
Abstract
Marine environments abound with opportunities to discover new species of fungi even in relatively well-studied ecosystems such as coral reefs. Here, we investigated the fungal communities associated with the canopy forming macroalga Sargassum ilicifolium(Turner) C. Argardh (1820) in Singapore. We collected eight S. ilicifolium thalli from each of eight island locations and separated them into three structures—leaves, holdfast and vesicles. Amplicon sequencing of the fungal internal transcribed spacer 1 (ITS1) and subsequent analyses revealed weak but significant differences in fungal community composition from different structures. Fungal communities were also significantly different among sampling localities, even over relatively small spatial scales (≤ 12 km). Unsurprisingly, all structures from all localities were dominated by unclassified fungi. Our findings demonstrate the potential of marine environments to act as reservoirs of undocumented biodiversity that harbour many novel fungal taxa. These unclassified fungi highlight the need to look beyond terrestrial ecosystems in well-studied regions of the world, and to fully characterize fungal biodiversity in hotspots such as Southeast Asia for better understanding the roles they play in promoting and maintaining life on our planet.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fungi are ubiquitous in almost all habitats on Earth. They have been documented in mesophotic coral ecosystems, deep-sea hydrothermal vents, arid, high altitude deserts and the cold Arctic and Antarctic (Robinson 2001; Burgaud et al. 2014; Gonçalves et al. 2016; Wainwright et al. 2017). Despite their ability to persist in many distinct habitats, our knowledge of their diversity is limited (Blackwell 2011; Richards et al. 2012; Peay 2016), especially in marine environments (Wainwright et al. 2017; Deshmukh et al. 2018). It is estimated that 93% of all fungal species remain unnamed and are waiting to be discovered (Nature Ecology and Evolution 2018), with conservative estimates of up to 10,000 unknown marine taxa (Amend et al. 2019a).
Fungi afford humans many benefits, from yeasts used in bread production, drugs that have medical applications (i.e. penicillin and lovastatin used to reduce cholesterol levels), to endophytic fungi that can break down polyurethane which is common in household wastes (Russell et al. 2011). Most of these discoveries have been made in terrestrial environments, and the unexplored marine environment represents a potential treasure trove of beneficial fungi. Research is showing that marine environments likely contain a high diversity of putatively novel taxa waiting to be explored (Ishino et al. 2016; Comeau et al. 2016; Picard 2017). Despite their oft-beneficial roles, fungi can also be harmful and have been implicated in the recent extinction of many species (Moree et al. 2014). Numerous fungal diseases have been identified which are detrimental to humans (e.g. fungal pneumonia), bats (e.g. white-nose syndrome) and amphibians (chytridiomycosis). Notably, pathogens are becoming more prevalent in marine environments (Weldon et al. 2004; Kim 2016). In particular, the fungus Aspergillus sydowii (Bainier & Sartory, 1926), black band disease and white plague type II have been implicated in coral mortality throughout the Caribbean and Atlantic (Rützler and Santavy 1983; Nugues et al. 2004; Moree et al. 2014).
Sargassum (Phaeophyceae), a large canopy-forming brown macroalga, is found throughout temperate and tropical seas. In the Caribbean, Sargassum beaching is becoming a frequent and increasingly problematic issue; several massive Sargassum beaching events since 2011 have negatively impacted local environments, fisheries and tourism (Wang and Hu 2016a, 2016b). Similar events have taken place in West Africa and northern Brazil (Oyesiku and Egunyomi 2015; Széchy et al. 2012). While no mass Sargassum beaching events have been observed in Singapore, Sargassum is abundant (Low 2015, Low et al. 2019; Yip et al. 2018) and is considered an important competitor for space in shallow habitats. Singapore’s high marine biodiversity (Low and Chou 1994; Huang et al. 2009) and location at the periphery of the hyperdiverse Coral Triangle (Allen and Werner 2000; Hughes et al. 2002) mean that fungal diversity could also be correspondingly high (Wainwright et al. 2018).
Sargassum shows distinct annual growth patterns in Singapore and dominates many reef flats where it comes into frequent contact with corals (Leong et al. 2018; Low et al. 2019). Macroalgae can damage corals through a variety of direct and indirect mechanisms, including abrasion, reduction of light, smothering or allelopathy (McCook 2001; Jompa and McCook 2003; Morrow et al. 2011; Rasher et al. 2011; Haas et al. 2011). Pratte et al. (2017) showed that coral-macroalgae interactions can alter the coral microbiome, which plays an important role in the health of corals and is sensitive to various other stresses (Ainsworth et al. 2010; Hernandez-Agreda et al. 2018). Any changes in the coral microbiome induced by contact with Sargassum are unlikely to be beneficial, in part because algae are reservoirs of pathogens harmful to corals (Nugues et al. 2004; Egan et al. 2013).
