
Abstract
For toxicological studies of B-type fumonisin in animals, high amounts of pure fumonisins are needed. In the past, several methods for the isolation and purification of fumonisins have been published, stating the problem of high losses of fumonisins during chromatography on solid phases. In this manuscript we describe a new approach based on liquid-liquid partition techniques using centrifugal partition chromatography in combination with ion exchange chromatography for the large-scale isolation of B-type fumonisins with good recovery rates, minimizing losses of fumonisins during the purification. A batch of cultures grown on solid media of 2 kg maize yields approximately 1 g of pure fumonisins with a purity of >98%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the first isolation and characterization of fumonisin B1 (FB1) and B2 (FB2) in 1988 (Gelderblom et al. 1988) and the following discovery of further fumonisins like FB3 and FB4 (Cawood et al. 1991), several methods for the isolation and purification of fumonisins have been published. The structures of the fumonisins are shown in Fig. 1. These isolation methods commonly start with the extraction of fumonisins from fungal cultures using methanol/water or acetonitrile/water mixtures. Subsequent purification steps make use of chromatographic techniques using a number of different solid phases or liquid-liquid extractions. Examples for larger scale approaches include the combination of chromatography on Amberlite XAD-2, Sephadex LH-20 and RP C18 (reverse phase material with a hydrocarbon chain length of 18 carbon atoms) (Gelderblom et al. 1988), Amberlite XAD-2, silica gel and RP C18 (Cawood et al. 1991; Vesonder et al. 1990) as well as RP C18 only (Eppley 2001). In some cases, anion or cation exchangers have been used; for example, an enrichment of fumonisins on a strong cation exchange (SCX) material, followed by liquid-liquid extraction and RP C18 clean-up (Plattner and Shackelford 1992), or enrichment on weak anion exchange (WAX) material (Sephadex A-25), chromatography on RP C18, followed again by medium-pressure liquid chromatography (MPLC) with WAX (Freudenschuss et al. 2003) have been described. During the purification of fumonisins two major problems may occur: (1) the loss of fumonisins due to the formation of methyl esters in methanolic solutions (Gelderblom et al. 1992); (2) the loss of fumonisins during liquid chromatography, most likely caused by irreversible binding of fumonisins to the solid phase, which in particular occurs with silica gel (Cawood et al. 1991; Gelderblom et al. 1992). Therefore, our approach avoids chromatographic techniques on solid phases as much as possible, focusing on liquid-liquid partition techniques, especially using centrifugal partition chromatography (CPC). Losses during the ion exchange chromatography step of the clean-up protocol have been minimized and allow good recovery rates. Utilization of methanol has been minimized to prevent the formation of esters.

Structures of important B-type fumonisins
CPC is a chromatographic technique which is based on the distribution of an analyte between two immiscible liquid phases (Armstrong 1988). It has been employed on a preparative scale for the isolation of several natural products (Marston et al. 1988; Hostettmann and Marston 1990). Due to the lack of a solid stationary phase, irreversible binding of an analyte can be avoided. Separation of the structurally very similar fumonisins B1, B2 and B3 could be achieved by optimization of the solvent systems used. Basic approaches for the preparation of solvent systems are described elsewhere (Berthod et al. 2005; Friesen and Pauli 2007).
The described method focused on the production of FB1, FB2 and FB3. Due to its much lesser occurrence, no method for the production of FB4 has been optimized.
Materials and methods
Chemicals
Pentane and ethanol were of technical grade, tert-butyl methyl ether and butan-1-ol of synthetic grade and acetonitrile and methanol of analytical grade. Other chemicals unless noted otherwise were of analytical grade. Strong anion exchange (SAX) material employed in the enrichment was Bondesil, 40 μm (Varian, Darmstadt, Germany). The water was demineralized by a MilliQ Gradient A10-System (Millipore, Schwalbach, Germany).
