
Abstract
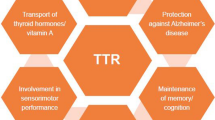
Transthyretin (TTR) (formerly, thyroxine binding prealbumin) is an evolutionarily conserved serum and cerebrospinal fluid protein that transports holo-retinol-binding protein and thyroxine. Its serum concentration has been widely used to assess clinical nutritional status. It is also well known that wild-type transthyretin and approximately 100 different mutants give rise to a variety of forms of systemic amyloid deposition. It has been suspected and recently established that TTR can suppress the Alzheimer’s disease phenotype in transgenic animal models of cerebral Aβ deposition. Thus, while TTR is a systemic amyloid precursor, in the brain it seems to have an anti-amyloidogenic effect. TTR is found in other organs as a result of local synthesis or transport, suggesting that it may have other, as yet undiscovered, functions. It is possible that its capacity to bind many classes of compounds allows it to serve as an endogenous detoxifier of molecules with potential pathologic effects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transthyretin (TTR) is a homotetrameric protein of 55 kDa found mainly in plasma and cerebrospinal fluid and previously thought to be synthesized only in liver, choroid plexus, retinal pigment epithelium, and pancreas [1–3]. In 1981, the International Union of Biochemists adopted the name “transthyretin” for the molecule previously designated as “thyroxine binding prealbumin” [4]. The implication of the action was that the physiologic role(s) of the protein was (were) sufficiently well known (transporter of thyroxine and retinol) to change the prior descriptive term to the more preferable functional nomenclature. It now appears that the assumption that the biology of TTR was completely understood was premature and may have led the scientific community to infer that further investigation was not necessary. Semantic clarity leading to undeserved observational certainty is a constant risk when trying to rationalize scientific nomenclature.
Until recently, TTR had its established clinical significance in two settings. TTR serum concentration has been utilized as a marker of nutritional/inflammatory status in a variety of conditions [5, 6]. More importantly, as an etiologic agent, TTR is one of the ∼30 human proteins associated with the amyloidoses, a group of disorders originally defined pathologically by the formation and aggregation of misfolded proteins which result in extracellular deposits that impair organ function [7]. The clinical syndromes associated with TTR aggregation are senile systemic amyloidosis (SSA), a late onset disease in which the wild-type protein deposits primarily in the heart, but also in the gut and carpal tunnel [8–10], and the familial amyloidotic polyneuropathies (FAP) and cardiomyopathies (FAC) in which point mutations in the gene encoding the protein result in deposition of aggregates in peripheral and autonomic nerves and heart, respectively [11]. FAP and FAC usually have an earlier onset than SSA presumably due to the decreased stability of variant TTR with respect to the wild-type protein, with the most aggressive variants depositing as early as the second decade of life [12].
In vitro studies have demonstrated that TTR forms amyloid fibrils through a process that involves tetramer dissociation followed by misfolding of the released monomers into an aggregation-prone conformation by which oligomers, soluble aggregates, insoluble amorphous aggregates, and amyloid fibrils are formed in a downhill polymerization process [13]. Whether the insoluble amorphous aggregates described first in transgenic animals [14] and then in humans [15] are in on- or off-pathway towards the amyloidogenic structure is still a matter of debate.
Tissue culture studies using cell lines derived from tissues that are targets of TTR deposition have shown that TTR monomers or small oligomers are cytotoxic to the cells, whereas the native tetramer, large soluble aggregates (>100 kDa), and amyloid fibrils are not. Furthermore, amyloidogenic TTR variants were toxic to the cells whereas non-amyloidogenic TTR was not [16]. These results support the notion that cell death in the protein misfolding diseases is caused not by amyloid fibrils, which are the end product of the aggregation process, but rather by the intermediate oligomers that are formed during that process. The results of earlier studies, in which either highly amyloidogenic mutant forms of TTR which did not occur in nature or tissue cultured cells which did not represent the disease associated in vivo targets were used, were in general agreement with such findings [15, 17].