On account of the various roles different algal structures (e.g. holdfast, vesicles and leaf-like laminae, which we refer to as leaves here) play in maintaining and promoting growth, we hypothesise that each structure will contain a different fungal community. For example, the grazing-resistant holdfast (Loffler et al. 2018) is not involved in nutrient uptake but functions to anchor and maintain contact with the substrate. The leaves, which are frequently observed with bite marks (Low 2015), perform photosynthesis, while gas-filled vesicles provide buoyancy. Considering the many vital roles fungi play within their hosts, differences in function of each structure may select for different fungal communities. In this study, as part of efforts to characterize and compare microbial communities on coral reefs, we investigated fungal diversity associated with Sargassum ilicifolium (Turner) C. Argardh (1820), the most abundant and widespread Sargassum species found in the waters of Singapore (Low 2015; Yip et al. 2018).
Materials and methods
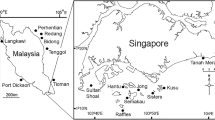
Eight entire Sargassum ilicifolium thalli were collected from each of eight sampling locations over 2 days in January 2018 (Fig. 1). All sampling sites are essentially homogeneous in terms of physical characteristics with comparable water quality throughout the year (Gin et al. 2000; Tun 2012; Chou et al. 2019; Tanzil et al. 2019). Individual thalli were separated into leaves, holdfast and vesicles. All tissues were surface sterilized by immersion in 1% NaClO for 2 min, 70% EtOH for 2 min and rinsed twice in sterile, DNA-free autoclaved water for 5 min. Tissues were disrupted in an Omni Bead Ruptor 24 (Omni International, Kennesaw, GA, USA) at 8 m/s for 2 min.

Map showing sampling locations in Singapore
DNA was extracted with a Qiagen DNeasy Powersoil kit following the manufacturer’s instructions. Because the mass of host DNA will be several orders of magnitude greater than that of fungal template, DNA concentration was not quantified. Fungal DNA amplification of the internal transcribed spacer 1 (ITS1) region was performed using the ITS1F (CTT GGT CAT TTA GAG GAA GTA A; Gardes and Bruns 1993) and ITS2 (GCT GCG TTC TTC ATC GAT GC; White et al. 1990) primers, which were modified to include Illumina adaptors, a linker and a unique barcode (see Smith and Peay 2014 for details of custom sequencing primers). Each reaction was performed in a total volume of 25 μl, containing 9 μl of template, with final concentrations of 0.25 U of KAPA 3G Enzyme (Kapa Biosystems, Inc., Wilmington, MA, USA), 0.3 μM of each primer, 1.5 mg/mL of BSA and KAPA Plant PCR Buffer. PCR cycling protocol was 95 °C for 3 min, followed by 35 cycles of 95 °C for 20 s, 53 °C for 15 s, 72 °C for 20 s with a final extension at 72 °C for 60 s. Negative PCR controls and DNA extraction blanks were included to detect potential contamination. PCR products were visualized on a 1% TBE buffer agarose gel, then normalized and cleaned using SequalPrep™ normalization plates (Invitrogen, Frederick, MD, USA). Purified PCR products were submitted for sequencing on the Illumina MiSeq platform (600 cycles, V3 chemistry, 300 bp paired-end reads) with a 15% PhiX spike at the Genome Institute of Singapore (GIS).
Raw reads were processed with ITSx (Bengtsson-Palme et al. 2013) (version 1.1b1) to remove adaptors and extract the ITS1 region. Quality scores were reassigned using the original fastq files and a custom R script. Due to the lower quality of reverse reads, only forward reads were used for all subsequent steps and analyses; discarding low-quality reverse reads is a common strategy that frequently gives better results than assembled reads (Pauvert et al. 2019). Forward reads were filtered based on quality scores and trimmed using the DADA2 package version 1.9.0 (Callahan et al. 2016) in R version 3.4.1. Reads were filtered to remove those with a max EE of 2, and subsequently truncated at the end of ‘a good quality sequence’ with the parameter truncQ = 2 (see benjjneb.github.io/dada2/ for detailed explanation of filtering parameters).
