 1). Tapwater sample B showed a similar trend, with poliovirus last being detected on day 16, 21, and 33 by first, second, and third passage, respectively (Fig. 2).

Effect of passage number on determining the survival of poliovirus in tapwater sample A determined by cytopathogenic effects (CPE) cell culture assay

Effect of passage number on the survival of poliovirus in tapwater sample B determined by cytopathogenic effects (CPE) cell culture assay
Viral Assay by CPE and ICC-PCR
Results of poliovirus survival could be obtained in a much shorter time using ICC-PCR relative to the CPE method (2 days compared to 16 days). In addition, ICC-PCR was more sensitive than single passage CPE assays (Fig. 3). ICC-PCR on first passage was able to detect poliovirus in the undiluted aliquots (103 MPN/ml) up to day 28 (Fig. 3). Two passages were required by cell culture to reach this same detection capability (Fig. 4). The reduced time for obtaining results was also evident in detection of the 10−1 diluted aliquots (102 MPN/ml) (Fig. 4). Poliovirus detection in the 10−1 diluted samples by second passage ICC-PCR was equivalent to third passage results of conventional cell culture (Fig. 5).

Comparison of poliovirus detection by ICC-PCR and cytopathogenic effects (CPE) cell culture assay in first passage. The undiluted stock contains 103 MPN/ml

Comparison of poliovirus detection by ICC-PCR and cytopathogenic effects (CPE) cell culture assay in second passage. The undiluted stock contains 103 MPN/ml