Apparatus
Centrifugal partition chromatography
For CPC runs, a Kromaton-200, Fast Centrifugal Partition Chromatograph (Kromaton Technologies, Angers, France) with a rotor volume of 200 ml was used, in combination with a preparative high-performance liquid chromatography (HPLC) pump (Wellchrom HPLC pump K-501; Knauer, Berlin, Germany).
Fourier transformation mass spectrometer
Measurement of exact masses was carried out on a LTQ Orbitrap XL mass spectrometer coupled to an Accela HPLC system. Data acquisition and processing was done with Xcalibur 2.07 SP1 (Thermo Fisher Scientific, Bremen, Germany).
Methods
Biosynthetic production and extraction
Fumonisins were produced on solid maize media. Baby food jars (n = 100) with a volume of 200 ml were loaded with approximately 20 g of crushed maize, moisturized by addition of 8 ml sucrose-casein suspension (38 g sucrose and 2.5 g casein in 1 l water), closed with magenta-B caps (Sigma-Aldrich, Deisenhofen, Germany) and autoclaved. Fusarium verticillioides MRC 826 was used for inoculation of the prepared jars and they were cultivated at room temperature (20–22°C) in the dark for 21 days. Afterwards, 80 ml methanol/water (70/30, v/v) was added to each jar and its contents homogenized with an Ultra Turrax (IKA, Staufen Germany) at 6,000 rpm. The homogenized culture suspension of 50 jars was filtered through a folded 580 × 600-mm filter sheet (Schleicher & Schüll, Dassel, Germany) with a 25-cm inner diameter (i.d.) funnel. The filter cake of 50 jars was split into two equally sized parts and each part was extracted a second time with 750 ml ethyl acetate/propan-2-ol/water (53/27/20, v/v/v). The second extractions were carried out in wide-necked polyethylene flasks closed with screw-caps. Filtration processes took place under a fume hood.
Liquid-liquid extraction
Crude extracts from fungal cultures were separated into portions of approximately 500 ml, consecutively transferred into a 1-l round-bottom flask and evaporated to near dryness. Evaporation was done using rotary evaporators with a water-bath temperature of 40°C at a pressure of 250 mbar, later decreased to 50 mbar. A solvent system of tert-butyl methyl ether/butan-1-ol/ethanol/1% formic acid (5/5/1/10, v/v/v/v) was prepared, which yielded two separate phases. For redissolving the crude extract 150 ml of the less dense (upper) phase and 200 ml of the denser (lower) phase were used per 10 g of the remaining residue. The solution was transferred into a separatory funnel and vigorously mixed. After separation, the upper phase was collected and the lower phase was extracted two more times with 100 ml of the upper phase per 10 g of employed residue. The combined less dense phases containing the fumonisins were evaporated to dryness.
Enrichment on SAX column
A glass column, 55 cm × 3.5 cm i.d., under methanol was filled with 100 g SAX material, 40 μm Bondesil SAX (Varian, Darmstadt, Germany) and flushed with 500 ml methanol. Aqueous phosphate buffer (0.1 mol/l pH 6) was prepared (13.6 g KH2PO4 was dissolved in water, set to pH 6 with 0.1 mol/l aqueous NaOH solution and filled up to 1 l) and 450 ml of this solution was used for preconditioning the column. A 100-ml volume of acetonitrile/water (6/4, v/v) mixture per 10 g of precleaned extract was used to dissolve the residue from the liquid-liquid extraction. The pH of this solution was brought to 6.0-7.0 using a 0.1 mol/l aqueous NaOH solution and a maximum of 150 ml of this solution was loaded onto the SAX column (eluate I). Afterwards the column was washed with 500 ml diluted phosphate buffer (20 mmol/l, pH 6) (eluate II). To elute the fumonisins from the SAX column, 800 ml of 1% aqueous acetic acid were used and the effluent monitored using pH test strips. After 100-150 ml (eluate III) the pH dropped to 3-4 and FB1, FB2 and FB3 were eluted (eluate IV). For regeneration, the column was washed with 400 ml of 