
Abstract
The presence of pathogenic viruses in drinking water is a major public health concern. Although viability RT-qPCR methods were developed to quantify infectious viruses, they may not always reflect viral infectivity, therefore leading to false-positive results. In this study, sodium deoxycholate (SD) pre-treatment was used to improve the efficiency of viability RT-qPCR methods with respect to exclusive quantification of infectious viruses. The ability of SD pre-treatment to enhance the penetration of three viability markers, namely, ethidium monoazide (EMA, 100 µM), propidium monoazide (PMA, 100 µM), and cis-dichlorodiammineplatinum (CDDP, 1000 µM), into heat-treated (90 °C for 1 min) Aichi virus at various concentrations (0.01–0.5%) was evaluated. The optimal SD concentration was found to be 0.1% for all markers. EMA/PMA/CDDP-RT-qPCR with 0.1% SD pre-treatment was significantly more effective than without SD pre-treatment in determining AiV inactivation after heat (50, 60, 70, 80, or 90 °C for 1 min) or chlorine treatment (1 mgCl2/L for 1, 2, 5, or 10 min). Among the viability RT-qPCR methods tested, CDDP-RT-qPCR with SD pre-treatment (SD-CDDP-RT-qPCR) was the most effective in reflecting viral infectivity. Performance testing of SD-CDDP-RT-qPCR in concentrated drinking water samples did not reveal any significant effects of SD-CDDP treatment. Thus, SD-CDDP-RT-qPCR could be a useful tool for monitoring infectious virus presence in drinking water.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ensuring microbial safety of drinking water is essential for protecting public health. Water supply systems currently rely on routine monitoring for bacterial indicators (e.g., E.coli and total coliforms); however, such monitoring is not enough to assess the presence or absence of infectious viruses. Recently, enteric viruses have been positively detected in drinking water in Japan (Haramoto et al. 2004), Brazil (Kluge et al. 2014), Korea (Lee and Kim 2002), and China (Yan et al. 2012). Additionally, outbreaks associated with the presence of enteric viruses in drinking water have been reported worldwide and they affect a large number of people (Beer et al. 2015; Brunkard et al. 2011; Craun et al. 2010; Kukkula et al. 1999; McAnulty et al. 1993; Pons et al. 2015). Thus, it is essential to monitor enteric virus presence in treated drinking water to ensure its microbial safety.
To date, cell culture assays are used for detecting infectious viruses; however, many types of viruses are not able to be cultivated due to the lack of available cell line. With low specificity for particular viruses, viral pathogen detected by cell culture assays should be confirmed by other approaches such as molecular or immunological assays (Hamza et al. 2011). Also, the results of cell culture assays should be interpreted with caution as one observed plaque does not necessarily mean one infectious virus particle; more than two virus particles have been reported to co-exist in one plaque (Teunis et al. 2005). In addition, cell culture assays are time-consuming, laborious, and expensive so they are not feasible or practical to routinely monitor viral presence in water environments. Recently, cell-culture-independent assays, such as quantitative real-time polymerase chain reaction (qPCR) or reverse transcription qPCR (RT-qPCR) are being used to quantify enteric virus load in drinking water as these methods are both rapid and sensitive (Haramoto et al. 2004; Kluge et al. 2014). However, these methods cannot be used to distinguish between infectious and noninfectious viruses and can lead to overestimation viral infection risk. Indeed, although enteric virus presence has been reported in drinking and tap water, their infectivity has not been investigated (Kluge et al. 2014; Yan et al. 2012). Therefore, it is important to overcome this limitation of conventional culture-independent assays to accurately quantify infectious virus load in drinking water and to assess viral infection risk associated with consuming contaminated drinking water.
Recently, viability (RT)-qPCR has been developed to determine viral infectivity, wherein water samples are pre-treated with viability dyes such as ethidium monoazide (EMA) or propidium monoazide (PMA), and subsequently detected by (RT)-qPCR. The underlying principle of this method is that viability dyes can only penetrate the damaged capsids of inactivated viruses and covalently bind to viral genomes after photoactivation to block subsequent qPCR amplification. Therefore, only viral genomes of intact viruses are detected by viability (RT)-qPCR. This method has been successfully used to determine potential infectivity of various enteric viruses (Fuster et al. 2016; Leifels et al. 2015; Prevost et al. 2016; Sangsanont et al. 2014), and is a promising tool to assess potential risk of viral infection in water samples. However, the efficacy of viability (RT)-qPCR was recently shown to depend on capsid structure of viruses and mode of their inactivation. Viability treatments are successful only if the viral capsid is damaged such that they permit the entry of the dyes (Fuster et al. 2016; Leifels et al. 2015). During such inactivation, viruses can undergo slighter alterations in capsid protein structure or damage to nucleic acids; both of which do not allow penetration of viability dyes into the inactivated virus particles, thus leading to failure of viability (RT)-qPCR method in estimating viral infectivity (Fuster et al. 2016; Leifels et al. 2015).
Several strategies have been introduced to improve viability (RT)-qPCR methods, such as using a longer PCR amplicon (Ho et al. 2016; McLellan et al. 2016), repeating viability treatment (Kralik et al. 2010), changing microtubes before photoactivation (Agusti et al. 2017), and using alternative viability markers such as PMAxx, PEMAX, or cis-dichlorodiammineplatinum (CDDP) (Randazzo et al. 2016, 2018; Soejima et al. 2016). CDDP is a platinum compound that is more effective and less expensive than the monoazide dyes for evaluating bacterial infectivity (Soejima et al. 2016); however, its applicability for discrimination among inactivated forms of infectious viruses is limited (Fraisse et al. 2018). From the above, it emerges that these strategies are not fully effective if the capsid structure of inactivated viruses remains intact despite inactivation and that it is important to improve viability marker penetration into inactivated viruses for successful viability (RT)-qPCR testing.
The applicability of viability (RT)-qPCR for testing drinking water is also relatively less well-established as environmental factors are known to interfere with viability treatments (Heise et al. 2016; Pisz et al. 2007; Taskin et al. 2011), and only a few published studies have reported on the use of viability (RT)-qPCR testing in actual drinking water samples (Canh et al. 2018; Prevost et al. 2016). Additionally, due to a low number of viruses present in drinking water, large volumes of water samples have to be concentrated to enrich the level of viruses before estimating viral load in drinking water samples. Importantly, such concentration of environmental water samples will also increase the concentration of inhibitory substances, along with target viruses, during the concentration processes which may have inhibitory effects on subsequent detection assays (Hata et al. 2015, 2017; Ijzerman et al. 1997). It is well known that environmental matrices can interfere with (RT)-qPCR (Hata et al. 2015, 2017), yet information on their effects on viability (RT)-qPCR methods remains limited (Canh et al. 2018; Fuster et al. 2016). Moreover, as viability (RT)-qPCR is a combination of viability treatment and (RT)-qPCR, therefore, such inhibitory effects may be more complicated than those on (RT)-qPCR alone.
