
Abstract
Inflammatory bowel disease (IBD) is an idiopathic prolonged ailment accountable for inflammatory conditions of the intestine. Moreover, arthritis is responsible for joints’ stiffness and painful inflammation. IBD shows certain articular extra-intestinal manifestations associating IBD with arthritis. IBD associated arthritis is found to be linked with ankylosing spondylitis (AS). The present study insights for the potential and putative drug targets and biomarkers of IBD associated with arthritis using in silico approaches. Microarray data analysis of datasets involving IBD affected and AS affected vs controls were done to explore the differentially expressed genes (DEGs). In majority of the datasets, the common DEGs found were sterile alpha motif domain containing 9 like (SAMD9L), inhibin beta A subunit (INHBA), transmembrane protein 45A (TMEM45A) and transmembrane and tetratricopeptide repeat containing 1 (TMTC1). The common functions and pathways found between the DEGs were control of macromolecule metabolism process, control of metabolic process, control of primary metabolic process, and control of protein metabolic process, cell differentiation, organ development, single-organism development process, multicellular organism development process, development of system, single–multicellular organism development process, developmental process, development of anatomical structure, multicellular organismal development process, control of biological process, cell proliferation, hematopoietic progenitor cell differentiation and immune system process. TMTC1 and INBHA were found to be more biologically significant genes according to the topological properties of the network. This study also suggests that TMTC1, INBHA, TMEM45A and SAMD9L DEGs and their accompanying pathways might have the potential to be exploited as drug targets and biomarkers in the diagnosis and/or treatment of IBD linked arthritis and warrants for further experimental validation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Inflammatory bowel diseases (IBD) are chronic idiopathic diseases responsible for inflammation of gastrointestinal tract [1]. There are two main classes of IBD, i.e., Crohn’s disease (CD), and ulcerative colitis (UC) [2]. Ulcerative colitis triggers inflammation in the colon that ultimately leads to diarrhoea followed by abdominal pain and tenesmus. However, Crohn’s disease causes inflammation in any part of the gastrointestinal tract and results into abdominal pain, weight loss, diarrhoea, anorexia, malaise, fistulae, strictures, and abscesses [3]. Also, IBD displays certain extra-intestinal manifestations [4] mainly articular associating IBD with arthritis. Arthritis associated with IBD (enteropathic arthritis) belongs to a group of seronegative spondyloarthropathies (SpA) that accompanies axial and peripheral joint disease along with inflammatory characteristics plus a negative rheumatoid factor according to the European Spondyloarthropathy Study Group (ESSG) criteria [5]. Nearly, 17–39% people suffer from IBD associated arthritis [6]. In IBD-associated arthritis, the peripheral arthropathies comprise peripheral arthritis, enthesitis and dactylitis, whereas axial arthropathies involve inflammatory back pain, sacroiliitis, and ankylosing spondylitis [7]. Ankylosing spondylitis (AS) is the most frequent and severe subtype of spondyloarthropathies that primarily affects the sacroiliac joints [8]. It is reported that IBD is associated with AS [9, 10]. However, no specific test is available that can diagnose arthritis linked with IBD, as it is based upon differential diagnosis patterns [11]. Hence, there is an urgent need to identify and evaluate the putative drug targets and biomarkers for refining the currently used diagnostic methods and therapies of IBD associated with arthritis.
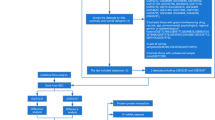
Computational analysis is a high-throughput procedure that assigns putative targets for the association and defines a directed pathway for identifying the experimental parameters of the study. For this type of analysis, high-throughput data of patient samples containing extensive quantitative evaluation of gene expression and/or transcriptomic information can be utilized. Databases such as ArrayExpress of European Bioinformatics Institute (EBI) [12] and Gene Expression Omnibus (GEO) database of National Centre for Biotechnology Information (NCBI) [13] maintains publically contributed gene expression data collected by high-throughput methodologies. Earlier studies reported inclusion of various computational tools for transcriptomic data analysis for correlating gene expression data with other or additional biological information [14, 15]. The major objective of the present study was to decipher important genes, functions and pathways between IBD and AS that might serve as potential triggers in IBD associated arthritis. Therefore, high-throughput transcriptomic datasets of IBD and AS were exploited to carry out a comparative transcriptomic analysis of the diseased and healthy subjects. Functional annotation pathway and network analysis of the differentially expressed genes (DEGs) have been performed providing a deep insight for putative drug targets and biomarkers of IBD associated with arthritis. To the best of our information this is the very first study suggesting potential association of IBD with arthritis providing evidence from in silico gene expression patterns and network topological analysis. A pictorial representation of the workflow of in silico transcriptional pathway and network analysis of the datasets applied in this study is shown in Fig. 1.