5% aqueous hydrochloric acid and the pH was still controlled by the use of pH test strips. The fraction was collected until the pH dropped to 1 (eluate V) contained FB2 and FB4. Remaining eluate (eluate VI) of the hydrochloric acid should be free of fumonisins. The mean relative amounts of fumonisins in each eluate are shown in Fig. 4. In most cases after the regeneration the column was still showing a red colour, most likely from bikaverin, a pigment produced by F. verticillioides (Kjær et al. 1971) and similar dyes. These can be removed by flushing the column with 400 ml of 5% hydrochloric acid in methanol. Afterwards the column was rinsed with at least 1 l demineralized water to remove excess hydrochloric acid. For longer storage, the acid-free column should be kept under methanol. Distribution of fumonisins between eluate IV and V may differ in relation to the relative amounts produced by the fungus. If eluate V is almost free from FB1 and FB3, it should be processed separately, avoiding unnecessary mixing of already separated fumonisins. Otherwise eluate V is combined with eluate IV. Eluate IV, containing the largest amount of fumonisins, is evaporated to near dryness and further purified with CPC. If absolute fumonisin levels in eluates II and III are high, these fractions can be evaporated to dryness and treated the same way as the crude extract from fungal cultures. This is especially useful if a continuous production of fumonisins is carried out.
Centrifugal partition chromatography
The further clean-up and separation of the fumonisins was carried out using CPC according to the scheme in Fig. 2 (see “Results and discussion” for further details). The following solvent systems were employed:
-
FB1 system: pentane/tert-butyl methyl ether/butan-1-ol/ethanol/1% aqueous formic acid (3.5/1.5/5.0/1.0/10.0, v/v/v/v/v), 80 fractions collected.
-
FB2 system: pentane/butan-1-ol/ethanol/1% aqueous formic acid (5.5/4.5/1.0/10.0, v/v/v/v), 50 fractions collected.
-
FB3 system: pentane/butan-1-ol/ethanol/1% aqueous formic acid (5.25/4.75/1.0/10.0, v/v/v/v), 50 fractions collected.

Scheme of solvent systems employed for the separation and purification of fumonisins
The solvent systems were mixed in a 2.5-l separatory funnel and the mixture was vigorously shaken. Each CPC run uses approximately 1 l of each phase. After phase separation, the rotor was loaded in the ascending mode with at least 300 ml of the lower (denser) phase to ensure a consistent filling. Afterwards the rotor was started and approximately 150 ml of the upper (less dense) phase was pumped through until the stationary phase was saturated with the mobile phase and no more bleeding of the stationary phase could be observed. For the observation of bleeding, the out-flow of the CPC system is collected in a graduated cylinder. Due to the quick separation of the two phases, the amount of both phases can be monitored. About 80 ml of the stationary (lower) phase are bled out of the rotor. All CPC separations were carried out in the ascending mode with a rotational speed of 1,000 rpm and a flow rate of 5 ml/min. Fractions were collected every 2 min (10 ml).
The first separation of the different fumonisins was done using the FB2 system. FB1 remains in the stationary phase, FB3 can be found in fractions 10-15 and FB2 in fractions 20-35. After evaporation to nearly dryness the fumonisins were further purified using the appropriate solvent system. If fractions containing fumonisins were still showing a slight red colour, another clean-up step using the same solvent system was performed. After removal of all solvents, the fumonisins were redissolved in water, deep frozen and freeze dried, yielding the fumonisins as a colourless fluffy solid. A batch of 100 baby-food jars (2 kg of maize substrate) yielded on the average 1 g of pure FB1, 200 mg FB2 and 100 mg FB3, determined gravimetrically after freeze drying.