Thus, the aim of this study was to improve the process of viability RT-qPCR for better determination of viral infectivity in drinking water samples. We analyzed the efficacy of using sodium deoxycholate (SD) pre-treatment to enhance penetration of three viability markers, (EMA, PMA, and CDDP), into inactivated Aichi virus 1 (AiV) and used heat and chlorination treatments to minimize false-positive RT-qPCR signals. SD was used as this surfactant is known to enhance permeability of bacterial membranes (Lee and Levin 2009; Wang et al. 2015). Additionally, the effects of concentrating drinking water samples on the performance of viability RT-qPCR methods were evaluated using spiked chlorinated AiV.
Materials and Methodologies
Virus Stock and Infectivity Assay
Aichi virus 1 (AiV), a human enteric virus belonging to the genus Kobuvirus and also a member of the family Picornaviridae, was used as it is a recommended and appropriate indicator of viral contamination in water environments (Kitajima and Gerba 2015). Further, the AiV was selected as a representative of human enteric viruses instead of poliovirus as currently, polio virus use is not recommended even in the laboratory due to the WHO global polio eradication program.
AiV was propagated in Vero cells in Eagle’s minimum essential medium (Nissui Pharmaceutical Co., Ltd, Tokyo, Japan). Culture media contained either 5% (for growth) or 1% (for maintenance) of fetal bovine serum (Sigma-Aldrich, Tokyo, Japan), 2 mM l-glutamine (Life Technologies, Tokyo, Japan), 1% antibiotic–antimycotic solution (Life Technologies), and 0.15% sodium bicarbonate (Life Technologies). AiV (200 µL) was added onto 75 cm2 tissue culture flasks (Iwaki, Asahi Glass Co., Ltd, Tokyo, Japan) with 80–90% confluent Vero cells and incubated for 3–4 days. After cells underwent complete cytopathic effects (CPE), the flasks were frozen and thawed three times to release the AiV particles and the released particles filtered through a 0.22-µm membrane filter (DISMIC-25CS, Advantec, Tokyo, Japan). This AiV stock was vigorously mixed with chloroform (1:1 ratio), centrifuged at 4000 g for 30 min, and the supernatant collected.
The supernatant was further purified by gel filtration in an Illustra Microspin S-300HR column (GE Healthcare, Tokyo, Japan) and used in heat inactivation experiments. For chlorine inactivation experiments, the AiV stock was purified using density gradient centrifugation, as follows. First, the 0.22-µm filtered AiV stock was centrifuged at 59,000 g for 6 h, the virus pellets resuspended in 20% iodixanol solution (2 mL) (OptiPrep™, Cosumo bio, Tokyo, Japan), then loaded onto 40% iodixanol solution (3 mL), and centrifuged at 160,000 g for 7 h. The band corresponding to the position of AiV was gently removed and finally dialyzed in Milli-Q water using a floatation dialysis device (100 kDa, 1 mL, Spectra/Por, USA).
To determine virus infectivity, AiV samples (1 mL, 10-fold serial dilutions, 1.0 × 100 to 1.0 × 107) were added onto confluent cells in 6-well plates (Iwaki, Asahi Glass Co., Ltd), and incubated for 2 h (gently shaken every 15 min). Next, the spiked cells were overlaid with a mixture of Eagle’s minimum essential medium (Nissui Pharmaceutical Co., Ltd) and EPI agar (Nacalai tesque, Tokyo, Japan) and the number of plaques counted after 2 days of incubation (37 °C, 5% CO2) when cells underwent complete CPE. The AiV stock concentration after purification was determined to be 106–107 PFU/mL.
SD Preparation and Pre-treatment Before Viability Treatment
SD powder (Wako Pure Chemical Industries, Tokyo, Japan) was dissolved in Milli-Q water to obtain a stock concentration of 10% (w/v). SD pre-treatment was performed as described by Wang et al., (2014). The SD stock solution was diluted and added to water samples to obtain a desired concentration (0.01–0.5%) in a total volume of 140 µL, mixed well, and incubated at 37 °C for 30 min. These SD pre-treated samples were subjected to viability treatments as described below.
Viability Treatments
EMA (powder-form, Invitrogen, Carlsbad, USA) or PMA (powder-form, Biotium Inc., Fremont, USA) was dissolved in 20% dimethyl sulfoxide (DMSO, Sigma-Aldrich) to obtain a concentration of 10 mM and stored at − 20 °C in the dark until further use. CDDP (powder-form, Sigma-Aldrich) was dissolved in DMSO to obtain a concentration of 100 mM and stored at − 20 °C until use. EMA, PMA, or CDDP stock solutions (1.4 µL) were added to water samples (140 µL) to obtain final concentrations of 100, 100, or 1,000 µM, respectively, which we predetermined as the most effective concentrations based on the reduction of the pure naked genome of AiV, successfully applied to distinguishing various enteric viruses (Escudero-Abarca et al. 2014; Fraisse et al. 2018; Graiver et al. 2010; Leifels et al. 2015; Sánchez et al. 2012).
EMA or PMA spiked samples were incubated at 4 °C for 30 min in the dark, followed by light exposure for 3 min at a distance of 15 cm from a 650 W halogen lamp (Selecon Pacific, Auckland, New Zealand). During exposure to light, the samples were placed on ice water to minimize unexpected effects. CDDP-spiked samples were incubated at 30 °C for 30 min. All viability treatment experiments were performed in 1.5-mL transparent microtubes.
Viral RNA Extraction and Reverse Transcription
Viral RNA was extracted from 140 µL of water samples using the QIAamp viral RNA minikit (Qiagen, Tokyo, Japan) according to the manufacturer’s instructions and was subjected to reverse transcription (RT) using a High Capacity cDNA reverse transcription kit (Applied Biosystems). A reaction mixture of 20 µL contained 10 µL of extracted RNA, 2 µL of 10 × RT buffer, 2 µL of 10 × random primers, 1 µL of MultiScribe™ reverse transcriptase (50 units/µL), 1 µL of RNase inhibitor, 0.8 µL of 25 dNTPs, and 3.2 µL of nuclease-free water, and the RT conditions were 25 °C for 10 min, 37 °C for 120 min, and 85 °C for 5 min.