The DADA2 algorithm was then used to estimate error rates from all filtered reads and infer exact sequence variants (ESVs). Chimeras were removed with de novo detection. Sequenced negative controls were used to identify possible contaminants using the decontam R package (Davis et al. 2017). A total of 196 reads were detected in our negative controls. Of these, 182 reads were of one sequence. BLAST results indicated that this sequence was Aspergillus penicillioides. This was identified by decontam as a potential contaminant and removed from the ESV table. Remaining ESVs were assigned taxonomy with the RDP classifier algorithm (Cole et al. 2007) against a training set based on the UNITE curated fungal amplicon database. Initial taxonomic assignments made using only the UNITE database returned greater than 90% fungal assignments. To increase confidence in our taxonomic assignments, we supplemented UNITE with appropriate outgroups, including 20,061 metazoan and 46,431 archaeplastid ITS1 sequences taken from the NCBI nt database. The inclusion of these outgroups resulted in the assignment of approximately 40% of reads to groups other than fungi, underscoring the importance of including appropriate outgroups when assigning taxonomy based on ITS amplicons. This approach ensured that the majority of non-fungal taxa were purged, eliminating their inclusion in all downstream analyses. Our complete taxonomic training set, including outgroups, is available online (https://github.com/gzahn/Sargassum_Fungi). ESVs not unambiguously assigned to fungi, those present as only singletons or doubletons, and sequences < 100 bp were removed.
Raw sequence counts were then converted to relative abundance (Gloor et al. 2017). Non-metric multi-dimensional scaling (nMDS) was performed on the Bray-Curtis dissimilarity matrix of samples using the phyloseq R package version 1.25.2 (McMurdie and Holmes 2013). A permutational multivariate analysis of variance (PERMANOVA) was performed on the ESV table with ‘location’ and ‘structure’ as predictors using the adonis function of the vegan R package version 2.5-2 (Okansen et al. 2016). Heatmaps were generated with R and Venn diagrams were built using the VennDiagram R package (Chen 2018).
All sequences associated with this work have been deposited at the National Center for Biotechnology Information under BioProject ID: PRJNA504438. All code, taxonomy databases, figures and output relating to this work can be found at https://github.com/gzahn/Sargassum_Fungi.
Results
Sequencing of the fungal ITS1 region generated 5,356,697 reads. Following filtering and quality control, 1,115 ESVs belonging to Fungi were retained for downstream analysis. The ITS1 sequences of fungal communities associated with S. ilicifolium revealed many fungal taxa that have yet to be discovered or named prior to this work. Dependent upon location, between 40 and 75% of the recovered taxa could not be assigned to a class (Fig. 2).

Bar plot showing relative abundance of fungal classes at each sampling location in Singapore
Non-metric multi-dimensional scaling (nMDS) showed no apparent structuring of fungal communities among sites, or between different anatomical structures (SI Fig. 3). However, PERMANOVA revealed weak but significant differences (P < 0.05) in community composition among locations and different structures (SI Table 1).
We were unable to assign the majority of the ESVs associated with S. ilicifolium to a class, but of those that could be assigned, Eurotiomycetes was the most abundant. The majority of the remaining ESVs were assigned to Agaricomycetes, Lecanoromycetes and Sordariomycetes (Fig. 2).
Four fungal orders, Eurotiales, Hypocreales, Lecanorales and Pleosporales, were frequently found throughout all surveyed structures, while several fungal orders appeared to be confined to certain structures. For example, Teloschistales appeared marginally less abundant in leaves compared to the holdfast and vesicles, while Capnodiales was most prevalent in the leaves and vesicles, suggesting structural specificity regarding fungal community composition (Fig. 3). Furthermore, of the 1,115 ESVs identified as belonging to Fungi, 88 were found shared throughout all structures, 61 were shared between the holdfast and leaves and 24 were shared between the vesicles and both the holdfast and leaves (Fig. 4). The holdfast contained the highest number of unique (not shared) ESVs at 535, followed by leaves at 218, while vesicles contained the fewest non-shared ESVs at 165.

Heatmap of order-level taxa distributed in each structure Deeper red indicates higher abundance

Venn diagram showing the number of ESVs unique to each sampled part and shared between parts
Aspergillaceae is the most common fungal family encountered at all islands and correspondingly we encounter the genus Aspergillus (see SI Figs. 4 and 5 for details of fungal family and genus assignments).
Discussion
Congruent with other work describing fungal biodiversity in remote and understudied locations (Archer et al. 2018), we have been unable to assign taxonomy to the vast majority of sequences in this study. This demonstrates that marine environments are likely reservoirs of yet-unknown fungi that should be considered when estimating global fungal diversity. At the very least, this inability to assign taxonomy represents a limitation of our most comprehensive databases to date. This is not entirely unexpected for marine fungi, given that most fungal research has focused on terrestrial environments, and thus fungi from other environments are likely underrepresented in current databases. Further examination of marine fungi will likely lead to the discovery of numerous new species.
