
Abstract
Background
Development of noninvasive imaging modalities to quantify amyloid burden over time is an unmet clinical need. Technetium pyrophosphate (99mTc-PYP) scintigraphy is a simple and widely available radiotracer useful to differentiate transthyretin from light-chain amyloidosis in patients with advanced cardiac amyloidosis. We examined the utility of serial 99mTc-PYP scanning to quantify amyloid burden over time in TTR cardiac amyloidosis (ATTR-CA).
Methods and Results
Twenty subjects with ATTR-CA (10 wild type, 10 mutant) underwent serial 99mTc-PYP planar cardiac imaging. Cardiac retention was assessed both semiquantitatively (visual score 0, no uptake to 3, uptake greater than bone) and quantitatively (region of interest drawn over the heart, copied, and mirrored over the contralateral chest) to calculate a heart-to-contralateral (H/CL) ratio. Index scan mean visual score and H/CL were 3.0 ± 0.2 and 1.79 ± 0.2, respectively, and after an average 1.5 ± 0.5 years follow-up, did not differ, 3.0 ± 0.2, P = .33 and 1.76 ± 0.2, P = .44. H/CL change was minimal, 0.03 ± 0.17, did not correlate with time between scans, r = 0.19, P = .43, and was observed despite obvious clinical progression (increase in troponin ≥ 0.1 ng/mL, BNP ≥ 400 pg/mL, NYHA class, and/or death).
Conclusions
Serial 99mTc-PYP scanning in subjects with advanced ATTR-CA does not show significant changes over an average 1.5 years of follow-up despite obvious clinical progression.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The systemic cardiac amyloidoses (CA) are an increasingly recognized and potentially treatable cause of heart failure with preserved ejection fraction (HFpEF) in older adults.1 While at least 3 different subtypes of cardiac amyloidosis share the general mechanism of extracellular fibrillary protein deposition leading to tissue and organ dysfunction, each has a different clinical course and treatment strategy.2 In light-chain (AL) cardiac amyloidosis, fibrils comprise immunoglobulin light chains produced by a clonal plasma cell population. Treatment involves chemotherapeutic agents and at times bone marrow transplantation aimed at eradicating the hyperproliferative plasma cell. In contrast, in the transthyretin cardiac amyloidoses (ATTR), misfolded monomers and dimers of normally tetrameric transthyretin protein (TTR) from either mutant TTR (ATTRm, also known as familial amyloid cardiomyopathy) or wild-type TTR (ATTRwt, also known as senile systemic amyloidosis) deposit in the myocardium, lead to diastolic and systolic dysfunction, arrhythmias, restrictive cardiomyopathy, and heart failure. Treatment involves conventional heart failure management and in select individuals, liver and/or heart transplantation. Over the past several years, however, a number of potentially disease-modifying treatment strategies have emerged and are now in human clinical trials for the treatment of ATTR cardiac amyloidosis.3
The gold standard for definitive diagnosis of cardiac amyloidosis is endomyocardial biopsy coupled with either immunohistochemistry or, even more specific, mass spectroscopy. Unfortunately, these diagnostic requirements are typically performed only in specialized centers, and do not provide sufficient information about the extent or distribution of cardiac amyloidosis, disease progression, or response to treatment, and in practice can lead to delayed care. Additionally, many older adults are reluctant to undergo invasive procedures.
A clinical unmet need in this arena is the development of a noninvasive imaging modality that can diagnose cardiac amyloid, differentiate AL from ATTR subtypes, quantify the extent of myocardial amyloid infiltration, and monitor disease progression and response to treatment. This quest for serial cardiac imaging using technetium pyrophosphate (99mTc-PYP) to quantify amyloid burden has been ongoing since the 1980s without significant advancement.4,5 Recently, quantitative 99mTc-PYP cardiac imaging was shown to be a useful method with 97% sensitivity and 100% specificity for differentiating AL vs ATTR cardiac amyloidosis in patients with advanced disease.6 However, the utility of this technique to monitor disease progression is unknown. In light of emerging disease-modifying therapies and the future need for disease monitoring over time, we sought to determine the utility of serial 99mTc-PYP scanning for ATTR cardiac amyloidosis. We hypothesized that 99mTc-PYP scanning would be able to identify significant changes in myocardial tracer retention over time that correlated with clinical disease progression.