Virus Quantification by qPCR
The 20 µL qPCR reaction mixture contained 5 µL of cDNA, 10 µL of TaqMan Gene Expression Master Mix (Applied Biosystems, Tokyo, Japan), 1 µL each of the forward and reverse primer (10 µM), 0.5 µL TaqMan probe (5 µM), and 2.5 µL of nuclease-free water. Sequences of the primer pairs and the TaqMan probe have been described in a previous study on AiV detection (Kitajima et al. 2013). The StepOnePlus real-time PCR system (Applied Biosystems) was used for real-time PCR analysis with cycling conditions of 95 °C for 10 min, followed by 50 cycles at 95 °C for 15 s, and 60 °C for 1 min. After each amplification, tenfold serial dilutions (1.0 × 100 to 1.0 × 104) of a plasmid DNA containing the target sequence were used to generate a calibration curve for quantification of viral genomes.
Heat Treatment
AiV samples (300 µL, 106 PFU/mL) in 1.5 mL microtubes were incubated at 50, 60, 70, 80, or 90 °C for 1 min in a dry block heater (Nissin, Tokyo, Japan), and samples were immediately placed on ice water while one sample was maintained at 4 °C as control.
Chlorine Treatment
A free chlorine stock solution (300 mg/L) was prepared by diluting sodium hypochlorite (NaOCl, 5%) in Milli-Q water. Free chlorine concentration was measured using the N,N-diethyl-p-phenylenediamine colorimetric method in a DR890 colorimeter (HACH, USA). Glassware was prepared by soaking them overnight in 100 mg/L chlorine solution, rinsing with Milli-Q water, and finally baking them at 250 °C for 3 h.
Milli-Q water (440 mL, pH around 6.5) was first mixed well with the AiV stock solution (440 µL, 3.3 × 107 PFU/mL) then divided into four aliquots of 110 mL each. Next, chlorine stock solution was added to these aliquots to obtain an initial concentration of 1.0 mg/L, while contact time was maintained at 1, 2, 5, or 10 min. From each aliquot, 10 mL of the sample was removed and used to measure free chlorine concentration, while the rest of the sample (100 mL) was immediately quenched by adding 1 mL of 400 mg/L sodium thiosulfate (4 mg/L final concentration). Quenched samples were then concentrated to approximately 1.0 mL volume by ultrafiltration (Centricon Plus-70, MWCO 30 kDa, Millipore, Tokyo, Japan) for subsequent virus quantification.
Determination of Optimal SD Concentration
The heated AiV samples (at 90 °C for 1 min) were pre-treated at final SD concentrations of 0.01, 0.05, 0.1, or 0.5%, and subjected to (1) RT-qPCR alone, (2) EMA treatment followed by RT-qPCR (EMA-RT-qPCR), (3) PMA treatment followed by RT-qPCR (PMA-RT-qPCR), and (4) CDDP treatment followed by RT-qPCR (CDDP-RT-qPCR). Samples without SD pre-treatment were also subjected to viability treatments and used as process control. All the samples were tested in triplicate.
Evaluation of AiV Inactivation by Heat Treatment
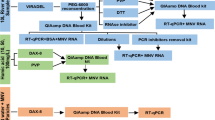
Samples heated to 50, 60, 70, 80, or 90 °C for 1 min were subjected to different detection methods, including (1) infectivity assay, (2) RT-qPCR alone, (3) viability RT-qPCR methods (EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR), and (4) viability RT-qPCR methods with SD pre-treatment (SD-EMA-RT-qPCR, SD-PMA-RT-qPCR, and SD-CDDP-RT-qPCR), as illustrated in Fig. 1. SD pre-treatment used the optimal concentration determined in the previous step. One sample at 4 °C (without the heat treatment) was used as control. All tests were conducted in triplicate.

Flow chart of the different detection methods used for evaluating AiV inactivation after heat and chlorine treatments. (1) Infectivity assay, (2) RT-qPCR alone, (3) viability RT-qPCR methods (EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR), and (4) viability RT-qPCR methods with SD pre-treatment (SD-EMA-RT-qPCR, SD-PMA-RT-qPCR, and SD-CDDP-RT-qPCR)
Evaluation of AiV Inactivation by Chlorine Treatment
Samples were chlorinated at 1.0 mg/L chlorine for 1 min, 2 min, 5 min, or 10 min, and subjected to different detection methods, including (1) infectivity assay, (2) RT-qPCR alone, (3) viability RT-qPCR (EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR), and (4) viability RT-qPCR with SD pre-treatment (SD-EMA-RT-qPCR, SD-PMA-RT-qPCR, and SD-CDDP-RT-qPCR), as illustrated in Fig. 1. SD pre-treatment used the optimal concentration determined in the previous step. One sample without choline treatment was used as control and all tests were conducted in triplicate.
Performance of the Viability RT-qPCR Methods in Real-World Samples
A total of 36 water samples (18 raw water and 18 treated water samples) were collected every two months from three full-scale drinking water treatment plants (A, B, C) in Japan, all of which use river water as the source. The sampling periods at plants A, B, and C were between May 2017 and March 2018, from December 2015 to October 2016, and from March 2016 to January 2017, respectively. The treatment processes at plants A, B, and C, sampling points, and average sample volumes are shown in Fig. 2. Treated water samples were collected after slow sand filtration in plant A, after rapid sand filtration in plant B, and after primary rapid sand filtration in plant C (Fig. 2). Raw and treated water samples (total 36 samples) were concentrated by a virus concentration method that used a negatively charged filter cartridge (Hata et al. 2015).

Treatment processes, sampling points, and average sample volumes from three full-scale drinking water treatment plants (A, B and C)
To evaluate the performance of viability RT-qPCR methods in real-world samples, fivefold dilutions of all raw water and treated concentrates spiked with the chlorinated AiV (at 1 mg/L chlorine for 5 min from the chlorine experiment above) were subjected to (1) RT-qPCR alone and (2) the most effective viability RT-qPCR (from previous section). Chlorinated AiV at 1 mg/L was used to detect the potential presence of inactivated viruses in actual drinking water where chlorination was employed. Positive controls were used in the absence of environmental matrices.
Additionally, to estimate the level of organic compounds in the concentrated raw and treated water samples, UV254 absorbance was measured on a UH5300 device (Hitachi, Tokyo, Japan).