In-silico transcriptional, pathway and network analysis of IBD and AS datasets
2 Results
2.1 Gene Expression Dataset Selection
One AS dataset and ten IBD datasets were obtained by following the given selection criteria (Table 1). To avoid inconsistencies for more robust comparative data analysis, all the datasets consisted of cases vs control, not undergoing any treatment, and utilized the same microarray platform.
2.2 IBD and AS Dataset Pre-Processing
Robust multi-array average (RMA) employed the RAW Affymetrix intensity values and achieved background correction and resulting the removal of local artefacts and noises. The mathematical values were log2 transformed (quantile normalized) for the elimination of the array effects and equalize the partition of probe intensities for each array. Further, a linear mathematical model was fit to the normalized values by integrating the probe intensities across the arrays for achieving an expression measure for the respective probes on each array, which is represented as
where, Yij signifies the normalized probe value corresponding to the ith GeneChip and the jth probe within the probe set, mi designates the log-scale expression for the probe set in the sample hybridized to the ith GeneChip, aj expresses the probe affinity effect for the jth probe within the probe set, and eij represents a random error term [16].
2.3 Statistical Analysis
Moderated t-statistics and log-odds of the differential expression by the empirical Bayes shrinkage categorized (ranked) the genes as per the evidences of the differential expression. The empirical Bayes shrinkage method reduced the sample variance for all the probes and resulting in a common value. This reduction in the sample variance resulted in the generation of statistical values for individual gene of all the datasets [17]. logFC is the log2 folds change estimated between the healthy subjects and diseased samples that distinguishes the up- and down-regulated values. Moreover, AveExpr is the average expression value given by all the arrays and channels. To curb the false discovery rate the p value was computed using Benjamini and Hochberg’s method. Adj.p. value is the adjusted p value for multiple testing and t is a moderated t-statistics showing the ratio of log2 expression level to its standard error. The B-statistics was employed to rank the genes to evaluate their differential expression. As an example, for a given gene g, limma fits a linear model and tests the null hypothesis H0:βg = 0,
where βg is the contrast of interest. To test this hypothesis, a moderated t-statistics \(\tilde{t}_{g}\) is constructed, as
where
and vg is the scaling factor of the variance estimates of \(\hat{\beta }_{g}\). This statistic is based on a hierarchical model, where the anonymous gene-level variances \(\sigma_{\text{g}}^{2}\) are modelled by a scaled inverse Chi-square distribution with d0 and \(s_{0}^{2}\) hyperparameters [17].
2.4 Identification of Differentially Expressed Genes (DEGs)
The statistical evaluation of the datasets resulted in the generation of differentially expressed genes, which respond to a signal and play an important role in the gene regulation [18]. To attain highly robust DEGs, log2 fold change and p values were taken into account. 1.5-fold variation and p value less than 0.05 were maintained as the cut-off criteria for identifying the up- and down-regulated genes [19] (Table 1) (Supplementary Information 1). 1.5-fold change was described by a log2 ratio of 0.58 (for up-regulation) or − 0.58 (for down-regulation). The p values below 0.05 were considered significant, which provide substantial potency related to the evidence. Hence, lesser p value represents stronger evidence. The annotation of the identified probes was supported by GeneAnnot database. The common DEGs were found in the maximum number of datasets of IBD and AS, and were present in sterile alpha motif domain containing 9 like (SAMD9L) inhibin beta A subunit (INHBA), transmembrane protein 45A (TMEM45A), and transmembrane and tetratricopeptide repeat containing 1 (TMTC1) (Table 2). These were utilized for further analysis as they might serve as robust targets for IBD related arthritis. SAMD9L was found in three datasets of UC, one dataset of CD, one dataset consisting of both UC and CD cohorts and one dataset of AS. INHBA, TMEM45A and TMTC1 were found in three datasets of UC, but not in the datasets of CD, whereas they were present in two datasets of both UC and CD cohorts and one dataset of AS. This shows that the above DEGs might be uniformly present in both CD and UC patient cohorts.
