
Abstract
Non-coding RNA (ncRNA) plays an important role in regulating biological activities of animals and plants, and the representative ones are microRNA (miRNA) and long non-coding RNA (lncRNA). Recent research has found that predicting the interaction between miRNA and lncRNA is the primary task for elucidating their functional mechanisms. Due to the small scale of data, a large amount of noise, and the limitations of human factors, the prediction accuracy and reliability of traditional feature-based classification methods are often affected. Besides, the structure of plant ncRNA is complex. This paper proposes an ensemble deep-learning model based on convolutional neural network (CNN) and independently recurrent neural network (IndRNN) for predicting the interaction between miRNA and lncRNA of plants, namely, CIRNN. The model uses CNN to explore the functional features of gene sequences automatically, leverages IndRNN to obtain the representation of sequence features, and learns the dependencies among sequences; thus, it overcomes the inaccuracy caused by human factors in traditional feature engineering. The experiment results show that the proposed model is superior to shallow machine-learning and existing deep-learning models when dealing with large-scale data, especially for the long sequence.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Eukaryotic genomes with broad transcription features can produce all kinds of RNA, and the studies have found that only about 1–2% of transcripts are involved in protein-coding [1], the vast majority of transcripts which are not involved in protein-encoding are called non-coding RNAs (ncRNAs) [2]. In recent years, the studies on quantity and function of ncRNA species are important fields in biology. ncRNA is divided into long non-coding RNA (lncRNA) and short non-coding RNA (sncRNA) according to whether the transcript length is greater than 200 nt [3]. lncRNA and microRNA (miRNA) [4] are the most two important types. As the gradual deepening of understanding of ncRNA, their function mechanism has drawn more and more people’s attentions. Their identification and functional inquiry have become a hot issue. Researchers find that the interaction between miRNA and lncRNA plays an important role in the regulation of gene expression, which is closely related to species’ evolution, embryonic development, material metabolism, and the occurrence of various diseases [5]. In-depth study of the interaction between miRNA and lncRNA will revolutionize the current understanding of cell structure and regulation and bring great scientific and medical value. Therefore, it is crucial to reveal the interaction between RNA molecules and explain their functions.
There are two main types of interactions between miRNA and lncRNA in plant: (1) as the precursor of miRNA, lncRNA can be spliced into shorter miRNA besides playing a direct role. For example, miR869a and miR160c can be sheared from lncRNAs npc83 and npc521 [6]; (2) as a target, lncRNA can be spliced by miRNA [7]. lncRNA regulates the balance of phosphate in vivo of plants by acting as a target for miRNA, weakening the inhibitory effect on genes of miRNA. lncRNA can also act as the decoy of miRNA, competing with mRNA to bind miRNA, to regulate the expression of these miRNA target genes, which is called “sponge effect” [8]. Two lncRNAs, slylnc0195, and slylnc1077 are found to act as decoys for miRNAs in the study of tomato yellow mosaic virus (TYMV) [9]. The expression of Slylnc0195 is significantly enhanced in tomato infected with TYMV, while miR166a is down regulated.
Studies have shown that the interaction between miRNA and lncRNA plays an important regulatory role in plant disease resistance, vernalization, cell differentiation, flowering and fruiting, cold resistance, drought resistance, and other biotic and abiotic stresses. Compared with human and animal, there are relatively few studies on the interaction between miRNA and lncRNA in plant. In addition, a few miRNAs and lncRNAs mechanism of action have been confirmed, which leads to insufficient experimental data on miRNA and lncRNA in the field of plant, making it difficult to meet the requirements of bioinformatics for in-depth analysis of the interaction between miRNA and lncRNA. Therefore, a large number of data on the interaction between miRNA and lncRNA related to plant growth and development are of vital significance for in-depth study of the functional mechanism of the interaction between miRNA and lncRNA in plant.