Analysis of fumonisins by HPLC-ELSD
Fractions of CPC runs were routinely checked by HPLC coupled with an evaporative light scattering detector (ELSD) before they were pooled. Samples for the determination of fumonisins were diluted with acetonitrile/water (25/75, v/v) to a total of 20–200 μg/ml of the fumonisins of interest. Separation was run on a 125 × 4-mm i.d., 7 μm LiChrosorb RP-select B (Merck, Darmstadt, Germany) using a binary gradient consistent of 1% aqueous formic acid (eluent A) and 1% formic acid in methanol (eluent B). The system used was a Shimadzu LC-20 AT pump and an ELSD (ELSD-LT; Shimadzu Corporation, Kyoto, Japan). HPLC parameters were as follows: flow rate 1 ml/min; injection volume 20 μl; start, 55% eluent B; linear gradient to 75% B in 7.5 min, afterwards linear gradient to 90% B in 2.5 min; these conditions were held for 5.5 min, followed by a return to starting conditions and equilibration for 9 min. The ELSD parameters were: air pressure 2.5 bar; nebulizer temperature 40°C. Standard solutions of 10 μg/ml, 50 μg/ml and 100 μg/ml FB1 were used to estimate fumonisin levels in the samples.
Analysis of purity of fumonisins by HPLC-ELSD
After purification the purity of fumonisins was checked by HPLC-ELSD. Analysis was run as described in the previous sub-section, “Analysis of fumonisins by HPLC-ELSD”, but using another gradient elution for better separation. Gradient parameters for the HPLC run were as follows: start, 55% eluent B; linear gradient to 75% B in 25 min, afterwards linear gradient to 90% B in 10 min; these conditions were held for 10 min, followed by a return to starting conditions and equilibration for 10 min. All peaks with an area larger than 5,000 counts were integrated. The area summed up from all impurities was set in relation to the area of the fumonisin to calculate its purity. Prior to the calculation of the sum of impurities, the area of the injection peak was corrected by the area of an injection peak of a blank run. All runs (blank and fumonisins) were done five-fold to minimize random errors.
Analysis of fumonisins by HPLC-electrospray ionization (ESI) tandem mass spectrometry (MS/MS)
For the detection and quantitation of fumonisins in strong-matrix contaminated samples, e.g. in crude extracts or to determine exact concentrations, HPLC-MS/MS in the multiple reaction monitoring (MRM) mode was employed. For the analysis, an Agilent 1100 HPLC-system coupled with an API 4000 QTrap mass spectrometer (Applied Biosystems, Darmstadt, Germany) was utilised. Data analysis was performed with the Analyst 1.4.2 software. Chromatographic separation was carried out on a 150 × 2-mm i.d., 3 μm, Hyperclone C8 3u BDS 130A column with a 4 × 3-mm i.d. guard column (Phenomenex, Aschaffenburg, Germany) using a binary gradient of 1% formic acid in acetonitrile (eluent A) and 1% aqueous formic acid (eluent B). HPLC parameters were as follows: flow rate, 300 μl/min; injection volume, 10 μl; column temperature, 40°C; gradient, start 35% eluent A, linear gradient to 95% eluent A in 8 min, holding this level for 2 min and afterwards returning to starting conditions and equilibration of the column for 5 min. Zero grade air was used as nebulizer gas (35 psi) and drying gas (45 psi) and nitrogen as curtain gas (30 psi) and collision gas (5 × 10-5 torr ≈ 6.65 × 10-5 kPa). Drying gas was heated to 350°C and an ionization voltage of 5,500 V in positive ionization mode was used. For each analyte, one MRM transition was measured for 50 ms (quantifier) and another one for 25 ms (qualifier) to ensure correct identification of analytes. MRM transitions were as follows with the according parameters entrance potential (EP), declustering potential (DP), collision energy (CE) and collision cell exit potential (CXP) given in parentheses: FB1: 722➔334 (EP 10 V, DP 121 V, CE 55 V, CED 8 V) and 722➔336 (EP 10 V, DP 121 V, CE 55 V, CXP 8 V), FB2 and FB3 706➔336 (EP 10 V, DP 121 V, CE 55 V, CXP 8 V) and 706➔354 (EP 10 V, DP 121 V, CE 55 V, CXP 8 V), FB4 690➔338 (EP 10 V, DP 121 V, CE 55 V, CXP 8 V) and 690➔320 (EP 10 V, DP 121 V, CE 55 V, CXP 8 V).