2.5 Functional Annotation and Pathway Analysis
DAVID tool and KEGG pathway were utilized for the functional annotation and pathway analysis of SAMD9L, INHBA, TMEM45A and TMTC1 (Table 3). Significant functions and pathways were obtained only in case of SAMD9L and INHBA. The common functions and pathways between SAMD9L and INHBA were control of macromolecule metabolic process, regulation of metabolic process, regulation of primary metabolic process, regulation of protein metabolic process, cell differentiation, organ development, single-organism developmental process, multicellular organismal development, system development, single–multicellular organism process, developmental process, anatomical structure development, multicellular organismal process, control of biological process, cell proliferation, hematopoietic progenitor cell differentiation and immune system process. These functions and pathways might serve as a trigger for pathophysiological analysis of IBD-related arthritis.
2.6 Network Analysis
The protein interactors among sterile alpha motif domain containing 9 like (SAMD9L), inhibin beta A subunit (INHBA), transmembrane protein 45A (TMEM45A) and transmembrane and tetratricopeptide repeat containing 1 (TMTC1) were used for STRING database that contains the protein interactions authenticated by numerous evidences from the experimental studies providing robustness to the retrieved interactions [20] (Fig. 2). These targets might serve as potential triggers for IBD-related arthritis (Supplementary Information 2). Below mentioned topological characteristics were found using Cytoscape tool [21].

Protein interaction network between SAMD9L, INBHA, TMTC1 and TMEM45A
-
1.
Clustering coefficient is described as the ratio of the number of the edges between the neighbours of a node and the maximum number of the edges that could perhaps occur between the neighbours of a node. Hence, clustering coefficient of a node is
$$n = N/M,$$where N is the number of edges between the neighbours of n, and M is the maximum number of the edges, which could probably exist between the neighbours of n.
-
2.
Topological coefficient is a relative estimate of the extent that a node shares the neighbours with other nodes.
$$Tn \, = {\text{ avg }}\left( {J\left( {n,m} \right)} \right)/kn,$$where Tn signifies the topological coefficient, and the value J(n, m) is the number of neighbours shared between the nodes n and m (plus one if n and m has a direct association).
-
3.
Stress represents the number of shortest paths passing through a node.
-
4.
Betweenness centrality is the extent of control that a node has over the interactions of other nodes.
$${\text{Cb}}\left( n \right) \, = \, \sum s \ne n \ne t \, \left( {\sigma st \, \left( n \right) \, / \, \sigma st} \right),$$
where Cb denotes the betweenness centrality; s and t are the nodes present in the network different from n node, σst signifies the number of shortest paths from s to t, and σst (n) is the number of shortest paths from s to t that n lies on.
-
5.
Closeness centrality is an estimate of the speedy flow of the information from a given node to other reachable nodes.
$$C_{c} \left( n \right) \, = 1/{\text{avg}}\left( {L\left( {n,m} \right)} \right),$$
where Cc(n) denotes the closeness centrality and L(n, m) signifies the length of the shortest path between the two nodes n and m.
Topological coefficient and clustering coefficient of TMTC1, INBHA, TMEM45A and SAMD9L were 0.14, 0.35, 0, 0 and 0.06, 0.35, 0 and 0, respectively. Greater the topological and clustering coefficients, more the neighbours of the node. The stress of TMTC1, INBHA, TMEM45A and SAMD9L was 6595, 3640, 0 and 0, respectively. Higher is the stress, greater is the number of the shortest paths passing via the node. The closeness centrality of TMTC1, INBHA, TMEM45A and SAMD9L was 0.48, 0.50, 0.34 and 0.23, respectively. More the closeness centrality, much faster the information spreads to other reachable nodes. The betweenness centrality of TMTC1, INBHA, TMEM45A and SAMD9L was 0.29, 0.15, 0 and 0, respectively. Greater is the betweenness centrality, higher is the strength of the control; the node exerts over the interactions of other nodes. The results indicate TMTC1 and INBHA to be more important interactors in the network (Table 4). The above network might be capable of providing deep understanding of the disease related biological processes, disease linked candidate genes, disease mechanistic processes and assists in the discovery of potential biomarkers [22] of IBD-associated arthritis.