Comparison of poliovirus detection by ICC-PCR and cytopathogenic effects (CPE) cell culture assay in third passage. The undiluted stock contains 103 MPN/ml
The greater sensitivity of ICC-PCR over conventional cell culture was evident considering poliovirus detection within the same passage (Fig. 4). In first passage, poliovirus in undiluted samples of Tapwater sample B was detected by ICC-PCR for 28 days versus 17 days by CPE. The first passage 10−2 diluted samples were ICC-PCR positive up to 7 days versus 4 days by CPE. In second passage, the 10−1 diluted samples were ICC-PCR positive for 28 days versus 17 days by CPE. The difference in sensitivities of detection between ICC-PCR and conventional cell culture decreased with each passage and by the third passage, the two methods agreed with respect to sensitivity (Fig. 4).
Linear regressions of poliovirus survival data were plotted to compare rates of inactivation (Table 1). The rate of inactivation determined by ICC-PCR analysis was always lower than the rate determined by conventional cell culture in each of the three passages. Furthermore, the rates of inactivation were not statistically significant when compared to the titers obtained by CPE. The rates of inactivation ranged from 0.097 to 0.110 for conventional cell culture analysis of tapwater sample A, 0.112 to 0.146 for conventional cell culture analysis of tapwater sample B, and 0.085 to 0.094 by ICC-PCR. The differences in inactivation rates for tapwater sample A analysis by CPE (P = 0.77), tapwater sample B analysis by CPE (P = 0.60), and ICC-PCR analysis of tapwater sample A (P = 0.91) were not significant by the f test. Thus, the rate of inactivation did not appear to change significantly with multiple cell passage. There was also no significant difference if the assays were performed by cell culture or ICC-PCR by the third passage.
Discussion
As expected, infectious poliovirus concentrations in tapwater decreased over time at room temperature (Melnick and Gerba 1980). This decrease in titer probably results from thermal degradation and/or enzymatic degradation by microorganisms (Melnick and Gerba 1980).
Performing more than one passage caused an increase in the measurable titer of poliovirus by CPE but did not significantly influence the rate of virus inactivation. In this study, the titer of poliovirus as determined by first passage was 10-fold lower than the titer in two subsequent passages. Thus, the concentration of laboratory grown viruses may be underestimated by enumeration with only a single passage regardless of the sensitivity of the cell line for the virus. However, inactivation rates were virtually the same regardless if the data from 1st, 2nd, or 3rd passage were used to calculate inactivation rates. Thus, results of this study confirm the validity of previous studies in which inactivation rates were determined solely by first passage.
Previous work citing ICC-PCR as a more rapid and sensitive method than conventional cell culture was confirmed in this study (Blackmer et al. 2000; Chapron et al. 2000; Reynolds et al. 1996, 2001). However, there was no difference in the overall detection sensitivities of each of each method (ICC-PCR and third-passage CPE assay). The exact reason or reasons for the improvement in the detection of infectious viruses by more than one passage is unknown. One explanation could involve subpopulations of virus particles initiating infection in later passages that might include “slow growing” viruses that do not infect and replicate in cells at a high enough titer to cause CPE until later passages. Viruses that replicate quickly to high numbers within cells, but do not produce CPE until later passages could also occur in such populations. The phenomenon of multiplicity reactivation (MR) may occur in such situations (Young and Sharp 1979) when simultaneous infection of the host cell by many damaged viral particles may allow for the formation of functional poliovirus RNA by random cleavage and recombination of the damaged poliovirus RNA. Genetic complementation, which is the compensation of one or more defective gene products of one virus by the functional form of the same gene product(s) of another virus without involving genotypic changes, could also play a role in this (Hellen and Wimmer 1995). Homologous interference is another possible explanation for the observed increase in conventional cell culture in second and third passages versus first passage. Infection of a cell by heat-inactivated poliovirus may have prevented subsequent infection of a cell by viable poliovirus as was illustrated by studies on heat inactivated and viable vaccinia virus (Galasso and Sharp 1961, 1963). Therefore, even if surviving viruses were present in first passage, conventional cell culture would have failed to reveal their presence if enough inactivated viruses caused interference. The concentration of the heat-damaged viruses, and thus interference, may have decreased with each passage due to repair of the adapting viruses in cell passage. This would allow more infection of cells by functional poliovirus and increased CPE. A final point of consideration is whether the greater titer in later passages was due to detection of viruses that were initially unabsorbed to cells in first passage and remained in cell culture fluid. These particles may have been carried over into second and third passages and initiated infection in these later passages.