Confirmation of the identity of fumonisins by Fourier transformation mass spectrometry (FTMS) experiments
FTMS data:
-
FB1: found m/z 722.3970 calculated for [C34H59NO15+H]+ 722.3958; MS/MS higher collision energy dissociation (HCD) at 35%: 722.3970 (47), 704.3845 (86), 686.3759 (61), 668.3653 (17), 546.3641 (13), 528.3535 (35), 510.3428 (28), 492.3323 (7), 370.3322 (29), 352.3215 (100), 334.3110 (78), 316.3047 (20), 299.2739 (3).
-
FB2: found m/z 706.4017 calculated for [C34H59NO14+H]+ 706.4008; MS/MS HCD at 35%: 706.4017 (29), 688.3913 (47), 670.3807 (19), 530.3690 (11), 512.3584 (30), 494.3478 (9), 372.3477 (5), 354.3371 (43), 336.3266 (100) 318.3161 (48), 301.2895 (2).
-
FB3: found m/z 706.4013 calculated for [C34H59NO14+H]+ 706.4008; MS/MS HCD at 35%: 706.4013 (43), 688.3907 (53), 670.3802 (21), 530.3686 (16), 512.3580 (21), 494.3474 (10), 372.3474 (5), 354.3369 (50), 336.3264 (100), 318.3159 (41), 301.2893 (4).
Results and discussion
For the large-scale isolation and purification of B-type fumonisins, a method combining liquid-liquid extraction, anion exchange chromatography and centrifugal partition chromatography was developed. In order to prevent formation of fumonisin methyl esters, methanol was employed only as an aqueous solution during extraction and quickly removed afterwards. In contrast to other methods, the use of methanol was avoided during further clean-up.
Fusarium verticillioides MRC 826, an isolate strain producing high amounts of fumonisins, was grown on solid maize media. Maize being a natural substrate for F. verticillioides showed very good production rates of fumonisins in fungal culture, as described elsewhere (Marín et al. 1999). The extraction of fumonisins was done using a methanol/water mixture as described in literature (Gelderblom et al. 1988; Cawood et al. 1991; Vesonder et al. 1990) because of the considerably lower cost of methanol compared with acetonitrile. Formation of methyl esters was monitored using HPLC-MS/MS and confirmed to be less than 2% based on the comparison of summed up areas of the peaks of monomethyl, dimethyl, trimethyl, and tetramethyl esters of FB1 to the area of the FB1 peak. A second extraction of the fungal cultures using a mixture of ethyl acetate, propan-2-ol and water as solvents improved the yields of all fumonisins by approximately 60% estimated by HPLC-MS/MS (Fig. 3)

Relative fumonisin contents in the first and second extraction steps from fungal cultures estimated by HPLC-MS/MS
A liquid-liquid separation in a separatory funnel was introduced in order to reduce the amount of small polar molecules, e.g. sugars and organic acids, in the crude extract. With less organic acids competing for binding sites, the capacity of the SAX column for fumonisins is enhanced. During liquid-liquid separation, only a very small amount (<1%) of fumonisins could be found in the lower phase. During chromatography using the SAX material, noticeable amounts of FB2 could be found in eluate II in addition to eluate IV and eluate V (Fig. 4). In order to minimize losses of FB2, this fraction can be evaporated to dryness and loaded on the SAX column for a second time or added to the crude extract of other batches during continuous production. Bikaverin and other reddish pigments are severely reduced during SAX chromatography, mostly adhering to the SAX material until removed with 5% hydrochloric acid in methanol.