3 Discussion
Chromosomal regions discovered by the linkage and association studies frequently contain candidate genes associated with a specific disease or a phenotype. Generally, the pathogenesis of a complex disease can be deciphered by the identification and characterization of the candidate gene(s) accountable for the complex interactions producing disease. Dearth of precise diagnostic technology demands the identification of putative drug targets and various biomarkers for improving the diagnostic process and therapeutic strategy of IBD-linked arthritis. Numerous reports have defined the application of computational pipelines for prioritizing the most potent and promising candidates [23,24,25,26,27,28,29]. Owing to the dearth of freely available public databases and citations of experimentally validated literature, the analyses of differential gene based on arthritic datasets were done earlier to check the disease-linked differentially expressed genes (DEGs) [30, 31] using statistical platforms. DEGs are the genes, which are expressed amidst varying physiological disease conditions. The transcriptomic analysis helps in the examination of the expression of DEGs leading to their deposition in the public databases. In general, the detection of DEGs can be performed by setting a threshold, and genes expressed above or below the set threshold limit are considered as potential biomarkers for the diagnosis and prognosis of the disease. Computational pipelines were initiated to narrow down the search space generated because of the accessibility of superfluous ‘omics’ data produced by the potential genes pre-selection employing automated recognition methods. These methods were mostly based on the measurement of the relatedness present among the genes and failed to recognize the phenotypic linkage as well as association of the genes with multiple diseases. To overcome this constraint, a combined strategy of transcriptomic profiling of differentially expressed genes accompanying network topology analysis was employed in the present study to investigate the underlying association of IBD with arthritis.
Database mining of the genome datasets in relation with IBD and AS was carried out for the generation of the primary data. Certain systemic variations of microarray are accountable for uncertainty in the analysis of the primary data. Thus, statistical algorithms implementing diverse approaches for the normalization and differential expression (DE) detection were tested. Normalization leads to noise reduction, artefacts removal, and the elimination of related technical errors [32]. Additionally, statistical evaluation reduced the sensitivity that was consistent with the expression levels. LogFC computation and B-statistics assisted in classifying the up- and down-regulated genes and calculated p value for the subset of genes expression reflecting the confidence in gene expression by monotonically correlating with the signal-to-noise ratios [33].
In majority of the datasets, the common DEGs found were sterile alpha motif domain containing 9 like (SAMD9L), inhibin beta A subunit (INHBA), transmembrane protein 45A (TMEM45A) and transmembrane and tetratricopeptide repeat containing 1 (TMTC1). Previously, published inflammatory bowel disease microarray data have showed the presence of SAMD9L [34,35,36,37,38], INHBA, TMEM45A and TMTC1 [34, 36, 38,39,40] in IBD patient cohorts. Similarly, the above genes have also been found in ankylosing spondylitis patients as reported by Smith et al. [41]. These studies reported the identification of SAMD9L, INHBA, TMEM45A and TMTC1, and suggested their role in IBD and AS disease conditions. These include metabolism, host defence, molecular transport, response to biotic and inflammatory stimulus, innate immune responses, chemotaxis, cell proliferation and tumour growth in pediatric UC; and altered gene expression profile of lamina propria cells in quiescent UC and IFN-γ gene regulation in AS. One of the identified DEGs according to our study, TMTC1, has been predicted to show favourable response to anti-TNF antibody therapy in IBD patients [42].
In our study, the common functions and pathways found between the DEGs were regulation of macromolecule metabolic process, control of metabolic process, regulation of primary metabolic process, control of protein metabolic process, cell differentiation, organ development, single-organism developmental process, multicellular organismal development, system development, single–multicellular organism process, developmental process, anatomical structure development, multicellular organismal process, regulation of biological process, cell proliferation, hematopoietic progenitor cell differentiation and immune system process. From the viewpoint of a gene/protein network, a candidate gene inclines to be surrounded by powerful differentially expressed neighbours contributing similar functions and phenotypic characteristics. Thus, studies of molecular interactions were carried out to detect and investigate the highly expressed neighbours between the TMTC1, INBHA, TMEM45A and SAMD9L. Exploration of betweenness and closeness centrality distinguishes the direct interactors and the shortest path between the nodes in the network to discover substantial differences between the etiological and palliative drugs [33]. TMTC1 and INBHA were found to be more biologically significant genes as compared to TMEM45A and SAMD9L according to the topological properties of the network. According to the published reports, the role of DEGs have been explored in molecular pathogenesis of IBD that makes it possible to discern UC and CD patient cohorts and portray the role of genetics in the diagnosis of IBD using the clinical diagnostic test as gold standard. The utility of DEGs as a diagnostic test was uncovered from pilot studies to precisely categorize UC or CD patients showing results akin to the capabilities of prevailing clinical diagnosis [43]. The DEGs identified from the present study are indicative of their potential role in molecular pathogenesis, aligning them as putative biomarkers for the diagnosis and prognosis of IBD-associated arthritis. The outcome of such studies paves an important workflow to conduct pilot scale investigations for future experimental validation.
