
Abstract
Results of recent genetic and immunologic studies have brought to the forefront several biologic pathways that allow for a better understanding of the mechanisms of tissue homeostasis, on the one hand, and inflammatory bowel disease (IBD) on the other. The explosion of research activity as a result of these newly identified targets is bringing the pathogenesis of these complex disorders into focus as well as creating new therapeutic opportunities. The greatest advances with perhaps the largest impact on our understanding of the etiology of Crohn’s disease are those related to bacterial sensing, such as through nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and its relationships to autophagy and the unfolded protein response as a consequence of endoplasmic reticulum stress. Interestingly, it appears as though these pathways, which are rooted in microbial sensing and regulation, are interrelated. Genetic studies have also renewed interest in previously studied pathways in IBD, such as the formation and function of the inflammasome and its relationship to interleukin (IL) 1-beta signaling. With the recent success of therapeutic agents designed to block tumor necrosis factor, the IL-12/23 pathways, and lymphocyte homing, insights have been gained into the biologic relevance and impact of these various inflammatory pathways in IBD. In this review, the exciting recent advances in these biologic pathways of IBD are discussed, particularly in light of their therapeutic relevance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many recent advances have been made in our understanding of the pathogenesis of inflammatory bowel disease (IBD). While the clinical phenotypes have been well characterized, the molecular basis behind this group of disorders is now coming into focus. In this review, we provide insights into some of the biologic pathways that have been the source of the most recent discoveries in IBD research and their relationship to the development of new therapeutic approaches to disease. Pathways for bacterial sensing and response, inflammasome regulation, inflammatory cytokines, and lymphocyte homing and recirculation will be discussed. In this article we do not extensively cover recent advances in genetics and the intestinal microbiota nor provide a comprehensive discussion of all of the immunologic advances which have been recently reviewed elsewhere [1–3].
Pathways of bacterial sensing and response
Genetic studies have brought nucleotide-binding oligomerization domain-containing protein 2 (NOD2), autophagy, and the endoplasmic reticulum (ER) stress responses to the forefront of IBD research. NOD2, which is expressed on macrophages, dendritic cells, and intestinal epithelial cells (IECs), is an intracellular sensor of bacterial peptidoglycan as well as of ligands from mycobacteria and viruses whose activation leads to NFκB induction and the release of inflammatory cytokines and chemokines. The common genetic variants of NOD2 found in patients with Crohn’s disease (CD) constitute the greatest known susceptibility to CD and abrogate this signaling [4]. While a complete understanding of how NOD2 leads to disease pathogenesis is not fully understood, it is known that NOD2 may also modulate Toll-like receptor (TLR) signaling [5–7] and affect the expression of α-defensins in Paneth cells [8, 9]. This decreased expression of defensins renders the host susceptible to model pathogens, as has been shown for Listeria monocytogenes [8].
The discovery that functional polymorphisms associated with ATG16L1 are also associated with CD [10] introduced the concept that autophagy also contributes to disease pathogenesis. This is a well-conserved regulatory process by which protein and organelle turnover occurs in cells by autodigestion through lysosomal degradation [11]. In periods such as starvation, oxidative stress, or infection, autophagy is increased. In studies of mice where the ATG16L1 gene is absent, autophagy of cell contents is impaired by a failure to assemble the proper destructive machinery [12]. The defense mechanism against invading Salmonella typhimurium is also rendered inadequate by dysfunctional autophagosomes in cultured human epithelial cells expressing the common variant of ATG16L1 found in CD [13]. Dysfunctional autophagy leads to phenotypic and transcriptional defects in Paneth cells, further suggesting that these secretory IECs may be particularly vulnerable to genetic mutations that affect these pathways and contribute to IBD.