Statistical Analyses
Microsoft Excel 2016 was utilized for all statistical analyses. The Student’s t-test was used to evaluate the difference between viability RT-qPCR with and without SD pre-treatment, and linear regression was used to evaluate the correlation between the UV254 absorbance and the inhibition of RT-qPCR. p values less than 0.05 were considered statistically significant.
Results
Determination of Optimal SD Concentration
The results of the experiments used to determine optimal SD concentration are provided in Fig. 3. The viral load of heated AiV, determined by EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR, in samples without SD pre-treatment (SD = 0%), was, respectively, 3.1, 3.0, and 4.6 log10, lower than those by RT-qPCR alone. The level of heated AiV viral load detected by EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR after 0.1% SD pre-treatment was, respectively, 1.0 log10, 1.0 log10, and 1.6 log10, less than that without SD pre-treatment; 0.1% SD pre-treatment did not influence RT-qPCR results (Fig. 3) or viral infectivity (Table 1). However, SD pre-treatment at concentrations greater than 0.1% resulted in RT-qPCR underestimation (1.6 log10), implying that a SD concentration of 0.1% was optimal for improving the efficacy of EMA, PMA, and CDDP treatments.

Optimization of SD concentration. Heated AiV samples (90 °C for 1 min) were pre-treated with various SD concentrations (0, 0.01, 0.05, 0.1, or 0.5%) and viral load determined by (1) RT-qPCR alone (PCR), (2) EMA treatment followed by RT-qPCR (EMA), (3) PMA treatment followed by RT-qPCR (PMA), and (4) CDDP treatment followed by RT-qPCR (CDDP). Error bars indicate standard deviation (n = 3 for each)
Evaluation of AiV Inactivation After Heat Treatment
AiV inactivation after heat treatment at 50, 60, 70, 80, or 90 °C for 1 min was determined using the infectivity assay, RT-qPCR alone, viability RT-qPCR without SD pre-treatment (including EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR), and viability RT-qPCR with 0.1% SD pre-treatment (including SD-EMA-RT-qPCR, SD-PMA-RT-qPCR, and SD-CDDP-RT-qPCR) (Fig. 4).

Heat inactivation of AiV at 50, 60, 70, 80, or 90 °C for 1 min. Results are expressed as log10 reduction, as determined by RT-qPCR alone (PCR), infectivity assay (PA), EMA-RT-qPCR (EMA), SD-EMA-RT-qPCR (SD-EMA), PMA-RT-qPCR (PMA), SD-PMA-RT-qPCR (SD-PMA), CDDP-RT-qPCR (CDDP), or SD-CDDP-RT-qPCR (SD-CDDP). Comparison of log10 reduction among a SD-EMA, b SD-PMA, and c SD-CDDP and other detection methods, respectively. The dotted and dashed lines indicate average log10 reduction limits of infectivity and PCR-based assays, which were 5.9 log10 PFU and 7.2 log10, respectively. Solid circles with asterisk symbol represent the negative results. Error bars indicate standard deviation (n = 3)
RT-qPCR alone showed no reduction of AiV viral load at any of the temperatures tested, while a reduction in infectivity was observed at 60 °C (3.3 log10), 70 °C (5.0 log10), and at both 80 °C and 90 °C (greater than 5.9 log10, below detection limits) (Fig. 4a–c). There was no difference in the reduction of AiV viral load (nearly 2.0 log10) between with and without SD pre-treatment at 60 °C, as determined by all viability RT-qPCR methods (Fig. 4a–c). However, at temperatures higher than 60 °C, EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR with SD pre-treatment resulted in an average reduction of, respectively, 4.0 log10, 4.0 log10 and 6.0 log10, which was 1.0 log10, 1.0 log10, and 2.4 log10 greater than that without SD pre-treatment, respectively (p < 0.05) (Fig. 4a–c).
Evaluation of AiV Inactivation After Chlorine Treatment
Figure 5 shows the extent of AiV inactivation after 1.0 mg/L chlorine treatment for 1, 2, 5, or 10 min, as determined using the infectivity assay, RT-qPCR alone, viability RT-qPCR without SD pre-treatment (including EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR), and viability RT-qPCR with SD pre-treatment (including SD-EMA-RT-qPCR, SD-PMA-RT-qPCR, and SD-CDDP-RT-qPCR).

Inactivation of AiV after chlorine treatment with 1.0 mg/L initial free chlorine of for 1, 2, 5, or 10 min. At least 0.94 mg/L of residual chlorine was estimated at any treatment time tested. Results are expressed as log10 reduction, as determined by RT-qPCR alone (PCR), infectivity assay (PA), EMA-RT-qPCR (EMA), SD-EMA-RT-qPCR (SD-EMA), PMA-RT-qPCR (PMA), SD-PMA-RT-qPCR (SD-PMA), CDDP-RT-qPCR (CDDP), or SD-CDDP-RT-qPCR (SD-CDDP). Comparison of log10 reduction among a SD-EMA, b SD-PMA, and c SD-CDDP and other detection methods, respectively. The dotted and dashed lines indicate average log10 limits of detection for the infectivity and PCR-based assays, namely, 5.5 log10 PFU and 7.7 log10 copies, respectively. Solid circles with asterisk symbol represent the negative results. Error bars indicate standard deviation (n = 3)
The loss in infectivity was 3.9 log10 for a treatment time of 1 min and was completely below the detection limit as treatment time increased to 10 min (residual chlorine ≥ 0.94 mg/L) (Fig. 5a–c). Despite such high loss in infectivity, RT-qPCR alone yielded a less than 2.7 log10 reduction even at the longest treatment time of 10 min (Fig. 5a–c). EMA-RT-qPCR showed less than 1.0, 2.6, and 4.3 log10 reduction at treatment times of 2, 5, and 10 min, respectively, which was significantly lower than the 2.3 log10, 3.2 log10, and 5.0 log10 reduction, respectively, obtained by SD-EMA-RT-qPCR (p < 0.05) (Fig. 5a). Similar tendencies were also observed when PMA-RT-qPCR and SD-PMA-RT-qPCR were compared and when CDDP-RT-qPCR and SD-CDDP-RT-qPCR were compared, indicating that these reductions due to SD pre-treatment were significantly higher than those without SD pre-treatment (p < 0.05) (Fig. 5b, c).