The vast majority of hosts recruit their microbiome constituents from the surrounding environment. This is thought to promote and increase adaptation to local environments (Amend et al. 2019b). Work is now showing that microbial community structure can be observed at small scales. For example, differences in fungal community can be seen on either side of Wallace’s line (Wainwright et al. 2018), and even within 200 m (Galand et al. 2009; Agogué et al. 2011). These small-scale patterns and the observation that the majority of hosts recruit their microbes from the local environment could be responsible for the significant differences we observe in fungal communities from different localities.
Fungal community composition also differs significantly between different structures. Given the distinct functions that each sampled part plays, this is not entirely unexpected. These differences could be responsible for the resistance to herbivory that holdfasts show in comparison to leaves that are frequently seen with numerous bite marks. Fungi play an active role in preventing herbivory in many plants (Mortensen 2013; García Parisi et al. 2014; Tanentzap et al. 2014), and though the current study has not investigated this, it is possible that fungi found in the holdfast secrete products that render it unpalatable to grazers, especially since the holdfast is known to be grazing resistant (Loffler et al. 2018). Kohlmeyer (1971) found a fungal pathogen associated with Sargassum from the Sargasso Sea that exclusively infects vesicles, and once infection develops, the vesicles are shed. Here, we have not identified Lindra thalassiae, the fungal pathogen responsible for the shedding of vesicles, but do observe similar fungal associations specific to certain structures that may help to drive fungal community differences among them. We also observe certain fungal orders preferentially associating with particular structures. For example, Capnodiales is found in the leaves and vesicles but is rare in the holdfast, while Teloschistales is found more commonly in the holdfast and vesicles relative to the leaves (Fig. 3). In support, each structure contains fungal ESVs that are unique and not shared between structures (Fig. 4).
Numerous terrestrial studies have shown that microbial communities differ strongly between plant part, and those communities can be further delineated into above- and belowground components (Ottesen et al. 2013; Junker and Keller 2015; Amend et al. 2019b). Our work in the marine environment shows that while various structures do have significantly different fungal communities, the relationship is weak and the different structures do not show the same abrupt differences in community structure seen in terrestrial plants. In terrestrial environments, it is probable that the majority of fungi associated with roots and other belowground structures recruit from the soil, whereas the atmosphere is likely to be a more important fungal reservoir for aboveground structures such as leaves. Sargassum has neither belowground parts nor specialized structures for nutrient uptake, and is completely surrounded by an essentially homogeneous aqueous environment. Consequently, we see less specialization of fungal communities in different algal parts.
Eurotiomycetes is the most common fungal class associated with S. ilicifolium in Singapore. This is the same fungal class that is most abundant in the seagrass Syringodium isoetifolium collected from Indonesia (Wainwright et al. 2018), and members of this class have also been found in Sargassum spp. collected elsewhere in Southeast Asia (Kawaroe et al. 2015). In addition to the Eurotiomycetes, we also find the fungal classes Dothideomycetes and Agaricomycetes at all localities and in all structures. These classes are frequently observed in marine environments (Gnavi et al. 2014; Rédou et al. 2015), a possible consequence of their adaptations to the marine realm, including spores that have long appendages to promote buoyancy as well as facilitate entrapment and adherence to marine substrates (Prasannarai and Sridhar 2001; Vijaykrishna et al. 2006).
Eurotiomycetes contains the genus Aspergillus, and members of this genus are known to cause aspergillosis. Aspergillus is frequently observed in our samples and also tends to be more abundant in comparison to other genera. Aspergillus sydowii has been identified as the pathogen responsible for the mass mortality of sea fans (Gorgonia spp.) throughout the Caribbean where up to 90% mortality has been reported (Alker et al. 2001). Although we have found Aspergillus in our samples, the short length of the ITS1 fragment used in this work prevents us from conclusively delineating species within this genus. Further work taking advantage of long-read DNA sequencing technology would allow the use of longer and more informative markers for improved taxonomic resolution, and consequently more precise species identifications (Tedersoo et al. 2018). This is particularly useful for identification of marine pathogens that would have implications for effectively managing marine systems, especially those associated with macroalgae (Egan et al. 2013). For example, if A. sydowii or other emerging fungal pathogens are identified, it may be possible to design and implement effective conservation measures to mitigate and even prevent future disease outbreaks (Gleason et al. 2017). The resources required to perform this type of monitoring are becoming more readily available and accessible to all, meaning that this approach to pathogen detection is feasible.