Methods
Patient Population
Patients with ATTR-related cardiac amyloidosis undergoing routine follow-up at the Columbia University Center for Advanced Cardiac Care participated in this study. The protocol was approved by the Columbia University IRB and subjects provided informed consent. 20 patients (10 ATTRwt and 10 ATTRm, including 8 with Val122Ile and 2 with Thr60Ala mutations) were enrolled. Inclusion criteria included a previous 99mTc-PYP scan demonstrating myocardial enhancement with heart-to-contralateral (H/CL) ratio ≥ 1.56 and evidence of TTR amyloid by standard criteria including (1) biopsy proven ATTR-CA (n = 17) or (2) documented amyloidogenic TTR mutation by DNA analysis and echocardiographically defined evidence of amyloid cardiomyopathy (thickness of the left ventricular septum or posterior wall >12 mm without another cause of hypertrophy) without evidence of a plasma cell dyscrasia (n = 3 confirmed Val122Ile mutation). Exclusion criteria included women of childbearing potential, minors, and inability to provide informed consent or lie still for 15 minutes under the camera.
Study Design
This was a single-center, blinded, prospective cohort design aimed at evaluating whether 99mTc-PYP serial scanning could be used to evaluate the progression of ATTR cardiac amyloidosis in 20 subjects previously diagnosed with the disease. All subjects with known intense myocardial enhancement on 99mTc-PYP scan (H/CL ≥ 1.5) and confirmed ATTR cardiac amyloidosis underwent a second 99mTc-PYP scan as described below. Scans were performed and interpreted by experienced nuclear cardiologists blinded to subjects’ clinical information (including amyloid subtype and mutation status) and prior 99mTc-PYP scan result.
Clinical, Laboratory, and Transthoracic Echocardiography Follow-Up
All subjects underwent routine follow-up at the Columbia Center for Advanced cardiac care. In additional to baseline and follow-up clinical examinations, patients underwent serial serum biomarker screening, including troponin I and brain natriuretic peptide (BNP), along with serial transthoracic echocardiography at the discretion of the treating cardiologist. Standard definitions of chamber quantification, valvular pathology, and myocardial function were read according to the American Society of Echocardiography definitions.7 In addition, myocardial contraction fraction (MCF), a novel measure of myocardial function, was calculated as the ratio of stroke volume to myocardial volume.8
Serial 99mTc-PYP Scintigraphy
The second 99mTc-PYP cardiac scan was performed similar to the baseline scan using the same dual-head Philips Precedence SPECT/CT camera (Philips Healthcare, Guildford, United Kingdom) equipped with low-energy, high-resolution collimators. The anterior and lateral planar images were acquired for a total of 750,000 counts with the heart centered in the field of view. For the second scan, patients received 10 mCi of 99mTc-PYP IV (baseline dose ranged 15-25 mCi) and planar images were obtained at 1 hour over 6-8 minutes duration for the 10 mCi dose and 3-4 minutes duration for the 15-25 mCi dose. The acquisition parameters used for planar imaging were 256 × 256 matrix with 1.46 zoom factor.
Cardiac retention was assessed with a semiquantitative visual score (range: 0, no uptake to 3, uptake greater than bone) and with and quantitative measure of heart retention calculated by drawing a region of interest (ROI) over the heart in the standard manner (Figure 1). A circular ROI was drawn over the heart, copied, and mirrored over the contralateral chest to normalize for background uptake in the ribs. Mean total heart ROI counts were measured and corrected for contralateral chest ROI counts by calculating a heart-to-contralateral (H/CL) ratio. In addition, a subjective interpretation of worsening was performed by comparing baseline and follow-up scans side-by-side with the reader blinded to visual score and H/CL ratio.