The particle:PFU ratio for laboratory grown poliovirus has been shown to be 100:1 (Margolin 1986; Richardson 1989). It has long been assumed that most of these particles were incapable of producing infection. Based on the 10-fold increase in viral titer from the first to second passage in this study, perhaps a greater proportion of these particles are complete and infectious. However, based on the results of this study, it is more likely due to the efficiency of the assay methodology, i.e., not all of the viruses attach to the monolayer during the first passage. This study indicates the non-infectious particles:PFU ratio for poliovirus is closer to 10:1.
The results of this study suggest that when assessing a treatment processes for virus removal or survival that virus removal rate or inactivation rate is preferred rather than just absence of the virus (i.e., 100% removal), since infectious virus may still remain if results are based on only one passage in cell culture. It is evident that multiple cell culture passages would be necessary to confirm the absence of infectious virus.
Conclusions
The results of this investigation validate the methodology of previous research conducted on the survival of viruses by a single passage using CPE to assess viability (Bitton 1999; Ma et al. 1994). ICC-PCR was observed to take less time to obtain results and was a more sensitive method than conventional cell culture, yielding similar inactivation rates as the CPE method. ICC-PCR is especially for slow enteric growing viruses, such as adenovirus, or when CPE is not useful easily observed or does not occur (e.g., human rotaviruses) (Ward et al. 1986). This study also demonstrated that more than one passage in cell culture is necessary to ensure the absence of virus in environmental samples.
References
Bigliardi, L., & Sansebastiano, G. (2006). Study on inactivation kinetics of hepatitis A and enteroviruses with peracetic acid and chlorine. New ICC/PCR method to assess disinfection effectiveness. Journal of Preventive Medicine and Hygiene, 47, 56–63.
Bitton, G. (1999). Water and wastewater disinfection. In G. Bitton (Ed.), Wastewater microbiology (pp. 137–168). New York, NY: Wiley-Liss, Inc.
Blackmer, F., Reynolds, K. A., Gerba, C. P., & Pepper, I. L. (2000). Use of integrated cell culture-PCR to evaluate the effectiveness of poliovirus inactivation by chlorine. Applied and Environmental Microbiology, 66, 2267–2268.
Chapron, C. D., Ballester, N. A., Fontaine, J. H., Frades, C. N., & Margolin, A. B. (2000). Detection of astrovirus, enterovirus, and adenovirus types 40 and 41 in surface waters collected and evaluated by the information collection rule and an integrated cell culture-nested PCR procedure. Applied and Environmental Microbiology, 66, 2520–2525.
Galasso, G. J., & Sharp, D. G. (1961). Heated vaccinia virus and plaque formation on L cell monolayers. Virology, 15, 376–378.
Galasso, G. J., & Sharp, D. G. (1963). Homologous inhibition with heated and ultraviolet-treated vaccinia virus cultures of L cells. Virology, 20, 1–3.
Hellen, C. U., & Wimmer, E. (1995). Translation of encephalomyocarditis virus RNA by internal ribosomal entry. Current Topics in Microbiology and Immunology, 203, 31–63.
Hurley, M. A., & Roscoe, M. E. (1983). Automated statistical analysis of microbial enumeration by dilution series. Journal of Applied Bacteriology, 55, 159–164.
Ma, J., Straub, T. M., Pepper, I. L., & Gerba, C. P. (1994). Cell culture and PCR determination of poliovirus inactivation by disinfectants. Applied and Environmental Microbiology, 60, 4203–4206.
Margolin, A. G. (1986). The development of a blot assay using gene probes for the detection of enteroviruses in water. Ph.D. Dissertation, The University of Arizona, Tucson, AZ.
Melnick, J. L., & Gerba, C. P. (1980). The ecology of enteroviruses in natural waters. CRC Critical Review in Environmental Contamination, 10, 65–93.
Reynolds, A. A. (2004). Integrated cell culture/PCR for detection of enteric viruses in environmental samples. Methods in Molecular Biology, 268, 69–78.
Reynolds, K. A., Gerba, C. P., Abbaszadegan, M., & Pepper, I. L. (2001). ICC/PCR detection of enteroviruses and hepatitis A virus in environmental samples. Canadian Journal of Microbiology, 47, 153–157.
Reynolds, K. A., Gerba, C. P., & Pepper, I. L. (1996). Detection of infectious enteroviruses by an integrated culture-PCR procedure. Applied and Environmental Microbiology, 62, 1424–1427.
Richardson, K. J. (1989). Use of nucleic acid probes and a nonradioactive labeling system for the detection of enteroviruses in water. Ph.D. Dissertation, The University of Arizona, Tucson, AZ.
Rodriguez, R. A., Pepper, I. L., & Gerba, C. P. (2009). Application of PCR-based methods to assess the infectivity of enteric viruses in environmental samples. Applied and Environmental Microbiology, 75, 297–307.
Schwab, K. J., De Leon, R., & Sobsey, M. D. (1996). Immunoaffinity concentration and purification of waterborne enteric viruses for detection by reverse transcriptase PCR. Applied and Environmental Microbiology, 62, 2086–2094.
Ward, R. L., Bernstein, D. I., Sherwood, J. R., Knowlton, D. R., & Schiff, G. M. (1986). Human rotavirus in volunteers: Determination of infectious dose and serological response to infection. Journal of Infectious Disease, 154, 871–880.
Young, D. C., & Sharp, D. G. (1979). Partial reactivation of chlorine-treated echovirus. Applied and Environmental Microbiology, 37, 766–773.
Acknowledgments
This work was supported in part by a grant from the University of Arizona-National Science Foundation Water and Environmental Technology (WET) Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mahalanabis, M., Reynolds, K.A., Pepper, I.L. et al. Comparison of Multiple Passage Integrated Cell Culture-PCR and Cytopathogenic Effects in Cell Culture for the Assessment of Poliovirus Survival in Water. Food Environ Virol 2, 225–230 (2010). https://doi.org/10.1007/s12560-010-9051-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-010-9051-4