Marine habitats abound with plentiful opportunities to discover novel fungi that may have applications for advancing human health and improving our standard of living (Balabanova et al. 2018). Research such as that performed here, and especially in diverse regions of the world, offers exceptional opportunities to advance our understanding of fungal diversity and ecology. By examining the fungal diversity associated with S. ilicifolium in the waters of Singapore, we improve our estimates and understanding of Earth’s biodiversity, particularly for the lesser-known yet biologically diverse coral reef habitats of Southeast Asia.
References
Agogué H, Lamy D, Neal PR, Sogin ML, Herndl GJ (2011) Water mass specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol Ecol 20:258–274. https://doi.org/10.1111/j.1365-294X.2010.04932.x
Ainsworth TD, Thurber RV, Gates DR (2010) The future of coral reefs: a microbial perspective. Trends Ecol Evol 4:233–240. https://doi.org/10.1016/j.tree.2009.11.001
Alker AP, Smith GW, Kiho K (2001) Characterization of Aspergillus sydowii (Thom et Church), a fungal pathogen of Caribbean Sea fan corals. Hydrobiologia 460:105–111. https://doi.org/10.1023/A:1013145524136
Allen GR, Werner TB (2000) Coral reef fish assessment in the “coral triangle” of southeastern Asia. Environ Biol Fish 65:209–214
Amend A, Burgaud G, Cunliffe M, Edgcomb VP, Ettinger CL, Gutiérrez MH, Heitman J, Hom EFY, Ianiri G, Jones AC, Kagami M, Picard KT, Quandt CA, Raghukumar S, Riquelme M, Stajich J, Vargas-Muñiz J, Walker AK, Yarden O, Gladfelter AS (2019a) Fungi in the Marine Environment: Open Questions and Unsolved Problems. mBio Mar 10(2):e01189–18
Amend AS, Cobian GM, Laruson AJ, Remple K, Tucker SJ, Poff KE, Antaky C, Boraks A, Jones CA, Kuehu D, Lensing BR, Pejhanmehr M, Richardson DT, Riley PP (2019b) Phytobiomes are compositionally nested from the ground up. PeerJ 7:e6609. https://doi.org/10.7717/peerj.6609
Archer S, Lee K, Caruso T, Maki T, Lee C, Cowan D, Maestre R, Pointing S (2018) Airborne microbial transport limitation to isolated Antarctic soil habitats. Nat Microbiol. https://doi.org/10.1038/s41564-019-0370-4
Balabanova L, Slepchenko L, Son O, Tekutyeva L (2018) Biotechnology potential of marine Fungi degrading plant and algae polymeric substrates. Front Microbiol 9:1527. https://doi.org/10.3389/fmicb.2018.01527
Bengtsson-Palme J et al (2013) Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol Evol. https://doi.org/10.1111/2041-210X.12073
Blackwell M (2011) The fungi: 1, 2, 3...5.1 million species? Am J Bot 98:426–438. https://doi.org/10.3732/ajb.1000298
Burgaud D, Meslet-Cladière L, Barbier G, Edgcomb VP (2014) Astonishing fungal diversity in deep-sea hydrothermal ecosystems: an untapped resource of biotechnological potential? In: Outstanding marine molecules: chemistry. Biology, Analysis, pp 85–98. https://doi.org/10.1002/9783527681501.ch04
Callahan BJ et al (2016) DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Chen H (2018) VennDiagram: generate high-resolution Venn and Euler Plots. R package version 1.6.20. https://CRAN.R-project.org/package=VennDiagram
Chou L, Huang MD, Tan KS, Toh TC, Goh BPL, Tun K (2019) Singapore. In: Sheppard CRC (ed) World seas: an environmental evaluation. Volume II: The Indian Ocean to the Pacific. Academic Press, London, pp 539–558. https://doi.org/10.1016/B978-0-08-100853-9.00031-2
Cole JR et al (2007) The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35(suppl 1):D169–D172. https://doi.org/10.1093/nar/gkl889
Comeau AM, Vincent WF, Bernier L, Lovejoy C (2016) Novel chytrid lineages dominate fungal sequences in diverse marine and fresh water habitats. Sci Rep 6:30120. https://doi.org/10.1038/srep30120
Davis NM, Proctor D, Holmes SP, Relman DA, Callahan BJ (2017) Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 6:226. https://doi.org/10.1186/s40168-018-0605-2
Deshmukh SK, Prakash V, Ranjan N (2018) Marine fungi: a source of potential anticancer compounds. Front Microbiol 8:2536. https://doi.org/10.3389/fmicb.2017.02536