In recent years, considerable effort has been devoted to developing computational methods for identifying associations in multiple biological data sets [10]. At present, in the prediction of the interaction between miRNA and lncRNA, many researchers have used shallow machine-learning methods to construct the prediction model through feature selection, but there are many problems such as less training data, large noise, and more human factors, making low reliability of the prediction results. In this paper, an ensemble deep-learning model CIRNN is proposed to predict the interaction between miRNA and lncRNA. This model uses the two-stage convolutional neural network (CNN) [11] to automatically learn sequence features and detect functional domains of nucleotide sequences, and then uses the two-layer independently recurrent neural network (IndRNN) [12] to learn the long-term dependence in functional domains to classify data. It obtains above 96% accuracy on Zea mays test set and better results on other plant data sets. This shows its good performance and generalization ability.
The rest of this paper is organized as follows. The model including the architecture of CIRNN is briefly introduced in Sect. 2. In Sect. 3, the results of experiments are analyzed and compared with shallow machine-learning and other deep-learning models. Section 4 concludes this paper and makes a preliminary discussion for future work.
2 Materials and Methods
2.1 Data Acquisition
The widely used and relatively rich Zea mays data set is selected for the experiment. Because of no public database of miRNA and lncRNA interaction pairs, we download 325 mature Zea mays miRNA sequences with high credibility from PNRD [13] (http://structuralbiology.cau.edu.cn/PNRD/), and 18,110 Zea mays lncRNA sequences from GreeNC [14] (http://greenc.sciencedesigners.com/wiki/Main_Page). The same sequences are removed, and 207 miRNAs and 17,684 lncRNAs are remained, as shown in Table 1.
2.2 Data Preprocessing
psRNATarget (http://plantgrn.noble.org/psRNATarget/) [15] is used as the miRNA–lncRNA interaction prediction tool in this paper. By analyzing the matching degree between miRNA and target sequences in plant, the target gene sequences that can interact with miRNA are identified. The filtered miRNAs and lncRNAs are imported into psRNATarget software for prediction, and a total of 18,241 miRNA–lncRNA interaction pairs are obtained as the positive data set. To better verify the performance of the model, it is necessary to construct a negative data set with strong interference ability.
Due to the small amount and short sequence length of miRNAs, the proportion of miRNA is relatively small in interaction pairs; therefore, the experiment mainly processes lncRNA sequences. Firstly, all lncRNAs are divided into two types, one is involved in the interaction, and another is not involved in interaction between lncRNA and miRNA. Then, Needleman Wunsch algorithm [16] is used to conduct similarity comparison between the two types of lncRNAs, and the lncRNA samples with similarity above 80% are removed [17]. Finally, lncRNAs which are not involved in the interaction between lncRNA and miRNA are randomly combined with all miRNAs to obtain the negative sample data set after similarity removal. To ensure the balance of positive and negative samples, a random sampling method is used to obtain the same number of negative samples as the positive samples. The positive and negative data sets are randomly shuffled to form the data set needed for the experiment, totaling 36,482 pieces.
For the inadequacy of the data and small sample size problems, we use the SMOTE algorithm [18] to increase the sample size by generating characteristic data that resemble the samples. Taking positive samples as an example, we randomly select a positive sample eigenvalue and determine the nearest positive sample eigenvalue, and then generate a new positive sample between the two samples. Finally, we repeat the above operations until the sample data amount is sufficient.
Because the maximum sequence length in the data set is over 8000 nt, this leads to the training time is too long. Meanwhile, there are only 216 sequences with length greater than 4000 nt. Therefore, we remove the sequences which length is greater than 4000 nt. The results verify that CIRNN’s accuracy hardly changes, but greatly reduces the training time after removing the data with the sequence length greater than 4000 nt.
Data set 1 is the original data set, and Data set 2 is the new data set after removing the length exceeding 4000 nt. We conduct three experiments, respectively, and the results are shown in Table 2. It can be seen that there is a small change in CIRNN’s accuracy, but the training time of each batch is shortened by more than half.
2.3 Model Description
Early data classification and predicting problems mainly use shallow machine-learning methods based on feature engineering, but due to its various disadvantages, researchers have begun to pay attention to deep-learning methods [19]. Recently, with the continuous development of deep learning, it has been widely used in image processing [20], sequence classification [21], natural language processing [22], biological information [23], computer vision [24], and other fields, and achieved good results.