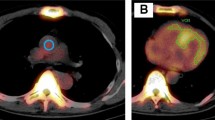
Semiquantitative and quantitative analysis of 99mTc-PYP myocardial uptake. Semiquantitative analysis (A): visual cardiac score, which was assigned 0–3 according to the scale detailed. The representative image demonstrates a visual cardiac score of 3. Quantitative analysis (B): heart-to-contralateral ratio was calculated by drawing a region of interest over the heart, copying and mirroring it to the contralateral chest and calculating the ratio of heart ROI mean counts to contralateral ROI mean counts. The representative image demonstrates a H/CL ratio 30/16 = 1.88. H/CL, heart-to-contralateral; ROI, region of interest
The reproducibility of the semiquantitative visual score, quantitative HCL ratio, and subjective interpretation was examined with inter- and intra-observer variability. For inter-observer variability (between readers), two nuclear cardiologists blinded to the patients’ clinical information (including mutation status and baseline 99mTc-PYP scan result) independently read 99mTc-PYP images assessing visual score, H/CL ratio, and subjective worsening. For intra-observer variability (within reader), two nuclear cardiologists read each scan twice >1 month apart assessing visual score and H/CL ratio.
Statistical Analyses
Demographic, laboratory, and imaging data were collected and analyzed with descriptive statistics using mean ± standard deviation for continuous variables and as relative percentages or median (range) for categorical variables. Statistical analyses were performed using Statistical Analysis Software version 9.4 (SAS Institute, Inc., Cary, NC). Continuous variables were tested for the assumption of normality using the Shapiro-Wilk test for normality. Comparisons between baseline and follow-up scans were analyzed using a student’s T test for paired comparisons for normally distributed continuous variables, a Wilcoxon signed-rank sum test for non-normally distributed continuous variables, Χ 2 test for categorical variables, and Fisher’s Exact Test for categorical variables with small cell counts on level of significance 5%. Comparisons between categorical and continuous variables were analyzed using logistic regression. In 20 paired subjects, this study was powered at 80% to detect a true H/CL difference of 0.13. Reproducibility of scans in terms of inter- and intra-observer variability was analyzed with a Kappa statistic for the visual score and subjective interpretation and with an interclass correlation coefficient (ICC) for the continuous H/CL ratio.9
Results
Demographics of Study Population
Twenty patients with transthyretin cardiac amyloidosis (10 ATTRwt and 10 ATTRm) were enrolled and completed the study protocol. Patients with ATTRm cardiac amyloidosis had the following TTR mutations: Val122Ile (n = 8) and Thr60Ala (n = 2). The majority of subjects were elderly men, mean age 74 ± 6 years old, with a racial distribution representative of a North American population with ATTRm (Table 1). Comorbid conditions included hypertension 58%, coronary artery disease 37%, diabetes 11%, chronic kidney disease 37%, and history of cerebral vascular accident 11%.10 Two patients were treated with Tafamidis, the TTR stabilizing agent. Mean duration of disease from symptom onset to time of first 99mTc-PYP scan was 5.0 ± 1.1 years. Mean time between scans was 1.5 ± 0.5 years.
Clinical, Biochemical, and Echocardiographic Data at Baseline and Follow-Up
At baseline, patients had a phenotype consistent with ATTR symptomatology described previously. Functionally, patients had a median baseline NYHA class 2 (range 2) with a mildly decreased left ventricular ejection fraction (LVEF) 42 ± 17%, increased wall thickness 1.7 ± 0.3 cm, and severely depressed myocardial contraction fraction 16.3 ± 7.3.