Egan S, Harder T, Burke C, Steinberg P, Thomas T (2013) The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol Rev 37:462. https://doi.org/10.1111/1574-6976.12011
Galand PE, Potvin M, Casamayor EO, Lovejoy C (2009) Hydrography shapes bacterial biogeography of the deep Arctic Ocean. Int Soc Microb Ecol 4(4):564–576. https://doi.org/10.1038/ismej.2009.134
García Parisi PA, Grimoldi AA, Omacini M (2014) Endophytic fungi of grasses protect other plants from aphid herbivory. Fungal Ecol 9:61–64. https://doi.org/10.1016/j.funeco.2014.01.004
Gardes M, Bruns TM (1993) ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizas and rusts. Mol Ecol 2:113–118. https://doi.org/10.1111/j.1365-294X.1993.tb00005.x
Gin KYH, Lin X, Zhang S (2000) Dynamics and size structure of phytoplankton in the coastal waters of Singapore. J Plankton Res 22:1465–1485. https://doi.org/10.1093/plankt/22.8.1465
Gleason FH, Gadd GM, Pitt JI, Larkum AWD (2017) The roles of endolithic fungi in bioerosion and disease in marine ecosystems. II. Potential facultatively parasitic anamorphic ascomycetes can cause disease in corals and molluscs
Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ (2017) Microbiome datasets are compositional: and this is not optional. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.02224
Gnavi G, Ercole E, Panno L, Vizzini A, Varese GC (2014) Dothideomycetes and Leotiomycetes sterile mycelia isolated from the Italian seagrass Posidonia oceanica based on rDNA data. Springerplus 3:508 http://www.springerplus.com/content/3/1/508
Gonçalves VN et al (2016) Fungi associated with rocks of the Atacama Desert: taxonomy, distribution, diversity, ecology and bioprospection for bioactive compounds. Environ Microbiol 18:232–235. https://doi.org/10.1111/1462-2920.13005
Haas AF et al (2011) Effects of coral reef benthic primary producers on dissolved organic carbon and microbial activity. PLoS One 6:e27973. https://doi.org/10.1371/journal.pone.0027973
Hernandez-Agreda A, Leggat W, Bongaerts P, Herrera C, Ainsworth TD (2018) Rethinking the coral microbiome: simplicity exists within a diverse microbial biosphere. mBio 9:e00812–e00818. https://doi.org/10.1128/mBio.00812-18
Huang D, Tun K, Chou LM, Todd PA (2009) An inventory of zooxanthellate scleractinian corals in Singapore including 33 new records. Raffles Bull Zool Sup 22:69–80
Hughes TP, Bellwood DR, Connolly SR (2002) Biodiversity hotspots, centres of endemicity, and the conservation of coral reefs. Ecol Lett 5:775–784. https://doi.org/10.1046/j.1461-0248.2002.00383.x
Ishino M, Kamauchi H, Takatori K, Kinoshita K (2016) Three novel phomactin-type diterpenes from a marine-derived fungus. Tetrahedron Lett 57:4341–4344. https://doi.org/10.1016/j.tetlet.2016.08.016
Jompa J, McCook LJ (2003) Coral–algal competition: macroalgae with different properties have different effects on corals. Mar Ecol Prog Ser 258:87–95. https://doi.org/10.3354/meps258087
Junker RR, Keller A (2015) Microhabitat heterogeneity across leaves and flower organs promotes bacterial diversity. FEMS Microbiol Ecol 91:1–9. https://doi.org/10.1093/femsec/fiv097
Kawaroe M, Sunuddin A, Hwangbo B, Shaumi A (2015) Characteristics and selulotic activities of endophytic fungi in macroalgae (Sargassum sp., Gracilaria sp., Gelidium sp., and Caulerpa sp.) from seagrass habitat in Pari Island, Thousand Islands, Jakarta. International Journal of Sciences: basic and applied research. pp 149-160
Kim JY (2016) Human fungal pathogens: why should we learn? J Microbiol 54:145–148. https://doi.org/10.1007/s12275-016-0647-8
Kohlmeyer J (1971) Fungi from the Sargasso Sea. Mar Biol 8:344–350
Leong RC et al (2018) Effect of coral-algal interactions on early life history processes in Pocillopora acuta in a highly disturbed coral reef system. Front Mar Sci. https://doi.org/10.3389/fmars.2018.00385
Loffler Z et al (2018) Holdfasts of Sargassum swartzii are resistant to herbivory and resilient to damage. Coral Reefs 37:1075–1084. https://doi.org/10.1007/s00338-018-01745-w
Low JKY (2015) Sargassum on Singapore’s reefs. PhD Thesis, National University of Singapore. Singapore. https://scholarbank.nus.edu.sg/handle/10635/118571. Accessed 08/03/2019