The above mentioned networks can provide advanced understanding of the disease-associated biological processes, related candidate genes, and mechanism of IBD-associated arthritis. The results of this study suggest novel drug targets and biomarkers of IBD-linked arthritis that can help in the improvement of the diagnostic pattern and/or the treatment regime in the near future [44,45,46].
In conclusion, this study depicts an effective computational approach for the identification of DEGs that might involve in IBD leading to arthritis. Overall, the current findings speculate a potential association of IBD with arthritis using in silico gene expression patterns and network topological analysis. DEGs such as TMTC1, INBHA, TMEM45A and SAMD9L along with their associated functions and pathways involving regulation of macromolecule metabolic process, regulation of metabolic process, regulation of primary metabolic process, regulation of protein metabolic process, cell differentiation, organ development, single-organism developmental process, multicellular organismal development, system development, single–multicellular organism process, developmental process, anatomical structure development, multicellular organismal process, regulation of biological process, cell proliferation, hematopoietic progenitor cell differentiation and immune system process may have the potential to be exploited as probable targets and biomarkers for the diagnosis and treatment of IBD linked arthritis in the near future. Also, this preliminary in silico study warrants for future larger experimental studies to validate our current findings.
4 Materials and Methods
4.1 Gene Expression Dataset Selection
European Bioinformatics Institute (EBI) ArrayExpress [12] source of high-throughput gene expression data that is also linked with National Centre for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database [13], was iterated to retrieve the gene expression profiles of IBD and AS. The inclusion and exclusion criteria for selecting the datasets were (1) datasets with IBD vs control and AS vs control, (2) patient cohorts not undergoing any treatment, (3) datasets published in journals (4) datasets whose transcriptional analysis has been performed using Affymetrix microarray (5) datasets having RAW files and (6) datasets containing replicates.
4.2 Pre-Processing of the Data
During the pre-processing step, screening of the differentially expressed genes (DEGs) within every dataset was done by accomplishing differential gene expression to reduce the complexity underlying because of the data redundancy. Hence, a robust multi-array average (RMA) data normalization of the expression data was performed to control the discrete hybridization intensities and to infer an accurate, significant and meaningful biological comparisons. It is a command-based Bioconductor R suite affy software package, generally needed for Affymetrix array analysis [47].
4.3 Statistical Analysis
For the statistical analysis, Limma, a package of Bioconductor R suite for the analysis of gene expression data arising from microarray employing statistical platform was utilized. Employment of linear models for evaluating the gene expression functions of the datasets from any quantitative gene expression technology (for, e.g., microarrays, etc.) is the distinctive characteristic of this package. The logFC, p.Value, adj.p.Val, Ave Expr, t- and B-value of every individual gene for all the three datasets were measured by employing moderated t-statistics along with log-odds of the differential expression by empirical Bayes shrinkage [48].
4.4 Identification of the Differentially Expressed Genes
The genes having 1.5-fold change and a p value less than 0.05 [p < 0.05 and FC ≥ 1.5 (|log2 FC| 0.58)] were chosen as top up- and down-regulated differentially expressed genes between the healthy and the affected (diseased) samples for all the datasets. The annotation of DEGs was done by the use of GeneAnnot database http://genecards.weizmann.ac.il/geneannot/index.shtml) that predicts the genes from GenBank (www.ncbi.nlm.nih.gov/genbank/), RefSeq (www.ncbi.nlm.nih.gov/refseq/) and Ensembl (http://www.ensembl.org/index.html) [49]. The common differentially expressed genes were found between the datasets of IBD and AS.