2.3.1 CNN and IndRNN Structure
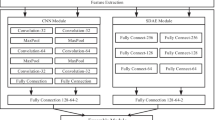
The most representative deep-learning models are CNN and recurrent neural network (RNN) [25]. Many existing deep-learning models are mostly their variants. CIRNN consists of CNN and IndRNN. CNN convolution layer can automatically extract feature information of data at different levels [26], and then sample and process the features through the pooling layer to obtain the features that are most suitable for classification. Afterwards, the feature information obtained is passed into the IndRNN layer to further learn the dependencies between features. The model uses Dropout layer to prevent overfitting. At the same time, Relu function is used as the activation function, because the Relu function has the advantages over sigmoid function in facilitating sparse and effectively reducing the gradient likelihood value [27]. To better extract and filtrate features, the model uses two-layer CNN. The specific structure of CNN is shown in Fig. 1.

CNN frame structure
IndRNN can learn long-term dependence between sequences. To better learn the dependencies between sequences, the model uses two-layer IndRNN. Different from traditional RNN, IndRNN is simple in structure and can be easily extended to different network architectures. Neurons in the same layer are independent, so that the behavior of each neuron can be analyzed without considering the influence of other neurons. It can solve the gradient disappearance and gradient explosion problems in traditional RNN with the deepening of network level, without loss of trainable loop connection ability and not involving gate parameters [28], and maintain long-term memory. Therefore, the gradient can be effectively propagated at different time steps. The network can be more in-depth and persistent, which enables multiple IndRNNs to be stacked up to build a deeper network, to better explore the cross-channel information and learn the dependence between data. The status update can be described as follows [12]:
where xt and ht are the input and hidden state at time step t, respectively. W and U are the weights of the current input and the recurrent input, and b is the bias of the neuron.where BN denotes standardized batch processing; W1, W2, and Recurrent + Relu represent the input weights and loop processing of each step with Relu as the activation function. By stacking this structure, a deeper IndRNN network can be built which is shown in Fig. 2.

IndRNN frame structure
2.3.2 CIRNN Structure
An ensemble deep-learning model CIRNN is proposed based on traditional CNN and IndRNN. The model is mainly divided into two parts. One part is the traditional CNN, which is a feedforward neural network that extracts features through convolution operation and then uses pooling layer to learn local features of data. Another part is IndRNN an extension of RNN. RNN has internal memory features and has internal feedback connection and feedforward adjustment between processing elements. Therefore, it has a good effect on processing sequence information. However, for sequence data, CNN only considers the correlation between continuous sequences and ignores the correlation between non-continuous sequences. Although RNN is suitable for processing sequence data, it is difficult to deal with the problem of long-term dependence of information. Furthermore, there are gradient disappearance and gradient explosion problems. CIRNN combines the advantages of CNN and IndRNN. In this way, feature information can be fully extracted and long-term dependence between sequences can be taken into account. The overall architecture is shown in Fig. 3.

Overall architecture of the proposed model
2.4 Experiment
The experiments are implemented in a Keras framework [29] based on TensorFlow 1.12.0 written in Python3.6.5 under Windows 10 system. Before the model is trained, we conduct data preprocessing, firstly. The bases A, T, C, and G are encoded as 1, 2, 3, and 4, respectively. Then, the embedding layer converts data after encoding into a matrix, which is presented to CNN. The convolution operation is carried out to extract feature information through convolution layers, and the important local feature information is filtered out by the maximum pooling operation. The vector matrix is transformed into a feature map as the input layer of IndRNN after activating by the Relu function. IndRNN is utilized to fully learn the dependence between features. Finally, using the dense layer to map the feature vector of the IndRNN output to a concrete number, and the sigmoid function is used to map the number to [0, 1], the predicted results are obtained. According to the loss between the real value and the predicted value, BP algorithm [30] is used to calculate the loss layer by layer to update the parameters. Dropout layer with a parameter of 0.5 is used to prevent overfitting. The learning rate of the model is set to 0.01, each batch of data is 128, and the stochastic gradient descent (SGD) algorithm is used to optimize the model.