Low JKY, Chou LM (1994) Coral reef fish in a sediment stressed environment. http://coralreef.nus.edu.sg/publications/Low1994LIPI_JSPS.pdf. Accessed 08 Nov 2018
Low JKY, Fong J, Todd PA, Chou LM, Bauman AG (2019) Seasonal variation of Sargassum ilicifolium (Phaeophyceae) growth on equatorial coral reefs. J Phycol 55:289–296. https://doi.org/10.1111/jpy.12818
McCook LJ (2001) Competition between coral and algal turfs along a gradient of terrestrial influence in the nearshorecentral Great Barrier Reef. Coral Reefs 19:419–425
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8(4):e61217. https://doi.org/10.1371/journal.pone.0061217
Moree WJ et al (2014) Microbiota of healthy corals are active against fungi in a light-dependent manner. ACS Chem Biol:2300–2308. https://doi.org/10.1021/cb500432j
Morrow KM, Paul VJ, Liles MR, Chadwick NE (2011) Allelochemicals produced by Caribbean macroalgae and cyanobacteria have species-specific effects on reef coral microorganisms. Coral Reefs 30:309–320. https://doi.org/10.1007/s00338-011-0747-1
Mortensen B (2013) Plant resistance against herbivory. Nat Educ Knowl 4:5
Nature ecology and evolution (2018) Fungus focus. Nat Ecol Evol 2:1675. https://doi.org/10.1038/s41559-018-0721-1
Nugues M, Smith GW, van Hooidonk RJ (2004) Algal contact as a trigger for coral disease. Ecol Lett 7:919–923. https://doi.org/10.1111/j.1461-0248.2004.00651.x
Okansen J et al (2016) Vegan: community ecology package. (version 2.4-0). Retrieved from https://CRAN.R-project.org/package=vegan
Ottesen AR, Peña AG, White JR, Pettengill JB, Li C, Allard S et al (2013) Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol 13:114. https://doi.org/10.1186/1471-2180-13-114
Oyesiku OO, Egunyomi A (2015) Identification and chemical studies of pelagic masses of Sargassum natans (Linnaeus) Gaillon and S. fluitans (Borgessen) Borgesen (brown algae), found offshore in Ondo State, Nigeria. Afr J Biotechnol 13:1188–1193
Pauvert C, Buée M, Laval V, Edel-Hermann V, Fauchery L, Gautier A, Lesur I, Vallance J, Vacher C (2019) Bioinformatics matters: the accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecol 41:23–33. https://doi.org/10.1016/j.funeco.2019.03.005
Peay KG (2016) Dimensions of biodiversity in the earth mycobiome. Nat Rev Microbiol 14:434–447. https://doi.org/10.1038/nrmicro.2016.59
Picard KT (2017) Coastal marine habitats harbor novel early-diverging fungal diversity. Fungal Ecol 23:1–13. https://doi.org/10.1016/j.funeco.2016.10.006
Prasannarai K, Sridhar KR (2001) Diversity and abundance of higher marine fungi on woody substrates along the west coast of India. Curr Sci India 81:304–311
Pratte ZA, Longo GO, Burns AS, Hay ME, Stewart FJ (2017) Contact with turf algae alters the coral microbiome: contact versus systemic impacts. Coral Reefs 37:1–13. https://doi.org/10.1007/s00338-017-1615-4
Rasher DB, Stout P, Engel S, Kubanek J, Hay ME (2011) Macroalgal terpenes function as allelopathic agents against reef corals. Proc Natl Acad Sci U S A 108:17726–17731
Rédou V, Navarri M, Meslet-Cladière L, Barbier G, Burgaud G (2015) Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl Environ Microbiol 81:3571–3583. https://doi.org/10.1128/AEM.04064-14
Richards TA, Jones DMM, Leonard G, Bass D (2012) Marine fungi: their ecology and molecular diversity. Annu Rev Mar Sci 4:495–522. https://doi.org/10.1146/annurev-marine-120710-100802
Robinson CH (2001) Cold adaptation in Arctic and Antarctic fungi. New Phytol 151:341–353. https://doi.org/10.1046/j.1469-8137.2001.00177.x
Russell JR et al (2011) Biodegradation of polyester polyurethane by endophytic fungi. Appl Environ Microbiol 17:6076–6084. https://doi.org/10.1128/AEM.00521-11
Rützler K, Santavy DL (1983) The black band disease of Atlantic corals. Mar Ecol. https://doi.org/10.1111/j.1439-0485.1983.tb00116.x
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS One 9:e90234. https://doi.org/10.1371/journal.pone.0090234