Recently, we and others have reported that TTR has a protective role in transgenic murine models of Alzheimer’s disease (AD) [18, 19] and that the absence of the TTR protein leads to a discrete behavioral phenotype, even in the absence of a pathologic Aβ gene, which has not been fully characterized [20]. The relationship of these observations to human AD has not yet been established.
In the course of studies directed at finding small molecules that could stabilize the native tetrameric TTR conformation to prevent amyloid formation, it has become apparent that TTR has the capacity to bind a variety of structures in its thyroxine (T4) binding pocket. It is possible that evolution has maintained such versatility in order to allow TTR to perform a multiplicity of biologic functions. Here, we will explore such a possibility.
Transthyretin in the brain
The notion that our knowledge of TTR function was incomplete was suggested in 1982 in a report showing that the characteristic plaques from brains of AD patients were reactive with an anti-TTR antibody [21]. Although that observation could not be confirmed in larger series of AD brains, the seed had been sown. A subsequent study indicated that some TTR alleles could inhibit Aβ fibril formation in vitro, but the mechanism was not defined [22, 23]. The finding was consistent with contemporaneous independent experiments showing that wild-type TTR could alleviate the pathologic impact of Aβ expression in muscle cells in transgenic C. elegans expressing both human TTR and Aβ genes [24]. However, despite clear evidence of the effect, these studies did not demonstrate an in vivo molecular interaction. A later analysis of brain RNA from mice expressing a human AD transgene showed increased cerebral transcription of the TTR gene, cerebral staining with an anti-TTR antibody, and an increase in AD pathology in the ipsilateral hemibrains of mice injected intraventricularly with an anti-TTR antibody [25]. However, none of the papers reporting these observations speculated on the possibility that TTR was synthesized anywhere but the choroid plexus. A recent report reiterated the long-standing view that TTR was only synthesized in the choroid plexus, largely based on a failure to demonstrate TTR mRNA transcription from material obtained from normal and AD model murine brains by laser-capture dissection, and the authors suggested that contamination with choroid plexus was responsible for findings from other laboratories [26]. However, several of the conflicting studies showed TTR transcripts in different regions of the brain that had been carefully dissected free of choroid plexus [27].
Using a combination of genetic and biochemical techniques, our laboratory has definitively shown that the TTR–Aβ interaction suggested by the earlier studies is both real and relevant. In animals transgenic for wild-type human TTR, the behavioral and neuropathologic effects of a simultaneously expressed human AD gene are almost fully suppressed. In those animals, the residual Aβ plaques and the bulk of cortical and hippocampal neurons stained with an antibody to TTR. Similarly, human AD brains showed both intraneuronal and vascular TTR staining [18]. While it was possible that the staining reflected neuronal uptake of choroid plexus-synthesized TTR, subsequent studies of primary cultured neurons from such animals showed both TTR transcription by quantitative PCR and intracellular TTR demonstrated by immunofluorescence confirming neuronal synthesis of the molecule (Li and Buxbaum, unpublished). Consistent with TTR’s salutary effect on pathogenesis in the Aβ transgenic mice, the absence of TTR in the Aβ transgenics results in acceleration of the pathologic phenotype in a gene dose-dependent manner [18, 19].
It has also become clear in data from several laboratories that mice in which the endogenous TTR gene has been silenced have a discrete behavioral deficit accompanied by neuropathologic changes [18, 28]. Dorsal ganglia from such animals appear to be deficient in the neuronal response to injury, and their developing brains show variation from normal in the representation of proliferating and apoptotic neuronal populations in the subventricular zone of the brain [29, 30]. While it is still uncertain exactly how TTR operates in normal neurons, these data suggest that its function goes beyond its systemic role as a carrier of T4 and retinol. It should be noted, however, that some of the defects in learning found in the TTR knockout mice appear to be overcome by the administration of retinoic acid [31].