3 Results and Discussion
3.1 Comparison with Shallow Machine-Learning Models
First, CIRNN is compared with shallow machine-learning methods on Zea mays data set, including classical machine-learning algorithms such as support vector machine (SVM) [31], logistic regression [32], random forest [33], and k-nearest neighbor (k-NN) [34].
Although deep learning automatically extracts features, the significant features may not be prominent in this process, which will result in a general but not optimal situation. Therefore, the performance of deep-learning methods may not be as good as shallow machine-learning methods. To verify the performance of the model, CIRNN is compared with shallow machine-learning models and other deep-learning models. In addition, we also applied CIRNN for other plants.
To ensure the accuracy and reliability of the experimental results, the experiments adopt ten-fold cross validation. The experimental data set is divided into 10 groups on average, 9 groups are used for training, and 1 group for verification. Performing experiment 10 times alternately, the average value of 10 experiments is taken as the final result.
For shallow machine-learning methods, we first conduct feature extraction. The main features extracted in this experiment are the primary structural features and secondary structural features of the sequence. k-mer is the common primary structural feature. A k-mer has k nucleotides, each of which can be one of A, T, C, and G. 1-mer (4 dimensions), 2-mer (16 dimensions), and 3-mer (64 dimensions) features of the sequence are extracted in the experiment. The sliding window with a length of k is used to match the above k-mer, with a sliding step size of 1. In addition to the k-mer features, the experiment also extracted the gap features of the sequence, including the first gap features (A*A, 64 dimensions) and the second gap features (A**A, 256 dimensions).
Secondary structure features determine the important functions of RNA molecules. Studies have shown that the more stable the structure of the RNA sequence is, the more free energy will be released when it folds itself to form the secondary structure; the more stable the secondary structure is, the more complementary base pairs it forms, and the higher the content of G and C. The base complementary pairing ratio (E1), G and C contents (E2) and the normalized minimum free energy (DM) of the sequences are extracted in the experiment. The experiment uses the ViennaRNA [35] toolkit to obtain the dot bracket form of the sequence secondary structure and the minimum free energy released by the formation of these secondary structures, specifically defined as follows:
where n_pairs represents the number of pairs of bases that can be paired in a sequence, L represents the sequence length, n_G and n_C represent the frequency of occurrence of G and C, and MFE represents the minimum free energy of a sequence.
A total of 407 dimensions are obtained including both primary structural features and secondary structural features. These features are fused to form 407 dimensional feature vectors. All feature vectors are combined into a vector set for model training and testing. The detail feature information is shown in Table 3.
In this paper, the four values of accuracy (ACC), Precision, Recall and F1 value (F1_score) are used as evaluation criteria for experimental results, which are defined as follows:
where TP represents the number of positive classes predicted to be positive classes, TN represents the number of negative classes predicted to be negative, FN represents the number of positive classes predicted to be negative, and FP represents the number of negative classes predicted to be positive. The experiments also adopt ten-fold cross validation, using 90% data as training data and 10% data as test data. The experimental results of CIRNN and shallow machine-learning models are shown in Fig. 4.

Performance of CIRNN compared with shallow machine-learning models
It can be seen from Fig. 4 that CIRNN reaches above 95% on both the four evaluation indicators; obviously, it is higher than other models, which proves CIRNN is superior to the shallow machine-learning models. Experimental results show that CIRNN performs better than shallow machine learning in the classification of miRNA–lncRNA interaction.
3.2 Comparison with Other Deep-Learning Models
In addition to comparisons with shallow machine-learning models, CIRNN is also compared with other deep-learning models, such as CNN, LSTM, CNN + LSTM, and IndRNN. We divide the Zea mays data set to 6 groups, and the maximum sequence length of each group, respectively, is 500 nt, 1000 nt, 1500 nt, 2000 nt, 2500 nt, and 3000 nt. Data distribution is shown in Fig. 5. 6 groups of data are put into each model for training and testing, and ten-fold cross validation is adopted; ACC is used as the evaluation indicator. The classification results are shown in Table 4.