4.5 Functional Annotation and Pathway Analysis
Functional annotation of the differentially expressed genes was done using DAVID (https://david.ncifcrf.gov/) tool, which is a visualization and integrated discovery tool links the gene probe ID’s with the functional terms by employing a clustering algorithm to evaluate the genes at their functional level and helps in elucidating their pertinent biological significance [50, 51]. Further, Kyoto Encyclopaedia of the Genes and Genomes (KEGG), repertoire of the information related to the genomic, enzymatic and biological pathway related data was mined for advanced level characterization of the DEGs [52,53,54].
4.6 Network Analysis
Search tool for the retrieval of interacting genes (STRING) (http://string-db.org/), a curated universal database comprising the interaction data, was used to evaluate the protein–protein interactions (PPIs) [20] of the target DEGs in Homo sapiens. STRING database contains around 9.6 million proteins from more than 2000 organisms. STRING provides the evidence of protein–protein interactions from previously established known experimental, pathway, genomic, orthology and co-expression data, which are available from a number of online databases such as HPRD, MINT, DIP, BIND, KEGG, BioGRID, Reactome, IntAct, GO, SGD, EcoCyc, FlyBase, OMIM, and PubMed. To evaluate the topological properties such as nodes, edges, closeness centrality, clustering coefficient, eccentricity, betweenness centrality, stress, degree, neighbourhood connectivity, number of directed edges, radiality and topological coefficient was determined for the visualization and analysis of the network using Cytoscape that aids in understanding the structure and underlying mechanisms of the network [21]. Figure 1 shows the pictorial representation of the workflow of the in silico transcriptional, pathway, and network analysis of IBD and AS datasets used in the present study.
References
Ruemmele FM (2016) Role of diet in inflammatory bowel disease. Ann Nutr Metab 68(1):33–41
Zhou N, Chen W, Chen S, Xu C, Li Y (2011) Inflammatory bowel disease unclassified. J Zhejiang Univ Sci B 12(4):280–286
Carter MJ, Lobo AJ, Travis SPL (2004) Guidelines for the management of inflammatory bowel disease in adults. Gut 53(5):V1–V16
Vavricka SR, Schoepfer A, Scharl M, Lakatos P, Navarini A, Rogler G (2015) Extraintestinal manifestations of inflammatory bowel disease. Inflamm Bowel Dis 21(8):1982–1992
Dougados M, Vanderlinden S, Juhlin R, Huitfeldt B, Amor B, Calin A, Cats A, Dijkmans B, Olivieri I, Pasero G, Veys E, Zeidler H (1991) The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum 34(10):1218–1227
Arvikar SL, Fisher MC (2011) Inflammatory bowel disease associated arthropathy. Curr Rev Musculoskelet Med 4(3):123–131
Brakenhoff LKPM, van der Heijde DM, Hommes DW, Huizinga TWJ, Fidder HH (2010) The joint-gut axis in inflammatory bowel diseases. J Crohn’s Colitis 4(3):257–268
El Maghraoui A (2011) Extra-articular manifestations of ankylosing spondylitis: prevalence, characteristics and therapeutic implications. Eur J Intern Med 22(6):554–560
Meuwissen SG, Dekker-Saeys BJ, Agenant D, Tytgat GN (1978) Ankylosing spondylitis and inflammatory bowel disease. I. Prevalence of inflammatory bowel disease in patients suffering from ankylosing spondylitis. Ann Rheum Dis 37(1):30–32
Jethwa H, Mann S (2013) Crohn’s disease unmasked following etanercept treatment for ankylosing spondylitis. BMJ Case Rep. https://doi.org/10.1136/bcr-2013-009166
Nagabhushanarao BV, Raman BS (2015) Arthritis predating inflammatory bowel disease. Am J Med Case Rep 3(1):16–20
Parkinson H, Sarkans U, Kolesnikov N, Abeygunawardena N, Burdett T, Dylag M, Emam I, Farne A, Hastings E, Holloway E, Kurbatova N, Lukk M, Malone J, Mani R, Pilicheva E, Rustici G, Sharma A, Williams E, Adamusiak T, Brandizi M, Sklyar N, Brazma A (2011) Arrayexpress update-An archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res 39(1):D1002–D1004
Barrett T, Edgar R (2006) Mining microarray data at NCBI’s Gene Expression Omnibus (GEO). Methods Mol Biol 338:175–190