Data from our laboratory suggest that at least one of the functions of TTR is to reduce the toxicity of the Aβ oligomers or decrease their formation, perhaps having a generic chaperone-like function within neurons as well as in the extracellular space of the brain. Several studies, although not all those reported, have found decreased CSF TTR levels in patients with AD [32, 33]. The mechanism underlying this phenomenon and its relevance to clinical AD is unclear. Experiments investigating the response of choroid plexus TTR synthesis to the inflammatory agent LPS suggest that TTR produced in the choroid does not behave as a negative acute phase reactant [34]. Neuronal regulation of TTR synthesis has not yet been thoroughly explored. If it is more like that in the liver than in the choroid (i.e., if it behaves as a negative acute phase reactant), it is plausible that the observed decrease of TTR in CSF is due to the inflammatory process related to AD. Alternatively, a decrease in TTR transcription might contribute to the onset of AD because the amount of TTR available is not sufficient to bind enough Aβ, either intra- or extra-cellularly to prevent the formation of, or to sequester, toxic Aβ oligomers.
In vitro studies have suggested that TTR can bind Aβ1–40 and Aβ1–42 monomers, aggregates smaller than 100 kDa, and fibrils, although the relative affinities of the smaller and larger aggregates have not been established [18, 35]. Binding of the aggregates is better than that of monomers and it is better at 37°C than at 25°C. Murine TTR binds better than the human homolog for reasons that are still unclear. It also appears that the affinity of TTR is greater for Aβ1–42 than for Aβ1–40. In any case, while the mechanism is still not definitively known, present data show that TTR excess suppresses the AD phenotype presumably via a direct interaction with Aβ, whereas the absence of TTR accelerates its development.
Transthyretin in the plasma
TTR in plasma is synthesized and secreted by the liver. It is known to transport about 15% of the total T4 (100 nM; 65% being transported by thyroxine binding globulin and 20% by albumin [36]) and retinol through the formation of a complex with retinol binding protein (RBP). The ratio RBP:TTR in plasma is around 0.3 in healthy individuals [37, 38] indicating that most of the circulating TTR (0.15–0.3 mg/ml; 3–6 μM) remains free of ligand, thus without an apparent function.
Extensive in vitro studies have established that small molecules of a diverse variety of scaffolds (stilbenes, flavones, benzoxazoles, tetrahydroquinolines, dihydropyridines, benzodiazepines, and phenoxazines), which are able to bind TTR in the T4 binding pocket, kinetically stabilize the native quaternary structure and preclude TTR disassembly, misfolding, and aggregation [39–43]. Consistent with the in vitro effect, tissue culture studies have demonstrated that such compounds inhibit amyloidogenic TTR-induced cytotoxicity in cells derived from targets of TTR deposition in vivo [16, 44]. These small molecules are typically composed of two differentially substituted aromatic rings connected by linkers of variable chemical composition. They usually have polar substituents on their aryl rings, which interact with carboxyl or hydroxyl groups located in the T4 binding site [42]. Why would a cavity accepting so many structures exist? It is possible that natural products with similar chemical characteristics are present in the circulation as a result of dietary intake or other environmental exposure. They may also be generated as catabolic products of naturally occurring secondary metabolites. We hypothesize that TTR in the plasma may bind such molecules which are then transported to hepatocytes and catabolized or to the kidneys where they can be excreted. In fact, TTR amyloid deposition in the kidney (where transcription is negligible) has been reported in individuals having certain unstable TTR variants (e.g., V30M TTR). This observation is consistent with the notion that TTR is stabilized by its ligands and would be most prone to disassembly and amyloid formation at the moment it releases its ligands to the target organs [45].
A detoxifying function has also been suggested for albumin, which is the most abundant protein in the plasma (35–50 mg/ml) and contains four hydrophobic binding regions, which are able to bind metal ions, fatty acids, drugs, and hormones, and to trap or prevent free radical formation [46]. There are many similarities between serum albumin and TTR including molecular weight (~60,000 Da), low isoelectric point (<5.0), the ability to bind aromatic (hydrophobic) compounds, high stability, etc. It is possible that TTR and albumin are functionally similar in removing potentially toxic dietary substances or by-products of metabolism. Their differences in secondary structures, albumin being mainly α-helix whereas TTR being predominantly β-sheet, might provide a spectrum of complementary affinities for a variety of compounds of many different structures that require detoxification and excretion. A recent review based on crystallographic data reports six different binding sites in human serum albumin, two in each of its three domains [47]. Several compounds known to bind TTR in its T4 pocket also bind albumin, among them T4 itself, non-steroidal anti-inflammatory drugs like indomethacin, diflunisal, or ibuprofen, or agents like bromophenol blue, salicylates, and furosemide, among others [39, 47–50]. It appears that albumin has relative low affinity for all its ligands, and its biologic effectiveness comes from its abundance, whereas TTR displays higher affinity for its ligands than albumin but is less abundant in serum. Excluding RBP, which has contacts in three of the TTR subunits, all the known interactions of TTR with other molecules take place via the T4 binding site. It is not yet known whether the interaction of TTR with Aβ occurs in that pocket, although unpublished data from our laboratory suggest that this is not the case. Thus, an unknown site of TTR might be available for further interactions.