During follow-up, clinical disease worsened significantly (Table 2). Obvious clinical progression defined as increase in troponin I ≥0.1 ng/mL, bran natriuretic peptide (BNP)≥ 400 pg/mL, New York Heart Association (NYHA) class, progression to heart transplantation and/or death was observed in 18 patients (90%). Between baseline and follow-up, mean NYHA class, creatinine, estimated glomerular filtration rate (eGFR), troponin I, and BNP worsened significantly. Notably, there were no significant changes in albumin, modified body mass index (mBMI), LVEF, myocardial wall thickness, or MCF. As of December 2014, ten patients (50%) died and one patient had progressed to require orthotopic heart transplantation (OHT) (mean time to death or OHT 3.5 ± 0.4 years) and these outcomes did not differ by mutation status (P = .99), tafamidis treatment (P = .4), or H/CL ratio (P = .4).
Serial 99mTc-PYP Cardiac Imaging
A representative example of 99mTc-PYP cardiac uptake between baseline and follow-up is shown in Figure 2. Cardiac retention reflected by semiquantitative visual score or quantitative H/CL ratio did not change between scans despite obvious progression of clinical disease (Table 3). Upon side-by-side comparison of baseline and follow-up scans, only Subject 17 (1 out of 20 subjects, 5%) was interpreted as having worsened subjectively, while the remainder (95%) had unchanged scans. There was no significant change between first and second scans in semiquantitative visual score (3 [range 0] and 3 [range 2]), P = .31] or H/CL ratio (1.79 ± 0.2 and 1.76 ± 0.2, P = .44). The mean change that did occur in H/CL ratio was minimal, 0.03 ± 0.18, and was not significantly correlated with duration between first and second scan (r = 0.19, P = .43). Of the 7 patients that showed an increase in H/CL ratio, 6 (3 ATTRm, 3 ATTRwt) had an increase >0.13 (the difference which this study was powered to detect) but displayed no difference in disease severity, biomarkers, or disease progression compared to the rest of the cohort. H/CL ratio change was not significantly different between subjects who experienced obvious disease progression compared to those who did not (Progressors: N = 18, H/CL change 0.4 ± 0.18, Non-Progressors: N = 2, H/CL change 0.7 ± 0.11, P = .4). In addition, H/CL ratio change between baseline and follow-up was not significantly different between TTR subtypes (ATTRm: −0.06 ± 0.02, ATTRwt: −0.002 ± 0.17, P = .4).

Serial 99mTc-PYP scintigraphy scanning for cardiac amyloidosis. Raw images of 99mTc-PYP cardiac uptake in a representative patient at baseline (A) and follow-up (B). Mean time between scans was 1.5 years. Individual H/CL ratios for the 20 study subjects are depicted at baseline and follow-up (C) with mean ± SD (red line). H/CL, heart-to-contralateral ratio; SD, standard deviation
99mTc-PYP semiquantitative visual score, subjective interpretation of disease progression, and quantitative H/CL ratio were highly reproducible. For the visual score, inter- and intra-observer variability demonstrated 100% agreement. The subjective interpretation of disease progression also demonstrated 100% agreement between readers. For the H/CL ratio, agreement was high with between reader ICC 0.94 (95% CI 0.81, 0.98) and within reader ICC 0.88 (95% CI 0.63, 0.96).