Fumonisin contents in the eluates during SAX enrichment determined by HPLC-MS/MS
In order to develop optimized solvent systems for CPC, the ARIZONA system has been employed. The classic ARIZONA system uses n-hexane, ethyl acetate, methanol and water as components. Because of the polar characteristics of fumonisins and the four free acid groups of the molecule, several changes in the selection of solvents have been made. Butan-1-ol is used as a “non-polar” solvent to increase the solubility of fumonisins in the organic phase. Instead of methanol, ethanol is used as a polar organic solvent to prevent the formation of methyl esters of fumonisins. Formic acid is added to the solvent system to ensure protonation of all acid functions of fumonisins, improving chromatography. While keeping the polar solvents constant at 1 part ethanol and 10 parts 1% formic acid, different mixtures of pentane, tert-butyl methyl ether and butan-1-ol have been tested with the test tube shake partition test following the guidelines of the ARIZONA system (Berthod 1991). Mixtures showing nearly equal distribution of the fumonisins in both phases have been used as solvent systems for the purification of the corresponding fumonisin. The FB2 system demonstrates the best separation for all fumonisins and was therefore employed as a first separation step, but with FB1 being found in the stationary phase accompanied by salts from the phosphate buffer no sufficient purity of FB1 could be achieved by using this solvent system only. Depending on the amounts of each fumonisin in the batch, one CPC run can be sufficient for complete separation of FB1, FB2 and FB3. If no complete separation can be achieved, e.g. due to a higher than normal amount of one of the fumonisins, additional CPC runs using the specific solvent system for the fumonisin can be used for further separation. In addition, impurities like pigments or salts from phosphate buffer can be removed with those specific CPC runs. Therefore, the second set of CPC runs using the appropriate FB1, FB2 or FB3 system is optional and normally employed only if production of reddish pigments in the batch is especially high and purified fumonisins show a slight rosy tint. Figure 2 gives an overview over the combination of CPC runs employed.
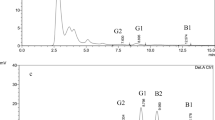
Fumonisins B1, B2 and B3 could be isolated in good purity (>98% by HPLC-ELSD). The identity of the products could be confirmed by FTMS and comparison of 1H and 13C NMR spectra (data not shown). NMR-spectra used for comparison were those published by Bezuidenhout et al. (1988), Plattner et al. (1992), Sydenham et al.(1996) and Månsson et al. (2010). Losses during clean-up on solid-phase material could be minimized as shown in Fig. 4, in which the distribution of fumonisins in the different fractions can be seen.
As the exact mode of action in the toxicology of fumonisins is still unknown and commercially available fumonisins are still very expensive, methods for the quick isolation of fumonisins in high purity are desirable. Our approach provides a fast and reliable method for the isolation of pure fumonisins, while minimizing the losses during clean-up caused by an irreversible binding and the formation of fumonisin methyl esters. On average, 500 mg of pure (>98% by ELSD) FB1 can be obtained per kilogram of maize culture.