Transthyretin in the eye
TTR is highly transcribed and translated in retinal pigment epithelium (RPE), a monolayer of cells with tight junctions that serves as blood barrier for the retina [51]. RPE is important to maintain many functions of the retina including phagocytosis of the outer segment of photoreceptor cells, ion homeostasis, and the visual cycle transforming all-trans-retinol into 11-cis-retinal. 11-cis-retinal is shuttled from RPE to the rod cell through the interphotoreceptor matrix (IPM), where it binds to the protein opsin to form the visual pigment, rhodopsin. Absorption of a photon of light catalyzes the isomerization of 11-cis-retinal to all-trans-retinal and results in its release. The isomerization triggers a cascade of events leading to the generation of an electrical signal, which is ultimately interpreted as vision by the visual cortex. Once released, all-trans-retinal is converted to all-trans-retinol, which can be transported across the IPM to the RPE to complete the visual cycle. The source of all-trans-retinol in RPE is ultimately the liver which secretes retinol bound to the TTR–RBP complex. In RPE, free retinol uptake seems to be mediated by STRA6, a retinol-RBP cell membrane receptor which is highly expressed in the basolateral side of these cells [52].
In the eye, TTR and RBP mRNAs are only found in RPE cells, where the proteins are synthesized and secreted together from the apical side of the cell layer, i.e., the IPM [1]. These proteins, however, have also been found in other cells of the retina. It has been speculated that TTR–RBP in the IPM might serve as transporters of retinol to other cells, like retinal amacrine and Muller cells, which contain cellular retinoic acid binding protein [53] and presumably require all-trans-retinol as a precursor. In vitro, the TTR:RBP secretion ratio of RPE to the IPM is 50:1 [54]. If the same ratio exists in vivo it implies that there is an excess of TTR in the IPM. Some authors suggest TTR might also function as a carrier of T4 during the development of the eye [54].
Deposition of amyloid TTR in the vitreous is found in individuals with FAP and apparently in at least one individual without a mutation, although the latter study did not definitively identify the vitreous amyloid as being reactive with an anti-TTR antibody [55]. Variant TTR deposition in the eye is found even after liver transplantation (which replaces a variant TTR-producing liver with a wild-type TTR liver [56]. It is also known that the metabolic exchange and equilibration between the systemic circulation and the vitreous humor is very slow, thus the source of variant TTR deposition in the vitreous is probably the RPE. It is possible that the glycosaminoglycan (GAG) hyaluronic acid in the vitreous promotes mutant TTR deposition as some GAGs do for other amyloidogenic proteins, e.g., gelsolin [57]. As yet, the role of TTR in the vitreous has not been explained.
TTR is a minor component of Drusen, extracellular deposits of material found in the Bruch’s membrane, which make contact with RPE and the choroid of the eye and are increased in age-related macular degeneration (AMD) [58]. It is possible that in this case the source of TTR is the systemic circulation rather than the RPE. Circulating RBP-charged TTR may be predisposed to dissociation at the time of delivery of retinol to the RPE with the subsequent generation of monomeric TTR, the aggregate precursor. Alternatively, TTR may be behaving as it appears to function in neurons, i.e., by binding protein aggregates, and it is trapped in the Drusen when its concentration is lower than that required to prevent Drusen formation or enhance their clearance.