Data distribution
We can see from Table 4, with the increase of sequence length, the accuracy of LSTM decreases significantly, and the accuracy of CNN + LSTM also decreases slightly. Only the accuracy of CIRNN and CNN remain unchanged, but the accuracy of CIRNN is significantly higher than CNN. The results show that CIRNN has a better performance in the accuracy of miRNA–lncRNA interaction than other deep learning models, especially when the sequence length is relatively long, the model shows good performance.
To further verify the performance of the model, we also compare the loss convergence rate of each model when the sequence length is 3000 nt. Figure 6 shows the comparison of the loss convergence rate in 25 iterations. We can see that CIRNN is superior to existing deep-learning models in both the convergence rate and the degree of convergence.

Loss ratio of different models
To verify the generalization ability of CIRNN, experiments are carried out on other several plants, including Arabidopsis Iyrata, Glycine Max, Setaria italica, Sorghum bicolor, Solanum tuberosum, and Brachypodium distachyon. miRNA and lncRNA data sets of these species are collected, in which miRNA is from PNRD database and lncRNA is from GreeNC database. The positive data set is obtained by psRNATarget software, and the negative data set is obtained by randomly binding miRNA with lncRNA without involving in the interaction of miRNA and lncRNA, and the similarity between the selected lncRNA and the positive set is greater than 70%. Samples with the same number of positive simples are randomly selected to form the final dat aset, which is put into the model for training and testing. Detailed information on experimental data sets and experimental results is shown in Tables 5 and 6.
As can be seen from Table 6, CIRNN has the highest accuracy. Its accuracy is better than other deep-learning models, indicating that the model has a good generalization ability and is suitable for most plants.
4 Conclusion
A deep-learning model CIRNN is proposed to predict the interaction between miRNA and lncRNA,based on the RNA sequence features of plant. The model can effectively solve the problems of gradient disappearance and explosion in the process of gradient propagation, and ensure the accuracy of classification. Moreover, the model is simple in structure, convenient to use and easy to expand. Meanwhile, CIRNN performs well in classification of the interactions between miRNA and lncRNA in plant. Compared with shallow machine-learning and other deep-learning models, the model has obvious advantages, that can be applied to other plants, and achieve good results. Meanwhile, the model has superior performance and good generalization ability, and can be widely used in the classification of plant miRNA–lncRNA interaction. To further explore the interaction mechanism of miRNA and lncRNA in plant, this research has laid the foundation. The accuracy of model classification can be further improved by adjusting the level of model structure and increasing the amount of data in the future.
References
Costa FF (2010) Non-coding RNAs: meet thy masters. BioEssays 32(7):599–608
Heo JB, Lee YS, Sung S (2013) Epigenetic regulation by long noncoding RNAs in plants. Chromosome Res 21(6–7):685–693
Liu YH, Diao HY, Yao YL et al (2016) Long noncoding RNA NEAT1 promotes glioma pathogenesis by regulating miR-449b-5p/c-Met axis. Tumor Biol 37(1):673–683
Ma R, Wang C, Wang J et al (2016) miRNA–mRNA Interaction Network in Non-small Cell Lung Cancer. Interdiscip Sci Comput Life Sci 8(3):209–219
Huang ZA, Huang YA, You ZH et al (2018) Novel link prediction for large-scale miRNA-lncRNA interaction network in a bipartite graph. BMC Med Genomics 11(6):113
Paraskevopoulou MD, Hatzigeorgiou AG (2016) Analyzing miRNA-lncRNA interactions. Methods Mol Biol 1402:271–286
Jalali S, Bhartiya D, Lalwani MK et al (2013) Systematic transcriptome wide analysis of lncRNA miRNA interactions. PLoS ONE 8(2):e53823
Thomson DW, Dinger ME (2016) Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17(5):272–283