Széchy MTM, Guedes PM, Baeta-Neves MH, Oliveira EN (2012) Verification of Sargassum natans (Linnaeus) Gaillon (Heterokontophyta: Phaeophyceae) from the Sargasso Sea off the coast of Brazil, western Atlantic Ocean. Checklist 8:638–641. https://doi.org/10.15560/8.4.638
Tanentzap AJ, Vicari M, Bazely DR (2014) Ungulate saliva inhibits a grass–endophyte mutualism. Biol Lett 10:20140460. https://doi.org/10.1098/rsbl.2014.0460
Tanzil JTI, Goodkin NF, Sin TM, Chen MT, Fabbro GN, Boyle EA, Lee AC, Toh KB (2019) Multi-colony coral skeletal Ba/Ca from Singapore’s turbid urban reefs: relationship with contemporaneous in-situ seawater parameters. Geochim Cosmochim Acta 250:191–208. https://doi.org/10.1016/j.gca.2019.01.034
Tedersoo L, Toomin-Klunderud A, Anslan S (2018) PacBio metabarcoding of Fungi and other eukaryotes: errors, biases and perspectives. New Phytol 217:1370–1385. https://doi.org/10.1111/nph.14776
Tun KPP (2012) Optimisation of reef survey methods and application of reef metrics and biocriteria for the monitoring of sediment-impacted reefs. PhD Thesis, Department of Biological Sciences, National University of Singapore
Vijaykrishna D, Jeewon R, Hyde KD (2006) Molecular taxonomy, origins and evolution of freshwater Ascomycetes. Fungal Divers 23:351–390
Wainwright BJ et al (2017) Fungi associated with mesophotic macroalgae from the ‘Au‘au Channel, west Maui are differentiated by host and overlap terrestrial communities. PeerJ 5:e3532. https://doi.org/10.7717/peerj.3532
Wainwright BJ et al (2018) Seagrass-associated fungal communities follow Wallace’s line, but host genotype does not structure fungal community. J Biogeograpy:1–9. https://doi.org/10.1111/jbi.13168
Wang M, Hu C (2016a) Mapping and quantifying Sargassum distribution and coverage in the Central West Atlantic using MODIS observations. Remote Sens Environ 183:350–367. https://doi.org/10.1016/j.rse.2016.04.019
Wang M, Hu C (2016b) Predicting Sargassum blooms in the Caribbean Sea from MODIS observations. Geophys Res Lettv 44:3265–3273. https://doi.org/10.1002/2017GL072932
Weldon C, du Preez LH, Hyatt AD, Muller R, Speare R (2004) Origin of the amphibian chytrid fungus. Emerg Infect Dis 10:2100–2105. https://doi.org/10.3201/eid1012.030804
White TJ, Bruns TD, Lee SB, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH (eds) PCR protocols: a guide to methods and applications. Academic Press, London, pp 315–322
Yip ZT et al (2018) Diversity and phylogeny of Sargassum (Fucales, Phaeophyceae) in Singapore. Phytotaxa. https://doi.org/10.11646/phytotaxa.369.3.3
Acknowledgments
We thank Jack Darcy for the R script that allows sequence quality information to be ‘reattached’ to the processed ITSx data. Full script can be found at https://github.com/gzahn/Sargassum_Fungi/blob/master/R/itsx_fastq_extractor.r
Funding
This study was funded by the National Research Foundation, Prime Minister’s Office, Singapore, under its Marine Science R&D Programme (MSRDP-P03) and The Wildlife Reserves Singapore Conservation Fund (WRSCF). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable permits, international, national and/or institutional guidelines required to perform the work were followed. All samples were collected under permit NP/RP15-009-2a.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities. The study is compliant with CBD and Nagoya protocols. All samples were collected under permit NP/RP15-009-2a.
Data availability
All sequences associated with this work have been deposited at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject/) under BioProject ID: PRJNA504438.
Additional information
Communicated by B. Beszteri
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 793 kb)
Rights and permissions
About this article

Cite this article
Wainwright, B.J., Bauman, A.G., Zahn, G.L. et al. Characterization of fungal biodiversity and communities associated with the reef macroalga Sargassum ilicifolium reveals fungal community differentiation according to geographic locality and algal structure. Mar. Biodivers. 49, 2601–2608 (2019). https://doi.org/10.1007/s12526-019-00992-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12526-019-00992-6