Transthyretin in the pancreas
TTR is produced in the islets of Langerhans; its mRNA has been detected in the glucagon-producing alpha cells. The protein is found to a lesser extent in the insulin- and amylin-producing beta-cells, perhaps as a result of endocytosis [3, 59]. TTR is stored in secretory vesicles in the alpha cells and perhaps released together with glucagon. It has been suggested that TTR may affect the processing of glucagon [60]. Non-fibrillar TTR immunostaining is increased in the beta cells of type 2 diabetes patients that have high amylin (IAPP) amyloid load. Interestingly, IAPP amyloid in the presence of TTR has different immunoreactivity than in the absence of TTR, suggesting that TTR might have a role in the formation or processing of these fibrils or the inhibition of those processes. Conversely, TTR amyloid has not been detected inside alpha cells or beta islets even in patients with familial TTR amyloid, nor in patients with IAPP amyloid. In FAP patients, TTR deposition was found only in the pancreatic exocrine parenchyma and surrounding connective tissue [61, 62].
There are some reports suggesting TTR may play a role in the pathogenesis of type 1 and type 2 diabetes, although the data are still difficult to interpret. In type 1 diabetes, related to the autoimmune destruction of beta cells, it seems that plasma TTR levels are lower than in controls, perhaps reflecting the inflammatory process producing the pathology [63, 64]. In type 2 diabetes where there is a reduced beta cell number and function, there are contradictory reports on the plasma TTR levels compared to controls. Some studies (with very small patient numbers) indicate that they are increased, while others suggest that they are decreased [65–67] (Buxbaum, unpublished). More studies with larger cohorts are necessary first to establish the facts and then to interpret them.
Transthyretin deposition
TTR deposition has been reported in heart, peripheral and autonomic nerve, kidney, vitreous, blood vessels, skin, gut, and other viscera, where TTR is not synthesized [11]. However, deposition in the liver, the main site of synthesis, apart from hepatic vessels, has not been described. It is possible that upon delivery of its cargo (retinol and T4) TTR is at its more vulnerable quaternary structure, i.e., the free tetramer which can disassemble into its monomers which can in turn misfold and aggregate. This notion would not be consistent with the fact that TTR circulates mainly as a free tetramer, unless there is a local increase in TTR concentration in the extracellular matrix (ECM) of these tissues and thus more monomeric TTR available for misfolding and aggregation. The effect of the ECM and its components on TTR aggregation and amyloid formation is a subject that has not received sufficient experimental attention although it has been studied intensively with respect to AA amyloid formation, deposition, and potential therapeutics [68, 69].
Summary
At this stage of our knowledge, it is certainly not appropriate to state that we know all there is to know about the biologic functions of TTR. The mechanism of its impact on normal neuronal function is not clear. While it may be independent of its function as a retinol carrier, sufficient data are not yet available to show that this is truly the case. The protein plays a role in pancreatic and retinal epithelial function. Whether it is critical in disease states affecting these organs is currently unknown, the available data being too fragmentary to draw definitive conclusions. What is quite striking is that TTR can be either an amyloid precursor or a defense against tissue amyloid fibrillogenesis depending on the cell of expression. The general mechanism of amyloid formation from either wild-type or mutant TTR precursor appears to be clear. The detailed or even general understanding of its functions in other organs is not yet in hand. Does it “chaperone” Aβ or pancreatic amyloidogenic precursors intra- or extracellularly? If so, what are the structural features that are responsible for those functions? It has often been said that the differences between law breakers and law enforcers is a very fine line; is the same true for amyloid precursors and molecules that are in fact anti-amyloidogenic?