Satpathy R, Konkimalla VB, Ratha J (2015) In-silico gene co-expression network analysis in Paracoccidioides brasiliensis with reference to haloacid dehalogenase superfamily hydrolase gene. J Pharm Bioallied Sci 7(3):212–217
Liu Y, Wang L, Wang B, Yue M, Cheng Y (2016) Pathway analysis based on attractor and cross talk in colon cancer. Dis Markers 2016(2016): Article ID 2619828. http://dx.doi.org/10.1155/2016/2619828
Bolstad BM, Irizarry R, Astrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19(2):185–193
Zhong D (2014) Identification of differentially expressed gene after femoral fracture via microarray profiling. Int J Genom 2014(2014): Article ID 208751. http://dx.doi.org/10.1155/2014/208751
Porkodi R, Savitha R (2015) Function and pathway analysis of differentially expressed genes in Alzheimer’s disease dataset using linear regression model. IJCST 6(4):226–230
Ning LF, Yu YQ, Guoji ET, Kou CG, Wu YH, Shi JP, Ai LZ, Yu Q (2015) Meta-analysis of differentially expressed genes in autism based on gene expression data. Genet Mol Res 14(1):2146–2155
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, Von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(D1):D447–D452
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504
Sevimoglu T, Arga KY (2014) The role of protein interaction networks in systems biomedicine. Comput Struct Biotechnol J 11(18):22–27
Bäuerl C, Llopis M, Antolín M, Monedero V, Mata M, Zúñiga M, Guarner F, Martínez GP (2013) Lactobacillus paracasei and Lactobacillus plantarum strains downregulate proinflammatory genes in an ex vivo system of cultured human colonic mucosa. Genes Nutr. 8(2):165–180
Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordás I, Esteller M, Ricart E, Piqué JM, Panés J, Salas A (2013) Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut 62(7):967–976
Hebels DGAJ, Sveje KM, de Kok MC, van Herwijnen MHM, Kuhnle GGC, Engels LGJB, Vleugels-Simon CBEM, Mares WGN, Pierik M, Masclee AAM, Kleinjans JCS, de Kok TMCM (2011) N-nitroso compound exposure-associated transcriptomic profiles are indicative of an increased risk for colorectal cancer. Cancer Lett 309(1):1–10
Xiong Q, Ancona N, Hauser ER, Mukherjee S, Furey TS (2012) Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res 22(2):386–397
Rybaczyk L, Rozmiarek A, Circle K, Grants I, Needleman B, Wunderlich JE, Huang K, Christofi FL (2009) New bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm Bowel Dis 15(7):971–984
Israeli E (2013) Common and specific signatures of gene expression and protein-protein interactions in autoimmune diseases. Isr Med Assoc J 15(5):261
Hughes AL (2005) Consistent across-tissue signatures of differential gene expression in Crohn’s disease. Immunogenetics 57(10):709–716
Xu Y, Huang Y, Cai D, Liu J, Cao X (2015) Analysis of differences in the molecular mechanism of rheumatoid arthritis and osteoarthritis based on integration of gene expression profiles. Immunol Lett 168(2):246–253
Frank M, Wang S, Aggarwal A, Knowlton N, Jiang K, Chen Y, McKee R, Chaser B, McGhee T, Osban J, Jarvis JN (2014) Disease-associated pathophysiologic structures in pediatric rheumatic diseases show characteristics of scale-free networks seen in physiologic systems: implications for pathogenesis and treatment. BMC Med Genom 2(1):9
Park T, Yi S-G, Kang S-H, Lee S, Lee Y-S, Simon R (2003) Evaluation of normalization methods for microarray data. BMC Bioinform 4(33):33
Sarajlić A, Janjić V, Stojković N, Radak D, Pržulj N (2013) Network topology reveals key cardiovascular disease genes. PLoS One 8(8):e71537
Gyorffy B, Molnar B, Lage H, Szallasi Z, Eklund AC (2009) Evaluation of microarray preprocessing algorithms based on concordance with RT-PCR in clinical samples. PLoS One 4(5):e5645
Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordás I, Esteller M, Ricart E, Piqué JM, Panés J, Salas A (2013) Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut 62(7):967–976
Ahrens R, Waddell A, Seidu L, Blanchard C, Carey R, Forbes E, Lampinen M, Wilson T, Cohen E, Stringer K, Ballard E, Munitz A, Xu H, Lee N, Lee JJ, Rothenberg ME, Denson L, Hogan SP (2008) Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol 181(10):7390–7399