Effect of Environmental Matrices on SD-CDDP-RT-qPCR Performance
The SD-CDDP-RT-qPCR method was the most effective in eliminating the false-positive RT-qPCR signals from inactivated AiV particles among all the viability RT-qPCR methods tested (Figs. 4, 5). Therefore, the performance of the SD-CDDP-RT-qPCR method was further evaluated in the presence of environmental matrices using a spiking test. Raw and treated water concentrates from the three drinking water treatment plants (plant A, plant B, and plant C) were spiked with chlorinated AiV (1.0 mg/L, 5 min) and viral load determined by RT-qPCR and SD-CDDP-RT-qPCR.
Figure 6a shows chlorinated AiV viral load detected in raw water concentrates by RT-qPCR and SD-CDDP-RT-qPCR. In seven raw water concentrate samples, including AR1, AR3, AR4, AR5, and AR6 (plant A) and BR1 and BR3 (plant B), the detection level of RT-qPCR was significantly lower than that of control (at least 1.0 log10, < 99% lower confidence limit for mean RT-qPCR control value, 7.8 log10 copies/mL), indicating that the RT-qPCR reaction was substantially inhibited by substances in concentrated raw water samples. Similarly, the performance of SD-CDDP-RT-qPCR was also affected because of the inhibitory effects on the RT-qPCR part of the method. Interestingly, no inhibition of the RT-qPCR process was seen in the rest of the concentrated raw water samples (n = 11) as there was no significant difference in RT-qPCR detection levels between control and raw water concentrates. However, AiV viral load detected by the SD-CDDP-RT-qPCR method in five out of these 11 raw water concentrates, namely BR2 (plant B) and CR1, CR2, CR5, and CR6 (plant C), were significantly higher than that of the control (0.4–1.4 log10 copies/mL, > 99% upper confidence limit for mean SD-CDDP-RT-qPCR control value, 4.0 log10 copies/mL), indicating that the higher AiV viral loads were caused by the inhibition on the SD-CDDP treatment.

Analysis of chlorinated AiV (1 mg/L for 5 min) spiked in raw and treated water concentrates from three drinking water treatment plants by RT-qPCR (PCR) and SD-CDDP-RT-qPCR (SD-CDDP). a Raw water concentrates and b treated drinking water concentrates. Blank columns indicate the limit of detection (3.3 log10 copies/mL). The dashed line indicates 99% lower confidence limit for mean PCR control value. The dotted line indicates 99% upper confidence limit for mean SD-CDDP control. Error bars present standard deviation (n = 6)
The profile of RT-qPCR and SD-CDDP-RT-qPCR analyses of treated water concentrates is illustrated in Fig. 6b. None of the treated water concentrate samples interfered with RT-qPCR or SD-CDDP-RT-qPCR, except for water concentrate samples from plant C (CT1–6), wherein a substantial inhibition of RT-qPCR was observed. The observed inhibition of SD-CDDP treatment could not be estimated due to the lack of a detection method.
Correlation Between RT-qPCR Inhibition and Organic Matter Concentration in the Concentrate Samples
The UV254 absorbance values of the raw and treated water concentrate samples were compared to inhibition of RT-qPCR (Fig. 7), and the results showed that RT-qPCR inhibition was positively correlated to the level of UV254 absorbance in raw water concentrates (R = 0.6; p < 0.05) and in treated water concentrates (R = 0.8; p < 0.01).

Correlation between the UV254 absorbance and the inhibition of RT-qPCR. a Raw water concentrates and b treated water concentrates collected from plants A, B, and C. Open square in a represents a RT-qPCR result that was excluded from the correlation analysis as it was below the detection limit
Discussion
The Effectiveness of SD Pre-treatment in Improving Penetration of Viability Markers
The current study evaluated the effectiveness of using SD, an anionic surfactant, to improve the efficacy of the viability markers, namely EMA, PMA, and CDDP. Our results show that 0.1% SD pre-treatment was optimal in enhancing the penetration of all viability makers into heat-inactivated AiV without affecting infectious virus load, and that viability RT-qPCR methods such as EMA-RT-qPCR, PMA-RT-qPCR, and CDDP-RT-qPCR, combined with 0.1% SD pre-treatment were significantly more effective in estimating the infectivity of AiV than methods without SD pre-treatment after heat and chlorine inactivation. It is important to note here that heat and chlorine may have altered capsid structure in dissimilar ways.
Previous studies have used SD to enhance permeability of dead bacterial cell membranes to monoazide dyes such as EMA and PMA (Lee and Levin 2009; Wang et al. 2014, 2015) as it can bind to lipids and proteins of cell membrane, and thereby lead to disruption of the membrane structure (Lichtenberg et al. 1983). Thus, it is possible that SD might have similarly helped enhance the permeability of the capsid of inactivated viruses to improve penetration of the viability markers (EMA, PMA, and CDDP). It has been previously reported that Triton X-100, in combination with EMA, PMA, or PtCl4 treatment prior to RT-qPCR, can be used to improve distinction between infectious and inactivated viruses (Coudray-meunier et al. 2013; Fraisse et al. 2018; Moreno et al. 2015; Randazzo et al. 2016). However, the use of Triton X-100 is not always effective as its efficiency is dependent on target viruses and disinfection procedures (Coudray-meunier et al. 2013; Fraisse et al. 2018; Fuster et al. 2016; Monteiro and Santos 2018).
Comparison of the Ability of the Different Viability Markers to Distinguish Between Infectious and Inactivated Viruses
We have only tested EMA at 100 µM, PMA at 100 µM, and CDDP at 1000 µM as they were determined to be the most effective concentrations, based on the reduction of the pure naked genome of AiV (data not shown). Additionally, CDDP was able to distinguish between infectious and heat or chlorine inactivated AiV more effectively than the monoazide dyes EMA and PMA (Figs. 4, 5). This can be explained as follows. The molecular weight of CDDP (300.05 g/mol) is lower than that of EMA (420.3 g/mol) or PMA (440.6 g/mol); therefore, CDDP could have more easily penetrated through the capsid of the inactivated viruses compared to the monoazide dyes. Next, CDDP chelates at a specific site in the G or C base of the genome (Lovejoy et al. 2008), whereas monoazide dyes bind only to G–C base pairs of double-stranded genomes (Fittipaldi et al. 2012). Therefore, compared to monoazide dyes, CDDP might be more effective in binding to genomes of single-stranded RNA viruses such as AiV. Considering the limitation test for a single virus type, further study is recommended to compare the efficiency of viability makers on different virus types. Additional advantages of CDDP use include no requirement of a light source, less time needed (no light exposure step), and lower cost compared to monoazide dyes (Soejima et al. 2016).