Genetic models have led to a third novel pathway in IBD which is the unfolded protein response (UPR). The UPR is induced by ER stress and caused by the accumulation of misfolded or unfolded proteins within the ER [14]. The UPR is controlled by three distinct pathways, among which the inositol-requiring enzyme (IRE)–X-box binding protein 1 (XBP1) pathway has been the most studied in CD and ulcerative colitis (UC). Deletion of XBP1 in intestinal epithelial cells leads to spontaneous enteritis, sensitivity to colitis induction by dextran sodium sulfate (DSS), increased ER stress in the IEC compartment, apoptosis of Paneth cells and reduction in the number of goblet cells, and finally impaired defense to Listeria monocytogenes [15]. Analysis of single nucleotide polymorphisms (SNPs) in the XBP1 locus has demonstrated an association between XBP1 and both UC and CD. Deep sequencing of the XBP1 gene in a large cohort of CD, UC and control patients have further shown that the association signal is likely derived from hypomorphic rare variants within the coding region of the gene [15]. This suggests that genetic risk factors within the proper environmental context may lead to spontaneous intestinal inflammation which emerges directly from abnormalities in IECs.
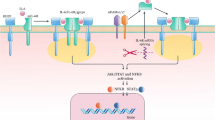
These pathways highlight the common importance for appropriate sensing and responding to commensal bacteria in the maintenance of homeostasis and the avoidance of unrestrained intestinal inflammation. Moreover, recent evidence further suggests that these aforementioned pathways may be convergent (Fig. 1). NOD2 and autophagy are linked through the recognition and clearance of invading intracellular bacterial pathogens. Epithelial cells that express the most common mutation found in NOD2 in association with CD display a profound defect in the autophagic destruction of intracellular bacteria [16]. Activation of NOD2 caused by these invading microbes appears to direct ATG16L1 to the plasma membrane adjacent to the point of bacterial entry in order to coordinate the clearance of the organism. In dendritic cells harboring either NOD2 or ATG16L1 CD susceptibility mutations, intracellular bacterial clearance was found to be similarly hampered by defective autophagy, which ultimately affected antigen presentation to CD4+ T cells [17].

Pathways that lead from intestinal homeostasis to inflammatory bowel disease. Homeostasis in the gut is altered by perturbations in the sensing of microbes by intestinal epithelia and subjacent immune cells by a variety of pattern recognition receptor systems, such as nucleotide-binding oligomerization domain-containing protein 2 (NOD2) and Toll-like receptors (TLR). This leads to cellular responses that place stress on immune and parenchymal cells, leading to adaptive unfolded protein responses (UPR) and autophagy. When maladapted, these pathways can lead to the inappropriate activation of inflammatory pathways (e.g. inflammasomes) and excessive release of inflammatory signals [e.g., interleukin (IL)-12/23, tumor necrosis factor (TNF), IL-1β, and TNF-like factor TL1A]. ER Endoplasmic reticulum
Autophagy is also linked to UPR and ER stress. ER stress induced in various cancer line cells results in the appearance of autophagosomes and autophagy machinery that are not observed when a c-Jun N-terminal kinase (JNK), downstream of the IRE1–XBP1 pathway, or IRE1 are absent [18, 19]. Evidence also exists to support the concept that impaired autophagy may lead to ER stress. This is the case in animal models of obesity that display decreased levels of autophagy proteins and henceforth have increased ER stress [20]. How precisely autophagy affects ER stress and vice versa in IECs needs to be established in intestinal model systems.
It must be pointed out that IECs are particularly susceptible to defects in autophagy and ER stress. While the exact reasons for this are unknown, it appears as though more complex requirements exist at this interface between the intestinal microbiota and the mucosal compartment. For example, IECs express an additional isoform of IRE1, called IRE1β, that may act to further protect IECs from ER stress in the setting of inflammation [21]. One can speculate therefore that the IECs have evolved to protect the host from bacteria or perhaps allow the host to be capable of adapting to local changes in the luminal environment as they are required.