References
Armstrong DW (1988) Theory and use of centrifugal partition chromatography. J Liq Chromatogr R T 11:2433–2446. doi:10.1080/01483918808076738
Berthod A (1991) Practical approach to high-speed counter-current chromatography. J Chromatogr A 550:677–693. doi:10.1016/S0021-9673(01)88574-5
Berthod A, Hassoun M, Ruiz-Angel MJ (2005) Alkane effect in the Arizona liquid systems used in countercurrent chromatography. Anal Bioanal Chem 383:327–340. doi:10.1007/s00216-005-0016-7
Bezuidenhout SC, Gelderblom WCA, Gorst-Allman CP, Horak RM, Marasas WFO, Spiteller G, Vleggaar R (1988) Structure elucidation of the fumonisins, mycotoxins from Fusarium moniliforme. J Chem Soc Chem Commun 1988:743–745. doi: 10.1039/C39880000743
Cawood ME, Gelderblom WCA, Vleggaar R, Behrend Y, Thiel PG, Marasas WFO (1991) Isolation of the fumonisin mycotoxins: a quantitative approach. J Agric Food Chem 39:1958–1962. doi:10.1021/jf00011a014
Eppley RM (2001) Preparatory isolation of mycotoxins from solid phase fungal cultures. In: Trucksess MW, Pohland AE (eds) Mycotoxin protocols, 1st edn. Humana Press, Totowa, pp 25–29
Freudenschuss M, Jaunecker G, Krska R (2003) Fumonisin B1—Isolation and analytical characterisation for the application as reference material. Mycotoxin Res 19:194–197. doi:10.1007/BF02942964
Friesen JB, Pauli GF (2007) Rational development of solvent system families in counter-current chromatography. J Chromatogr A 1151:51–59. doi:10.1016/j.chroma.2007.01.126
Gelderblom WCA, Jaskiewicz K, Marasas WFO, Thiel PG, Horak RM, Vleggaar R, Kriek NPJ (1988) Fumonisins—novel mycotoxins with cancer-promoting activity produced by Fusarium moniliforme. Appl Environ Microb 54:1806–1811
Gelderblom WCA, Marasas WFO, Vleggaar R, Thiel PG, Cawood ME (1992) Fumonisins: isolation, chemical characterization and biological effects. Mycopathologia 117:11–16. doi:10.1007/BF00497273
Hostettmann K, Marston A (1990) Liquid-liquid partition chromatography in natural product isolation. Anal Chim Acta 236:63–76. doi:10.1016/S0003-2670(00)83300-0
Kjær D, Kjær A, Pedersen C, Bu’Lock JD, Smith JR (1971) Bikaverin and norbikaverin, benzoxanthentrione pigments of Gibberella fujikuroi. J Chem Soc C 1971:2792–2797. doi: 10.1039/J39710002792.
Månsson M, Klejnstrup ML, Phipps RK, Nielsen KF, Frisvad JC, Gotfredsen CH, Larsen TO (2010) Isolation and NMR characterization of fumonisin B2 and a new fumonisin B6 from Aspergillus niger. J Agric Food Chem 58:949–953. doi:10.1021/jf902834g
Marín S, Magan N, Serra J, Ramos AJ, Canela R, Sanchis V (1999) Fumonisin B1 production and growth of Fusarium moniliforme and Fusarium proliferatum on maize, wheat, and barley grain. J Food Sci 64:921–924. doi:10.1111/j.1365-2621.1999.tb15941.x
Marston A, Borel C, Hostettmann K (1988) Separation of natural products by centrifugal partition chromatography. J Chromatogr 450:91–99. doi:10.1016/S0021-9673(00)90718-0
Plattner RD, Shackelford DD (1992) Biosynthesis of labeled fumonisins in liquid cultures of Fusarium moniliforme. Mycopathologia 117:17–22. doi:10.1007/BF00497274
Plattner RD, Weisleder D, Shackelford DD, Peterson R, Powell RG (1992) A new fumonisin from solid cultures of Fusarium moniliforme. Mycophatologia 117:23–28. doi:10.1007/BF00497275
Sydenham EW, Thiel PG, Vleggaar R (1996) Physicochemical data for some selected Fusarium toxins. J AOAC Int 79:1365–1379
Vesonder R, Peterson R, Plattner RD, Weisleder D (1990) Fumonisin B1: isolation from corn culture, and purification by high performance liquid chromatography. Mycotoxin Res 6:85–88. doi:10.1007/BF03192147
Acknowledgments
We thank Wentzel C.A. Gelderblom for supplying the Fusarium verticillioides strain MRC 826.
Conflict of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hübner, F., Harrer, H., Fraske, A. et al. Large scale purification of B-type fumonisins using centrifugal partition chromatography (CPC). Mycotoxin Res 28, 37–43 (2012). https://doi.org/10.1007/s12550-011-0114-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12550-011-0114-7