Limitations of Viability RT-qPCR Methods in Distinguishing Between Infectious and Inactivated Viruses
The reduction in AiV viral load after the heat and chlorine treatments determined by viability RT-qPCR methods (with and without SD pre-treatment) in most cases was lower than the loss of infectivity. In some cases (heat treatment at 80 °C and chlorine treatment at the contact time of 10 min), infectivity assay resulted in a negative result while a positive result was still obtained by viability RT-qPCR methods (Figs. 4c, 5b, c), suggesting that viability RT-qPCR methods overestimated viral infectivity. This result is consistent with those from previous studies, indicating that the use of various viability markers (including EMA, PMA, PEMAX, PMAxx, and PtCl4) cannot completely eliminate a positive RT-qPCR signal from inactivated viruses (Fraisse et al. 2018; Fuster et al. 2016; Leifels et al. 2015; Randazzo et al. 2016, 2018). This observation is related to the fact that loss of viral infectivity is not always caused by loss of capsid integrity (Hamza et al. 2011; Li et al. 2014).
Next, we tested the capacity of EMA, PMA, and CDDP markers to block RT-qPCR amplification of the pure naked genome of AiV (around 108 copies/mL), and the results showed that none of viability markers could completely eliminate the RT-qPCR signal (data not shown). This result suggests that the overestimation of viral infectivity, determined by viability RT-qPCR methods, might have also resulted from the ability of the viability markers to bind to nucleic acids; this binding depends on targeted genome regions, secondary structures on the genome regions, amplicon length, and high viral concentration in solutions (Coudray-meunier et al. 2013; Ho et al. 2016; Monteiro and Santos 2018; Prevost et al. 2016).
Performance of SD-CDDP-RT-qPCR Under the Presence of Environmental Matrices
In the SD-CDDP-RT-qPCR method, RT-qPCR succeeds SD-CDDP treatment; therefore, it is important to investigate the effects of environmental matrices on the efficiency of RT-qPCR and on the effectiveness of SD-CDDP treatment. RT-qPCR was considerably inhibited only in raw water concentrates from plant A and treated water concentrates from plant C, even though similar volumes of raw and treated water samples, 20–85 L and 500–560 L, respectively, were collected from plants A, B, and C. It is possible that these water samples contained certain inhibitory substances that were selectively co-concentrated during the virus concentration processes (Canh et al. 2018; Hata et al. 2017). The presence of inhibitory substances in treated water samples from plant C might be related to the water treatment processes themselves. Indeed, treated water samples were collected after coagulation sedimentation and rapid sand filtration in plant C, whereas they were collected after slow sand filtration in plant A, and after coagulation-sedimentation, ozonation, biological activated carbon, and rapid sand filtration in plant B. Therefore, it is possible that, compared to those in plant A and plant B, the water treatment processes in plant C did not effectively remove inhibitory substances in their water source. In addition, although SD-CDDP treatment effectively eliminated the RT-qPCR signal of inactivated virus particles in raw and treated water concentrates, some of them could have lowered the effectiveness of SD-CDDP treatment. Therefore, we suggest implementation of process control protocols to confirm the efficiency of RT-qPCR and the effectiveness of SD-CDDP treatment when using SD-CDDP-RT-qPCR to evaluate environmental water samples.
We also show that the inhibition of RT-qPCR was positively correlated to UV254 absorbance in both raw and treated water concentrates. This result points to the presence of organic compounds in the water samples, which could possibly inhibit the RT-qPCR reaction. Additionally, the correlation equation for treated water concentrates (y = 0.33x + 0.18) showed a higher slope than that of the raw water concentrates (y = 0.12x + 0.47), indicating that the UV254 absorbance is more representative of the inhibitory substances present in the treated water samples compared to those in the raw water samples.
Conclusions
Our results show that pre-treatment with 0.1% SD is optimal for enhancing the performance of viability markers (EMA, PMA and CDDP) by excluding a majority of the false-positive RT-qPCR signals from heat or chlorination inactivated AiV. Among the viability RT-qPCR methods tested, SD-CDDP-RT-qPCR most effectively reflected viral infectivity. The performance of SD-CDDP-RT-qPCR can be influenced by environmental factors from concentrated raw and treated water samples, but their influence on RT-qPCR was greater than on SD-CDDP treatment. Viability RT-qPCR methods combined with 0.1% SD pre-treatment, especially SD-CDDP-RT-qPCR, can be used to provide a more accurate estimate on the presence of infectious viruses in drinking water, and is a useful tool for assessing the potential risk of viral infections in drinking water.
References
Agusti, G., Fittipaldi, M., & Codony, F. (2017). False-positive viability PCR results: An association with microtubes. Current Microbiology, 74(3), 377–380. https://doi.org/10.1007/s00284-016-1189-3.
Beer, K. D., Gargano, J. W., Roberts, V. A., Hill, V. R., Garrison, L. E., Kutty, P. K., et al. (2015). Surveillance for waterborne disease outbreaks associated with drinking water —United States, 2011–2012. Morbidity and Mortality Weekly Report, 64(31), 3260–3267.
Brunkard, J. M., Ailes, E., Roberts, V. A., Hill, V., Hilborn, E. D., Craun, G. F., et al. (2011). Surveillance for waterborne disease and outbreaks associated with recreational water use and other aquatic facility-associated health events—United States, 2007–2008. Morbidity and Mortality Weekly Report, 60(12), 39–73. doi:ss6012a4 [pii].
Canh, D. V., Kasuga, I., Furumai, H., & Katayama, H. (2018). Impact of various humic acids on EMA-RT-qPCR to selectively detect intact viruses in drinking water. Journal of Water and Environment Technology, 16(2), 83–93. https://doi.org/10.2965/jwet.17-044.
Coudray-meunier, C., Fraisse, A., Martin-latil, S., Guillier, L., & Perelle, S. (2013). Discrimination of infectious hepatitis A virus and rotavirus by combining dyes and surfactants with. BMC Microbiology, 13(216), 1–16. https://doi.org/10.1186/1471-2180-13-216.
Craun, G. F., Brunkard, J. M., Yoder, J. S., Roberts, V. A., Carpenter, J., Wade, T., et al. (2010). Causes of outbreaks associated with drinking water in the United States from 1971 to 2006. Clinical Microbiology Reviews, 23(3), 507–528. https://doi.org/10.1128/CMR.00077-09.