There is further evidence that the bacterial sensing and regulation pathways that affect CD are linked to a common biologic pathway. The animal studies described above that result in hypomorphic NOD2, ATG16L1, and XBP1 function impair IEC Paneth cell function, resulting in deficient α-defensin production and impaired anti-microbial responses. Also gleamed from these studies is the notion that IECs and macrophages with hypomorphic NOD2, ATG16L1, and XBP1 function exhibit an excessive inflammatory response [e.g., tumor necrosis factor-alpha (TNF-α) production] in response to microbial products. Similarly, myeloid cells expressing the CD variants for NOD2 or lacking ATG16L1 respond to bacterial stimuli with an increased inflammasome response. Therefore, the consequence of genetic abnormalities in these biologic pathways may lead to inappropriate stimulation of anti-microbial and inflammasome systems that in turn perpetuate a chronic inflammatory environment. These defects have significant consequences for the relationship between the host and commensal microbiota and likely explain many previous findings in this area. For example, CD patients have been shown to harbor increased bacteria within IECs [22], including adherent-invasive Escherichia coli [23, 24]. Interestingly, this bacterium was found to be increased in autophagosomes of ATG16L1-deficient cells [25]. Furthermore, alterations in these pathways may result in overall changes to the composition of the microbiota. With the development of improved molecular techniques, including 16 rRNA sequencing and metagenomic sequencing, it is now possible to characterize complex microbiomes [26]. Such studies have produced evidence indicating that the intestinal microbiomes of patients with IBD may be substantially altered. Subgroups of patients with IBD appear to contain fewer overall bacterial genes, particularly those in the “protective” Firmicutes and Bacteroidetes phyla [27]. Whether these changes in the microflora cause inflammation or are a consequence of inflammation has not yet been determined, and current knowledge may suggest that the etiology of inflammation lies somewhere in between.
Lymphocyte homing and recirculation
Recent studies from experimental animal models of IBD, genome-wide association studies (GWAS), and success in the treatment of patients with CD with therapeutic antibodies to α4-integrins have brought forward the importance of homing pathways which coordinate the movement of lymphocytes into the site of intestinal inflammation. A meta-analysis of GWAS data in patients with CD [28] has identified an association between disease and CCR6. Chemokine receptor and ligand interactions are important for the process of migration of lymphocytes from the vasculature to the tissue microenvironment in inflammation. Chemokine receptor 6 (CCR6) is expressed on Th17 cells and interacts with its ligand CCL20 on IECs. CCL20 expression is increased in inflamed intestinal segments of patients with IBD [29], and the Th17 cells in Ccr6 −/− mice exhibit abnormal migration patterns. These mice consequently develop less severe DSS-induced colitis [30]. However, it is important to note that these mice are also more susceptible to trinitrobenzene sulfonic acid (TBS)-induced colitis, suggesting different roles of this regulatory chemokine pathway in different models of intestinal inflammation. These seemingly disparate results in two models of experimental colitis also underscore the potential for expression of this chemokine receptor and ligand on different inflammatory cells in different tissue compartments which carry out different functions. In this regard, CCR6 is also expressed on Tregs [31]. Nonetheless, with the success of a blocking antibody directed against CCL20 [32] and with the results of a GWAS implicating the CCR6 locus in CD, it is conceivable that pharmacologic blockade of this pathway may be a therapeutic possibility in select patients.
With this in mind, natalizumab is being increasingly used to treat patients with moderate to severe CD based on a large clinical trial that showed therapeutic responses in a select group of patients [33]. This drug is a fully humanized monoclonal antibody directed towards the α4 integrin subunit contained in the mucosal integrin α4β7. Integrins are a group of heterogeneous transmembrane molecules expressed on multiple surfaces, including leukocytes where they serve as adhesion molecules for endothelium. They are composed of an alpha and beta subunit and the α4β7 integrin homes leukocytes (except for neutrophils) to inflamed tissue compartments of the gut. The natalizumab pharmacologic approach may be problematic due to unintended blockade of integrin-driven leukocytes homing to tissues outside of the gut, such as the brain. Although clinical trials show this agent to be efficacious in the treatment of multiple sclerosis through the blockade of α4β1 on brain-homing lymphocytes, this may result in progressive multifocal leukoencephalopathy, polyoma virus infection [34]. This infection occurs in 1:1000 patients receiving natalizumab, including those receiving this drug for CD. Therefore, clinical trials are underway to investigate gut-specific antibodies targeting components of this integrin pathway (α4β7, MADCAM1, the endothelial ligand for α4β7, and the β7 subunit). It is also possible that blockade of lymphocyte homing may result in an undesired deficiency of regulatory cells, such as Tregs; therefore, more specific targets aimed at integrins expressed on pro-inflammatory immune cells are most desired.