Discussion
The results of this study show that in patients with advanced cardiac amyloidosis, clinical disease progressed over an average 1.5 years follow-up in the absence of discernible changes in myocardial tracer retention on 99mTc-PYP scanning. An important overarching goal for the application of nuclear technologies to noninvasively image cardiac amyloidosis is to define the temporal relationship between cardiac fibril deposition, clinical symptoms, and cardiac radiotracer uptake through the lifespan of an affected individual. Elaboration of these dynamic processes with reliable noninvasive cardiac imaging techniques will be relevant, not just for tracking disease progression over time, but also for monitoring response to treatments in individual patients as amyloid therapies emerge from clinical trials.11-17
Currently, the molecular mechanism responsible for 99mTc-PYP binding to amyloid fibrils in the myocardium is unknown. One theory is that 99mTc-PYP may bind to amyloid deposits in a calcium-dependent manner. Calcium is known to enhance the affinity of human serum amyloid protein P (SAP) to several different types of amyloid fibrils as well as its ability to self-aggregate and resist proteases.18-21 Perhaps different amounts of calcium in different tissues may explain varying degrees of 99mTc-PYP tissue-specific uptake. Another hypothesis is that the intensity of 99mTc-PYP binding relates to the duration over which amyloid deposition has occurred in the affected tissue. This may explain the known fact that 99mTc-PYP myocardial enhancement is significantly higher among patients with ATTR,6 whose disease course is typically indolent compared to patients with AL, where toxic fibrils accumulate over a shorter time period.
In the current study, the observation of unchanged 99mTc-PYP cardiac uptake between scans yields several possible interpretations about the pathophysiology and dynamics of cardiac amyloid deposition and its relationship with 99mTc-PYP radiotracer uptake. One interpretation is that 99mTc-PYP cardiac uptake does not mirror clinical disease progression and, therefore, this test is best suited to aid in the diagnosis of ATTR cardiac amyloidosis but not to track clinical disease progression. It should be noted, however, that previous reports of bone isotopes detecting cardiac amyloid in patients with familial amyloid polyneuropathy (FAP) prior to echocardiographic manifestations22,23 suggest that myocardial uptake of bone isotopes in TTR amyloid can occur in early stages of disease, when it is assumed that small amount of amyloid is present, and thus could potentially increase with further amyloid deposition. The results of the current study may not reflect the expected temporal changes in radiotracer uptake based on technical limitations of detection not allowing for resolution of clinically relevant increases in amyloid in the context of the intense myocardial uptake, indicative of advanced clinical disease, seen at baseline in our subjects. Thus, it is possible that detection of changes in myocardial uptake of bone isotopes such as 99mTc-PYP is dependent on the stage of disease and rate of TTR deposition and may not be useful to track disease progression in advanced ATTR cardiac amyloid. Importantly, the degree of 99mTc-PYP myocardial uptake by H/CL ratio was not related to the 99mTc-PYP dose injected (P = .73). An alternative interpretation of the current findings is that in this advanced disease population, the rapid disease progression observed over just 1.5 years follow-up is disproportionate to any putative increase in myocardial amyloid deposition and may be due to alternative processes.
Mechanisms by which already deposited amyloid fibrils may exacerbate symptoms of congestive heart failure in patients with advanced disease include apoptosis, inflammation, oxidative stress, fibrosis, and direct cytotoxicity.24-27 These insults to myocardial structure and function are likely to occur throughout the natural history of ATTR but may predominate in advanced disease stages of ATTR cardiac amyloid. Therefore, experimental treatments designed to mitigate non-amyloidogenic processes including inflammatory responses and others previously mentioned may have the potential to provide additional benefit to patients with advanced disease.
Irrespective of the underlying explanation for these findings, the current data suggest that in advanced disease followed over a short time period relative to the lifespan of an individual, any increase resulting from additional amyloid deposition into a heart already burdened with extensive amyloid deposition falls below the limit of detection of quantitative serial 99mTc-PYP scanning. Further investigation is required to understand the sensitivity of 99mTc-PYP cardiac imaging in relationship to amyloid changes, and the physiologic relevance of those changes, during all stages of disease progression in patients with ATTR.