Escudero-Abarca, B. I., Rawsthorne, H., Goulter, R. M., Suh, S. H., & Jaykus, L. A. (2014). Molecular methods used to estimate thermal inactivation of a prototype human norovirus: More heat resistant than previously believed? Food Microbiology, 41, 91–95. https://doi.org/10.1016/j.fm.2014.01.009.
Fittipaldi, M., Nocker, A., & Codony, F. (2012). Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. Journal of Microbiological Methods, 91(2), 276–289. https://doi.org/10.1016/j.mimet.2012.08.007.
Fraisse, A., Niveau, F., Hennechart-collette, C., Coudray-meunier, C., Martin-latil, S., & Perelle, S. (2018). International Journal of Food Microbiology Discrimination of infectious and heat-treated norovirus by combining platinum compounds and real-time RT-PCR. International Journal of Food Microbiology, 269, 64–74. https://doi.org/10.1016/j.ijfoodmicro.2018.01.015.
Fuster, N., Pinto, R. M., Fuentes, C., Beguiristain, N., Bosch, A., & Guix, S. (2016). Propidium monoazide RTqPCR assays for the assessment of hepatitis A inactivation and for a better estimation of the health risk of contaminated waters. Water Research, 101, 226–232. https://doi.org/10.1016/j.watres.2016.05.086.
Graiver, D., Saunders, S. E., Topliff, C. L., Kelling, C. L., & Bartelt-Hunt, S. L. (2010). Ethidium monoazide does not inhibit RT-PCR amplification of nonviable avian influenza RNA. Journal of virological methods, 164(1–2), 51–54. https://doi.org/10.1016/j.jviromet.2009.11.024.
Hamza, I. A., Jurzik, L., Überla, K., & Wilhelm, M. (2011). Methods to detect infectious human enteric viruses in environmental water samples. International Journal of Hygiene and Environmental Health, 214(6), 424–436. https://doi.org/10.1016/j.ijheh.2011.07.014.
Haramoto, E., Katayama, H., & Ohgaki, S. (2004). Detection of noroviruses in tap water in japan by means of a new method for concentrating enteric viruses in large volumes of freshwater. Applied and Environmental Microbiology, 70(4), 2154–2160. https://doi.org/10.1128/AEM.70.4.2154.
Hata, A., Inaba, M., Katayama, H., & Furumai, H. (2017). Characterization of natural organic substances potentially hindering RT-PCR-based virus detection in large volumes of environmental water. Enviromental Science & Technology, 51, 13568–13579. https://doi.org/10.1021/acs.est.7b00306.
Hata, A., Katayama, H., & Furumai, H. (2015). Organic substances interfere with reverse transcription-quantitative PCR-based virus detection in water samples. Applied and Environmental Microbiology, 81(5), 1585–1593. https://doi.org/10.1128/AEM.03082-14.
Heise, J., Nega, M., Alawi, M., & Wagner, D. (2016). Propidium monoazide treatment to distinguish between live and dead methanogens in pure cultures and environmental samples. Journal of Microbiological Methods, 121, 11–23. https://doi.org/10.1016/j.mimet.2015.12.002.
Ho, J., Seidel, M., Niessner, R., Eggers, J., & Tiehm, A. (2016). Long amplicon (LA) -qPCR for the discrimination of infectious and noninfectious phix174 bacteriophages after UV inactivation. Water Research, 103, 141–148. https://doi.org/10.1016/j.watres.2016.07.032.
Ijzerman, M. M., Dahling, D. R., & Fout, G. S. (1997). A method to remove environmental inhibitors prior to the detection of waterborne enteric viruses by reverse transcription-polymerase chain reaction. Journal of virological methods, 63(1–2), 145–153. https://doi.org/10.1016/S0166-0934(96)02123-4.
Kitajima, M., & Gerba, C. P. (2015). Aichi Virus 1. Environmental Occurrence and Behavior. https://doi.org/10.3390/pathogens4020256.
Kitajima, M., Hata, A., Yamashita, T., Haramoto, E., Minagawa, H., & Katayama, H. (2013). Development of a reverse transcription-quantitative pcr system for detection and genotyping of aichi viruses in clinical and environmental samples. Applied and Environmental Microbiology, 79(13), 3952–3958. https://doi.org/10.1128/AEM.00820-13.
Kluge, M., Fleck, J. D., Soliman, M. C., Luz, R. B., Fabres, R. B., Comerlato, J., et al. (2014). Human adenovirus (HAdV), human enterovirus (hEV), and genogroup A rotavirus (GARV) in tap water in southern Brazil. Journal of Water and Health, 12(3), 526–533. https://doi.org/10.2166/wh.2014.202.
Kralik, P., Nocker, A., & Pavlik, I. (2010). Mycobacterium avium subsp. paratuberculosis viability determination using F57 quantitative PCR in combination with propidium monoazide treatment. International Journal of Food Microbiology, 141, S80–S86. https://doi.org/10.1016/j.ijfoodmicro.2010.03.018.
Kukkula, M., Maunula, L., Silvennoinen, E., & von Bonsdorff, C. H. (1999). Outbreak of viral gastroenteritis due to drinking water contaminated by Norwalk-like viruses. The Journal of infectious diseases, 180(6), 1771–1776. https://doi.org/10.1086/315145.
Lee, J., & Levin, R. E. (2009). Discrimination of viable and dead Vibrio vulnificus after refrigerated and frozen storage using EMA, sodium deoxycholate and real-time PCR. Journal of Microbiological Methods, 79(2), 184–188. https://doi.org/10.1016/j.mimet.2009.08.014.
Lee, S., & Kim, S. (2002). Detection of infectious enteroviruses and adenoviruses in tap water in urban areas in Korea. Water Research, 36, 248–256.
Leifels, M., Jurzik, L., Wilhelm, M., & Hamza, I. A. (2015). Use of ethidium monoazide and propidium monoazide to determine viral infectivity upon inactivation by heat, UV- exposure and chlorine. International Journal of Hygiene and Environmental Health, 218(8), 686–693. https://doi.org/10.1016/j.ijheh.2015.02.003.
Li, D., De Keuckelaere, A., & Uyttendaele, M. (2014). Application of long-range and binding reverse transcription-quantitative PCR to indicate the viral integrities of noroviruses. Applied and Environmental Microbiology, 80(20), 6473–6479. https://doi.org/10.1128/AEM.02092-14.