Valiollahi E, Farsi M, Kakhki AM (2014) Sly-miR166 and Sly-miR319 are components of the cold stress response in Solanum lycopersicum. Plant Biotechnol Rep 8(4):349–356
Chen J, Peng H, Han G et al (2018) HOGMMNC: a higher order graph matching with multiple network constraints model for gene–drug regulatory modules identification. Bioinformatics 35(4):602–610
Gu JX, Wang ZH, Kuen J (2018) Recent Advances in Convolutional Neural Networks. Pattern Recogn 77:354–377
Li S, Li W, Cook C, et al (2018) Independently recurrent neural network (IndRNN): building a longer and deeper RNN. In: IEEE conference on computer vision and pattern recognition. https://arxiv.org/abs/1803.04831
Yi X, Zhang Z, Ling Y et al (2015) PNRD: a plant non-coding RNA database. Nucleic Acids Res 43(D1):D982–D989
Andreu PG, Antonio HP, Irantzu Anzar ML et al (2016) GREENC: a Wiki-based database of plant lncRNAs. Nucleic Acids Res 44(D1):D1161–D1166
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39(suppl):W155–W159
Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48(3):443–453
Hinton GE, Osindero S, Teh YW (2006) A fast learning algorithm for deep belief nets. Neural Comput 18(7):1527–1554
Douzas G, Bacao F (2019) Geometric SMOTE a geometrically enhanced drop-in replacement for SMOTE. Inf Sci 501:118–135
Li C, Bovik AC, Wu X (2011) Blind image quality assessment using a general regression neural network. IEEE Trans Neural Networks 22(5):793–799
Krizhevsky A, Sutskever I, Hinton GE (2017) Imagenet classification with deep convolutional neural networks. Adv Neural Inf Process Syst 60(6):84–90
Wang L, Yang J, Liu H et al (2016) Research on a self-adaption algorithm of recurrent neural network based chinese language model. Fire Control Command Control 41(5):31–34
Alipanahi B, Delong A, Weirauch MT et al (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33(8):831–838
Jin KH, Mccann MT, Froustey E et al (2017) Deep convolutional neural network for inverse problems in imaging. IEEE Trans Image Process 26(9):4509–4522
Campos Victor, Sastre F, Yagues Maurici et al (2017) Distributed training strategies for a computer vision deep learning algorithm on a distributed GPU cluster. Procedia Comput Sci 108:315–324
Shi H, Xu M, Li R (2018) Deep learning for household load forecasting-a novel pooling deep RNN. IEEE Trans Smart Grid 9(5):5271–5280
Zhou C, You W, Ding X (2010) Genetic algorithm and its implementation of automatic generation of Chinese songci. J Softw 21(3):427–437
Yarotsky D (2017) Error bounds for approximations with deep Relu networks. Neural Netw 94:103–114
An FP (2018) Human action recognition algorithm based on adaptive initialization of deep learning model parameters and support vector machine. IEEE Access 6:59405–59421
Manaswi, Kumar N (2018) Deep learning with applications using Python || understanding and working with Keras. https://springerlink.bibliotecabuap.elogim.com/chapter/10.1007/978-1-4842-3516-4_2
Liu T, Yin S (2017) An improved particle swarm optimization algorithm used for BP neural network and multimedia course-ware evaluation. Multimed Tools Appl 76(9):11961–11974
Cherkassky V, Ma Y (2004) Practical selection of SVM parameters and noise estimation for SVM regression. Neural Netw 17(1):113–126
Hung H, Jou ZY, Huang SY (2017) Robust mislabel logistic regression without modeling mislabel probabilities. Biometrics 74(1):145–154
Lu X, Wang P, Niyato D (2014) Wireless Networks with RF Energy Harvesting: a Contemporary Survey. IEEE Commun Surv Tutor 17(2):757–789
Xu A, Chen J, Peng H et al (2019) Simultaneous interrogation of cancer omics to identify subtypes with significant clinical differences. Front Genet 10:236
Lorenz R, Bernhart SH, Honer Christian, zu Siederdissen CH et al (2011) ViennaRNA package 2.0. Algorithms Mol Biol 6(1):26
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 61872055 and 31872116).This paper was recommended by CBC2019.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, P., Meng, J., Luan, Y. et al. Plant miRNA–lncRNA Interaction Prediction with the Ensemble of CNN and IndRNN. Interdiscip Sci Comput Life Sci 12, 82–89 (2020). https://doi.org/10.1007/s12539-019-00351-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-019-00351-w