Future research directions might also investigate the utility of other advanced cardiac imaging modalities such as 18F-florbetapir positron emission tomography (PET) for diagnosing and following cardiac amyloidosis over time. Recently, a small pilot study using PET demonstrated significant myocardial radiotracer uptake of 18F-florbetapir in 9 patients with cardiac amyloidosis (4 ATTR, 5 AL) compared to controls, suggesting this imaging modality may also be a promising technique to image cardiac amyloidosis28. Its sensitivity and specificity for diagnosing cardiac amyloid, utility in serial scanning, and cost-effectiveness compared to 99mTc-PYP cardiac imaging, however, need further study in larger cohorts garnered from multi-center collaborations.
Limitations
This was a small single-center study that included primarily a North American ATTR population with advanced cardiac amyloidosis and only two mutations represented. However, to the best of our knowledge, this is the only study to date that specifically focused on the utility of serial 99mTc-PYP cardiac imaging to monitor ATTR disease progression over time. We were powered to detect a mean H/CL difference of 0.13 (7% change); however, no change was detected. The mean time between scans of 1.5 ± 0.5 years may have been too short a period in which to observe meaningful change in 99mTc-PYP myocardial uptake but the worsening clinical condition of subjects over this time makes this unlikely. The generalizability of these results to other populations is, therefore, unknown. Most of these subjects enrolled had severe phenotypes with markedly thickened LV walls, which although similar to other cohorts of patients with cardiac amyloidosis, may be characterized by enhanced uptake of 99mTc-PYP, which may have led to signal saturation and subsequent inability to discriminate subtle changes between scans.
While additional studies are needed to determine the role of 99mTc-PYP for early disease detection in genotype-positive, phenotype-negative ATTR patients, these data demonstrate that serial 99mTc-PYP scanning in subjects with advanced ATTR cardiac amyloidosis does not show significant changes over an average 1.5 years of follow-up despite obvious clinical progression.
New Knowledge Gained
As 99mTc-PYP scanning has become more standard in the evaluation and diagnosis of subjects with cardiac amyloidosis, to the best of our knowledge, no other study has reported on the utility of serial 99mTc-PYP scanning in this number of patients followed over time. This is the first study to perform serial scans using the H/CL ratio quantitative score and demonstrate that in a population of patients with advanced ATTR cardiac amyloidosis, myocardial 99mTc-PYP uptake did not change even though clinical disease progressed markedly. The next logical step to evaluate the utility of serial 99mTc-PYP scanning would be to study it in a population of patients with mild-moderate disease who are followed over a longer time period.
Abbreviations
- AL:
-
Amyloid light-chain
- ATTR:
-
Transthyretin amyloidosis
- EF:
-
Ejection fraction
- HFpEF:
-
Heart failure preserved ejection fraction
- ROI:
-
Region of interest
- 99mTc-PYP:
-
Technetium pyrophosphate
References
Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765–74.
Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–12.
Castano A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: Emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20:163–78.
Hongo M, Hirayama J, Fujii T, et al. Early identification of amyloid heart disease by technetium-99m-pyrophosphate scintigraphy: A study with familial amyloid polyneuropathy. Am Heart J. 1987;113:654–62.
Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM. Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J. 1982;103:468–73.
Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6:195–201.
Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovas Imaging. 2015;16:233–71.
King DL, El-Khoury Coffin L, Maurer MS. Myocardial contraction fraction: A volumetric index of myocardial shortening by freehand three-dimensional echocardiography. J Am Coll Cardiol. 2002;40:325–9.
Lachin JM. The role of measurement reliability in clinical trials. Clin Trials. 2004;1:553–66.
Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: A clinical overview. Nat Rev Cardiol. 2010;7:398–408.
Safety and efficacy of tafamidis in patients with transthyretin cardiomyopathy (ATTR-ACT); 2014. http://www.clnicaltrials.gov/show/NCT01994889.)
Maurer M, JDP, Rosas GR, Mandel FS, Aarts J. Interim analysis of long-term, open-label tafamidis treatment in transthyretin amyloid cardiomyopathy after up to 5 years of treatment. In: International symposium on amyloidosis; 2014; Indianapolis, USA.
Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819–29.