Lichtenberg, D., Robson, J., R., & Dennis, A., E (1983). Solubilization of phospholipids by detergents: structural and kinetic aspects. Biochim Biophys, 737, 285–304.
Lovejoy, K. S., Todd, R. C., Zhang, S., McCormick, M. S., D’Aquino, J. A., Reardon, J. T., et al. (2008). cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proceedings of the National Academy of Sciences of the United States of America, 105(26), 8902–8907. https://doi.org/10.1073/pnas.0803441105.
McAnulty, J. M., Rubin, G. L., Carvan, C. T., Huntley, E. J., Grohmann, G., & Hunter, R. (1993). An outbreak of Norwalk like gastroenteritis associated with contaminated drinking water at a caravan park. Australian Journal of Public Health, 17(1), 36–41. https://doi.org/10.1111/j.1753-6405.1993.tb00102.x.
McLellan, N. L., Lee, H., & Habash, M. B. (2016). Evaluation of propidium monoazide and long-amplicon qPCR as an infectivity assay for coliphage. Journal of Virological Methods, 238, 48–55. https://doi.org/10.1016/j.jviromet.2016.10.004.
Monteiro, S., & Santos, R. (2018). Enzymatic and viability RT-qPCR assays for evaluation of enterovirus, hepatitis A virus and norovirus inactivation: Implications for public health risk assessment. Journal of Applied Microbiology, 124(4), 965–976. https://doi.org/10.1111/jam.13568.
Moreno, L., Aznar, R., & Sánchez, G. (2015). Application of viability PCR to discriminate the infectivity of hepatitis A virus in food samples. International Journal of Food Microbiology, 201, 1–6. https://doi.org/10.1016/j.ijfoodmicro.2015.02.012.
Pisz, J. M., Lawrence, J. R., Schafer, A. N., & Siciliano, S. D. (2007). Differentiation of genes extracted from non-viable versus viable micro-organisms in environmental samples using ethidium monoazide bromide. Journal of Microbiological Methods, 71(3), 312–318. https://doi.org/10.1016/j.mimet.2007.09.015.
Pons, W., Young, I., Truong, J., Jones-Bitton, A., McEwen, S., Pintar, K., & Papadopoulos, A. (2015). A systematic review of waterborne disease outbreaks associated with small non-community drinking water systems in Canada and the United States. PLoS ONE, 10(10), 1–17. https://doi.org/10.1371/journal.pone.0141646.
Prevost, B., Goulet, M., Lucas, F. S., Joyeux, M., Moulin, L., & Wurtzer, S. (2016). Viral persistence in surface and drinking water: Suitability of PCR pre-treatment with intercalating dyes. Water Research, 91, 68–76. https://doi.org/10.1016/j.watres.2015.12.049.
Randazzo, W., Khezri, M., Ollivier, J., & Le, F. S. (2018). International Journal of Food Microbiology Optimization of PMAxx pretreatment to distinguish between human norovirus with intact and altered capsids in shell fi sh and sewage samples. International Journal of Food Microbiology, 266, 1–7. https://doi.org/10.1016/j.ijfoodmicro.2017.11.011.
Randazzo, W., Lopez-Galvez, F., Allende, A., Aznar, R., & Sanchez, G. (2016). Evaluation of viability PCR performance for assessing norovirus infectivity in fresh-cut vegetables and irrigation water. International Journal of Food Microbiology, 229, 1–6. https://doi.org/10.1016/j.ijfoodmicro.2016.04.010.
Sánchez, G., Elizaquível, P., & Aznar, R. (2012). Discrimination of infectious hepatitis A viruses by propidium monoazide real-time RT-PCR. Food and Environmental Virology, 4(1), 21–25. https://doi.org/10.1007/s12560-011-9074-5.
Sangsanont, J., Katayama, H., Kurisu, F., & Furumai, H. (2014). Capsid-damaging effects of UV irradiation as measured by quantitative PCR coupled with ethidium monoazide treatment. Food and Environmental Virology, 6(4), 269–275. https://doi.org/10.1007/s12560-014-9162-4.
Soejima, T., Minami, J., Xiao, J., & Abe, F. (2016). Innovative use of platinum compounds to selectively detect live microorganisms by polymerase chain reaction. Biotechnology and Bioengineering, 113(2), 301–310. https://doi.org/10.1002/bit.25711.
Taskin, B., Gozen, A. G., & Duran, M. (2011). Selective quantification of viable Escherichia coli bacteria in biosolids by quantitative PCR with propidium monoazide modification. Applied and Environmental Microbiology, 77(13), 4329–4335. https://doi.org/10.1128/AEM.02895-10.
Teunis, P. F. M., Lodder, W. J., Heisterkamp, S. H., & De Roda Husman, A. M. (2005). Mixed plaques: Statistical evidence how plaque assays may underestimate virus concentrations. Water Research, 39(17), 4240–4250. https://doi.org/10.1016/j.watres.2005.08.012.
Wang, L., Li, P., Zhang, Z., Chen, Q., Aguilar, Z. P., Xu, H., et al. (2014). Rapid and accurate detection of viable Escherichia coli O157: H7 in milk using a combined IMS, sodium deoxycholate, PMA and real-time quantitative PCR process. Food Control, 36(1), 119–125. https://doi.org/10.1016/j.foodcont.2013.08.011.
Wang, L., Ye, C., Xu, H., Aguilar, Z. P., Xiong, Y., Lai, W., & Wei, H. (2015). Development of an SD-PMA-mPCR assay with internal ampli fi cation control for rapid and sensitive detection of viable Salmonella spp., Shigella spp. and Staphylococcus aureus in food products. Food Control, 57, 314–320. https://doi.org/10.1016/j.foodcont.2015.04.016.
Yan, X., Xing, Y., Yong, M., & Zhang, L. (2012). Real-time PCR detection of enteric viruses in source water and treated drinking water in Wuhan, China. Current Microbiology, 65(3), 244–253. https://doi.org/10.1007/s00284-012-0152-1.
Acknowledgements
This research was supported by the Bureau of Waterworks Tokyo Metropolitan Government and the Health Labor Sciences Research Grant (H28-Kenki- Ippan-005) by the Ministry of Health, Labor and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Canh, V.D., Kasuga, I., Furumai, H. et al. Viability RT-qPCR Combined with Sodium Deoxycholate Pre-treatment for Selective Quantification of Infectious Viruses in Drinking Water Samples. Food Environ Virol 11, 40–51 (2019). https://doi.org/10.1007/s12560-019-09368-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-019-09368-2