References
Cavallaro T, Martone RL, Dwork AJ, Schon EA, Herbert J (1990) The retinal pigment epithelium is the unique site of transthyretin synthesis in the rat eye. Invest Ophthalmol Vis Sci 31:497–501
Herbert J, Wilcox JN, Pham KC, Fremeau RT, Zaviani M, Dwork A, Soprano D, Makover A, Goodman DS, Zimmerman EZ, Roberts JL, Schon EA (1986) Transthyretin: a choroid plexus-specific transport protein in human brain. Neurology 36:900–911
Jacobsson B, Collins VP, Grimelius L, Pettersson T, Sandstedt B, Carlstrom A (1989) Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J Histochem Cytochem 37:31–37
Goodman DS (1986) Statement regarding nomenclature for the protein known as prealbumin, which is also (recently) called transthyretin. In: Glenner GG, Osserman EF, Benditt EP, Calkins E, Cohen AS, Zucker-Franklin D (eds) Amyloidosis. Plenum, New York, pp 287–288
Ingenbleek Y, Young VR (2002) Significance of transthyretin in protein metabolism. Clin Chem Lab Med 40:1281–1291
Potter MA, Luxton G (2002) Transthyretin measurement as a screening tool for protein calorie malnutrition in emergency hospital admissions. Clin Chem Lab Med 40:1349–1354
Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD (2007) A primer of amyloid nomenclature. Amyloid 14:179–183
Pitkanen P, Westermark P, Cornwell GGIII (1984) Senile systemic amyloidosis. Am J Pathol 117:391–399
Kyle RA, Gertz MA, Linke RP (1992) Amyloid localized to tenosynovium at carpal tunnel release. Immunohistochemical identification of amyloid type. Am J Clin Pathol 97:250–253
Rocken C, Saeger W, Linke RP (1994) Gastrointestinal amyloid deposits in old age. Pathol Res Pract 190:641–649
Buxbaum JN (2007) Transthyretin and the transthyretin amyloidoses. In: Uversky VN, Fink A (eds) Protein misfolding, aggregation, and conformational diseases. Springer, Santa Cruz, pp 259–283
Jacobson DR, McFarlin DE, Kane I, Buxbaum JN (1992) Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet 89:353–356
Hurshman AR, White JT, Powers ET, Kelly JW (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43:7365–7381
Teng MH, Yin JY, Vidal R, Ghiso J, Kumar A, Rabenou R, Shah A, Jacobson DR, Tagoe C, Gallo G, Buxbaum J (2001) Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest 81:385–396
Sousa MM, Cardoso I, Fernandes R, Guimaraes A, Saraiva MJ (2001) Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol 159:1993–2000
Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: transthyretin monomers and non-native oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA 101:2817–2822
Andersson K, Olofsson A, Nielsen EH, Svehag SE, Lundgren E (2002) Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem Biophys Res Commun 294:309–314
Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T (2008) Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci USA 105:2681–2686
Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ (2007) Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). J Neurosci 27:7006–7010
Sousa JC, Grandela C, Fernandez-Ruiz J, de Miguel R, de Sousa L, Magalhaes AI, Saraiva MJ, Sousa N, Palha JA (2004) Transthyretin is involved in depression-like behaviour and exploratory activity. J Neurochem 88:1052–1058
Shirahama T, Skinner M, Westermark P, Rubinow A, Cohen AS, Brun A, Kemper TL (1982) Senile cerebral amyloid. Prealbumin as a common constituent in the neuritic plaque, in the neurofibrillary tangle, and in the microangiopathic lesion. Am J Pathol 107:41–50
Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D (2004) Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid 11:1–9
Schwarzman AL, Goldgaber D (1996) Interaction of transthyretin with amyloid beta-protein: binding and inhibition of amyloid formation. In: Bock GR, Goode JA (eds) The nature and origin of amyloid fibrils. Wiley, New York, pp 146–164
Link CD (1995) Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA 92:9368–9372
Stein TD, Johnson JA (2002) Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci 22:7380–7388
Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA (2006) Transthyretin and Alzheimer’s disease: where in the brain? Neurobiol Aging 28:713–718
Hovatta I, Schadt EE, Libiger O, Schork NJ, Lockhart DJ, Barlow C, Zapala MA, Broide RS (2007) DNA variation and brain region-specific expression profiles exhibit different relationships between inbred mouse strains: implications for eQTL mapping studies. Genome Biol 8:R25
Sousa JC, Marques F, Dias-Ferreira E, Cerqueira JJ, Sousa N, Palha JA (2007) Transthyretin influences spatial reference memory. Neurobiol Learn Mem 88:381–385
Fleming CE, Saraiva MJ, Sousa MM (2007) Transthyretin enhances nerve regeneration. J Neurochem 103:831–839
Richardson SJ, Lemkine GF, Alfama G, Hassani Z, Demeneix BA (2007) Cell division and apoptosis in the adult neural stem cell niche are differentially affected in transthyretin null mice. Neurosci Lett 421:234–238
Brouillette J, Quirion R (2008) Transthyretin: a key gene involved in the maintenance of memory capacities during aging. Neurobiol Aging 29:1721–1732
Serot JM (1997) Cerebrospinal fluid transthyretin: aging and late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry 63:506–508
Hansson SF, Andreasson U, Wall M, Skoog I, Andreasen N, Wallin A, Zetterberg H, Blennow K (2009) Reduced levels of amyloid-beta-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. J Alzheimers Dis 16:389–397
Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, Sousa N, Correia-Neves M, Palha JA (2009) Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab 29:921–932
Liu L, Murphy RM (2006) Kinetics of inhibition of beta-amyloid aggregation by transthyretin. Biochemistry 45:15702–15709
Schreiber G, Richardson SJ (1997) The evolution of gene expression, structure and function of transthyretin. Comp Biochem Physiol 116B:137–160
Shoji S, Nakagawa S (1988) Serum prealbumin and retinol-binding protein concentrations in Japanese-type familial amyloid polyneuropathy. Eur Neurol 28:191–193
Filteau SM (2000) Use of the retinol-binding protein: transthyretin ratio for assessment of vitamin A status during the acute-phase response. Br J Nutr 83:513–520
Baures PW, Peterson SA, Kelly JW (1998) Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem 6:1389–1401
Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW (2004) Diflunisal analogues stabilize the native state of transthyretin Potent inhibition of amyloidogenesis. J Med Chem 47:355–374
Johnson SM, Connelly S, Wilson IA, Kelly JW (2009) Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem 52:1115–1125
Johnson SM, Connelly S, Wilson IA, Kelly JW (2008) Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem 51:6348–6358
Johnson SM, Connelly S, Wilson IA, Kelly JW (2008) Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem 51:260–270
Reixach N, Adamski-Werner SL, Kelly JW, Koziol J, Buxbaum JN (2006) Cell based screening of inhibitors of transthyretin aggregation. Biochem Biophys Res Commun 348:889–897
White JT, Kelly JW (2001) Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc Natl Acad Sci USA 98:13019–13024
Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E (2008) The antioxidant properties of serum albumin. FEBS Lett 582:1783–1787
Varshney A, Sen P, Ahmad E, Rehan M, Subbarao N, Khan RH (2009) Ligand binding strategies of human serum albumin: how can the cargo be utilized? Chirality. doi:10.1002/chir.20709
Steinrauf LK, Cao Y, Hamilton J, Murrell J, Liepnieks JJ, Benson MD (1991) Preparation and crystallization of human transthyretin (prealbumin) variants. Biochem Biophys Res Commun 179:804–809
Stockigt JR, Lim CF, Barlow JW, Wynne KN, Mohr VS, Topliss DJ, Hamblin PS, Sabto J (1985) Interaction of furosemide with serum thyroxine-binding sites: in vivo and in vitro studies and comparison with other inhibitors. J Clin Endocrinol Metab 60:1025–1031
Larsen PR (1972) Salicylate-induced increases in free triiodothyronine in human serum. Evidence of inhibition of triiodothyronine binding to thyroxine-binding globulin and thyroxine-binding prealbumin. J Clin Invest 51:1125–1134
Pfeffer BA, Becerra SP, Borst DE, Wong P (2004) Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol Vis 10:23–30
Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H (2007) A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315:820–825
Gaur VP, De Leeuw AM, Milam AH, Saari JC (1990) Localization of cellular retinoic acid-binding protein to amacrine cells of rat retina. Exp Eye Res 50:505–511
Ong DE, Davis JT, O’Day WT, Bok D (1994) Synthesis and secretion of retinol-binding protein and transthyretin by cultured retinal pigment epithelium. Biochemistry 33:1835–1842
Barouch FC, Benson MD, Mukai S (2004) Isolated vitreoretinal amyloidosis in the absence of transthyretin mutations. Arch Ophthalmol 122:123–125
Ando Y, Terazaki H, Nakamura M, Ando E, Haraoka K, Yamashita T, Ueda M, Okabe H, Sasaki Y, Tanihara H, Uchino M, Inomata Y (2004) A different amyloid formation mechanism: de novo oculoleptomeningeal amyloid deposits after liver transplantation. Transplantation 77:345–349
Suk JY, Zhang F, Balch WE, Linhardt RJ, Kelly JW (2006) Heparin accelerates gelsolin amyloidogenesis. Biochemistry 45:2234–2242
Mullins RF, Russell SR, Anderson DH, Hageman GS (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14:835–846
Cras-Meneur C, Inoue H, Zhou Y, Ohsugi M, Bernal-Mizrachi E, Pape D, Clifton SW, Permutt MA (2004) An expression profile of human pancreatic islet mRNAs by serial analysis of gene expression (SAGE). Diabetologia 47:284–299
Westermark GT, Westermark P (2008) Transthyretin and amyloid in the islets of Langerhans in type-2 diabetes. Exp Diabetes Res 2008: Article ID 429274
Ando Y, Yi S, Nakagawa T, Ikegawa S, Hirota M, Miyazaki A, Araki S (1991) Disturbed metabolism of glucose and related hormones in familial amyloidotic polyneuropathy: hypersensitivities of the autonomic nervous system and therapeutic prevention. J Auton Nerv Syst 35:63–70
Nagasaka T, Togashi S, Watanabe H, Iida H, Nagasaka K, Nakamura Y, Miwa M, Kobayashi F, Shindo K, Shiozawa Z (2009) Clinical and histopathological features of progressive-type familial amyloidotic polyneuropathy with TTR Lys54. J Neurol Sci 276:88–94
Itoh N, Hanafusa T, Miyagawa J, Tamura S, Inada M, Kawata S, Kono N, Tarui S (1992) Transthyretin (prealbumin) in the pancreas and sera of newly diagnosed type I (insulin-dependent) diabetic patients. J Clin Endocrinol Metab 74:1372–1377
Refai E, Dekki N, Yang SN, Imreh G, Cabrera O, Yu L, Yang G, Norgren S, Rossner SM, Inverardi L, Ricordi C, Olivecrona G, Andersson M, Jornvall H, Berggren PO, Juntti-Berggren L (2005) Transthyretin constitutes a functional component in pancreatic beta-cell stimulus-secretion coupling. Proc Natl Acad Sci USA 102:17020–17025
Dowling P, Shields W, Rani S, Meleady P, Henry M, Jeppesen P, O’Driscoll L, Clynes M (2008) Proteomic analysis of conditioned media from glucose responsive and glucose non-responsive phenotypes reveals a panel of secreted proteins associated with beta cell dysfunction. Electrophoresis 29:4141–4149
Sundsten T (2006) The use of proteomics in identifying differentially expressed serum proteins in humans with type 2 diabetes. Proteome Sci 4:22. doi:10.1186/1477-5956-4-22
Sundsten T, Ostenson CG, Bergsten P (2008) Serum protein patterns in newly diagnosed type 2 diabetes mellitus—influence of diabetic environment and family history of diabetes. Diabetes Metab Res Rev 24:148–154
Kisilevsky R (2000) The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000. Amyloid 7:23–25
Kisilevsky R, Szarek WA, Ancsin J, Vohra R, Li Z, Marone S (2004) Novel glycosaminoglycan precursors as antiamyloid agents: part IV. J Mol Neurosci 24:167–172
Acknowledgements
This work was supported by National Institute on Aging Grants R01 AG30027 (to J.N.B.) and R01 AG19259 (to J.N.B.), the W. M. Keck Foundation (to J.N.B.) and The American Heart Association BGIA award 0865061F (to N.R.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Buxbaum, J.N., Reixach, N. Transthyretin: the servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101 (2009). https://doi.org/10.1007/s00018-009-0109-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0109-0