Phase 2 study to evaluate ALN-TTRSC in patients with transthyretin (TTR) cardiac amyloidosis; 2014. https://www.clinicaltrials.gov/ct2/show/NCT01981837.)
Tolerability and efficacy of a combination of doxycycline and TUDCA in patients with transthyretin amyloid cardiomyopathy; 2013. https://www.clinicaltrials.gov/ct2/show/NCT01855360?term=NCT01855360&rank=1
Quarta CCF SR, Suhr SD, Obici OB, Perlini L, Lindqvist S, Koyama P, Sekijima J, Zeldenrust Y, Yamashita SR, Horibata T, Miller Y, Gorevic F, Merlini P, Ando G, Ikeda Y, Ruberg S, Berk F. The prevalence of cardiac amyloidosis in familial amyloidotic polyneuropathy with predominant neuropathy: The diflunisal trial. In: International Symposium on Amyloidosis; 2014; Indianapolis, USA. p. 88-9.
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18:315–9.
Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin Exp Immunol. 1979;38:284–93.
Hawkins PN, Myers MJ, Lavender JP, Pepys MB. Diagnostic radionuclide imaging of amyloid: Biological targeting by circulating human serum amyloid P component. Lancet. 1988;1:1413–8.
Nielsen EH, Sorensen IJ, Vilsgaard K, Andersen O, Svehag SE. Calcium-enhanced aggregation of serum amyloid P component and its inhibition by the ligands heparin and heparan sulphate. An electron microscopic and immunoelectrophoretic study. APMIS. 1994;102:420–6.
Hamazaki H. Ca(2+)-dependent binding of human serum amyloid P component to Alzheimer’s beta-amyloid peptide. J Biol Chem. 1995;270:10392–4.
Puille M, Altland K, Linke RP, et al. 99mTc-DPD scintigraphy in transthyretin-related familial amyloidotic polyneuropathy. Eur J Nucl Med Mol Imaging. 2002;29:376–9.
Russo M, Mazzeo A, Stancanelli C, et al. Transthyretin-related familial amyloidotic polyneuropathy: description of a cohort of patients with Leu64 mutation and late onset. J Peripher Nerv Syst. 2012;17:385–90.
Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66:3095–101.
Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101:2817–22.
Nagasaka T. Familial amyloidotic polyneuropathy and transthyretin. Sub-Cell Biochem. 2012;65:565–607.
Benson MD. Pathogenesis of transthyretin amyloidosis. Amyloid. 2012;19(Suppl 1):14–5.
Dorbala S, Vangala D, Semer J, et al. Imaging cardiac amyloidosis: A pilot study using (1)(8)F-florbetapir positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41:1652–62.
Acknowledgments
We would like to acknowledge the patients with cardiac amyloidosis who participated in this study and who continue to hope for methods to improve outcomes including earlier and more efficient diagnosis and better therapeutics.
Conflicts of interest
Dr. Maurer’s institution, Columbia University Medical Center, receives funding for research and serving on advisory boards and DSMBs from Pfizer, Inc, Alnylam Pharmaceuticals Inc, ISIS Pharmaceuticals and Prothena Inc.
Disclosure
Alnylam Pharmaceuticals provided a grant to support the imaging performed in this study. Dr. Maurer’s institution, Columbia University Medical Center, receives funding for research and serving on advisory boards and DSMBs from Pfizer, Inc, Alnylam Pharmaceuticals Inc, ISIS Pharmaceuticals and Prothena Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
See related editorial, doi:10.1007/s12350-015-0295-0.
Rights and permissions
About this article

Cite this article
Castaño, A., DeLuca, A., Weinberg, R. et al. Serial scanning with technetium pyrophosphate (99mTc-PYP) in advanced ATTR cardiac amyloidosis. J. Nucl. Cardiol. 23, 1355–1363 (2016). https://doi.org/10.1007/s12350-015-0261-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12350-015-0261-x