Bjerrum JT, Hansen M, Olsen J, Nielsen OH (2010) Genome-wide gene expression analysis of mucosal colonic biopsies and isolated colonocytes suggests a continuous inflammatory state in the lamina propria of patients with quiescent ulcerative colitis. Inflamm Bowel Dis 16(6):999–1007
Funke B, Autschbach F, Kim S, Lasitschka F, Strauch U, Rogler G, Gdynia G, Li L, Gretz N, Macher-Goeppinger S, Sido B, Schirmacher P, Meuer SC, Roth W (2009) Functional characterisation of decoy receptor 3 in Crohn’s disease. Gut 58(4):483–491
Olsen J, Gerds TA, Seidelin JB, Csillag C, Bjerrum JT, Troelsen JT, Nielsen OH (2009) Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm Bowel Dis 15(7):1032–1038
Carey R, Jurickova I, Ballard E, Bonkowski E, Han X, Xu H, Denson LA (2008) Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis 14(4):446–457
Smith JA, Barnes MD, Hong D, DeLay ML, Inman RD, Colbert RA (2008) Gene expression analysis of macrophages derived from ankylosing spondylitis patients reveals interferon-gamma dysregulation. Arthritis Rheum 58(6):1640–1649
Wang Y, Zhu X, Zhen N, Pan Q, Li Y (2016) Gene expression profile predicting the response to anti-TNF antibodies therapy in patients with inflammatory bowel disease: analyses of GEO datasets. Int J Clin Exp Med 9(12):23397–23406
Huett A, Xavier RJ (2008) Neither hide nor hair: the difficulty of identifying useful disease biomarkers. Gastroenterology 134(7):2164–2168
Verma A, Rathi B, Sharda S (2016) Identification of novel inhibitors against potential targets of Campylobacter jejuni. Int J Pharm Pharm Sci 8(2):312–316
Verma A, Goel N, Laxmi V, Rathi B, Sharda S (2016) Elucidating vital drug targets of Salmonella enterica utilizing the bioinformatic approach. JPSR. 8:71–78
Verma A, Rathi AB, Shrada S (2016) Identification of novel drug targets against Campylobacter jejuni using metabolic network analysis. Int J Pharm Biol Sci 7(7):56–62
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10):R80
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47
Chalifa-Caspi V, Yanai I, Ophir R, Rosen N, Shmoish M, Benjamin-Rodrig H, Shklar M, Stein T, Shmueli O, Safran M, Lancet D (2004) GeneAnnot: comprehensive two-way linking between oligonucleotide array probesets and GeneCards genes. Bioinformatics 20(9):1457–1458
Huang DW, Lempicki RA, Sherman BT (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57
Bhardwaj T, Somvanshi P (2015) Plant Systems Biology: Insights and Advancements. In: Barh D, Khan M, Davies E (eds) PlantOmics: the omics of plant science. Springer, New Delhi
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44(D1):D457–D462
Srivastava A, Somvanshi P, Mishra BN (2013) Reconstruction and visualization of carbohydrate, N-glycosylation pathways in Pichia pastoris CBS7435 using computational and system biology approaches. Syst Synth Biol 7(1–2):7–22
Bhardwaj T, Somvanshi P (2017) Pan-genome analysis of Clostridium botulinum reveals unique targets for drug development. Gene 623:48–62
Acknowledgements
The authors are grateful to Amity Institute of Biotechnology, Amity University Noida Campus, UP and Department of Biotechnology, TERI University, New Delhi India for providing the facility and technical support during the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the study and experiments: AV, PS, SH, BR, SS. Performed the experiments: AV, PS, SS. Analysed the data: PS, SH, BR. Contributed reagents/materials/analysis tools: PS, SH, SS, BR. Wrote the paper: AV, PS, SH, BR, SS. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Funding
No direct or indirect financial support was available for this research work.
Conflict of interest
The authors declare no conflicts of interest exist.
Rights and permissions
About this article

Cite this article
Verma, A., Somvanshi, P., Haque, S. et al. Association of Inflammatory Bowel Disease with Arthritis: Evidence from In Silico Gene Expression Patterns and Network Topological Analysis. Interdiscip Sci Comput Life Sci 11, 387–396 (2019). https://doi.org/10.1007/s12539-017-0272-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12539-017-0272-1