While the process of homing lymphocytes to sites of inflammation has been extensively studied, a relatively new area of investigation in IBD is the process of resolving inflammation. It appears as though this is a coordinated and active process in order to prevent excessive and collateral damage, to provide defense against pathogens that may take advantage of this environment, and to rid the tissue of unwanted inflammatory cells, necrotic cells, and other debris. The family of lipid-derived signaling molecules, including the lipoxins, resolvins, and protectins, play an increasingly recognized role in this process, and their functions have been recently reviewed [35].
In this light, it is important to highlight an interesting pathway associated with the resolution of intestinal inflammation that is linked to an environmental factor. Short chain fatty acids (SCFAs) are derived from the microbial fermentation of dietary fibers and bind to G-coupled receptor 43 (GPR43) that shows enhanced activity on certain immune cells, such as neutrophils, and has been linked to receptors associated with the innate immune response [36]. Interestingly, Gpr43 −/− mice exhibit a markedly delayed recovery to DSS-induced colitis due to a lack of response of their neutrophils to SCFAs, which normally induce the migration of neutrophils out of the inflamed tissues. This mechanism is highly similar to that of resolvins, which utilize an orthologous receptor, chem23, which is structurally similar to GPR43. When SCFAs are cultured with isolated human peripheral blood mononuclear cells, there is an increased production of anti-inflammatory prostaglandin-E2 and a decreased expression of lipopolysaccharide (LPS)-induced inflammatory cytokines [37]. From a clinical perspective, SCFAs in the form of n-butyrate have been found to be increased in the stools of patients with IBD, suggesting a possible inadequate response in the gut [38]. In another study, a 4-week treatment with germinated barley foodstuff was found to reduce clinical activity scores compared with placebo and to cause an increase of beneficial bacteria in the colon such as Bifidobacterium [39].
Inflammasome regulation
Inflammasomes are composed of an intracellular complex of proteins that assemble in myeloid cells and play an important role in the innate immune system through recognition of damage-associated molecular patterns. Activation of the inflammasome ultimately results in the formation of cytokines, such as interleukin (IL)-1β, IL-18, and IL-33, to carry out various inflammatory and regulatory immune functions [40]. In general, these proteins are made as nonfunctional pro-proteins that must be cleaved by caspase-1 derived from the inflammasome to become active. In IBD, perturbation of the intestinal epithelial barrier might activate and potentiate the action of the inflammasomes that are poised to recognize pathogens and other “warning signals”. NOD-like receptor family pyrin containing 3 (NLRP3) encodes cryopyrin which activates caspase-1 to ultimately coordinate IL-1β production. Gain-of-function mutations in the nucleotide–oligomerization binding domain (NOD) component of NLRP3 results in the overproduction of IL-1β and consequently rare fever-associated inflammatory diseases, including familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disease [41]. Interestingly, a recent candidate gene approach study identified NLRP3 as a susceptibility factor for CD [42], resulting in yet more interest being directed towards the inflammsome in IBD. In contrast to the three rare febrile diseases just mentioned, LPS-stimulated monocytes harboring CD susceptibility SNPs in NLRP3 show a decreased production of IL-1β. This is analogous to susceptibility mutations in NOD2 that result in a loss of function and decreased IL-1β. A recent study examined the inflammasome function composed of another NOD-like receptor family pyrin member, NLRP6. In this study, Nlrp6 −/− mice harbored decreased levels of IL-18 in intestinal tissues and developed a dysbiosis with an accumulation of Bacteroidetes sp. that could be transmissible to wild-type mice and that conferred increased susceptibility to DSS-induced colitis [43].
These studies show how the loss of inflammasome function and, consequently, the production of cytokines, such as IL-1β, under homeostatic conditions result in a disabled relationship between the epithelial barrier and the commensal microbiota. This accumulated data also raise the possibility that defective processing of IL-1β contributes to the initiation of IBD. Alternatively, IBD is a chronic process, which could suggest that continued and inappropriate activation of caspase-1 and subsequent production of active IL-1β could perpetuate IBD. There is evidence gleamed from the study of intestinal specimens of patients with IBD that indeed IL-1β and IL-18 may be elevated in patients with certain forms of IBD [44–46]. Furthermore, animal studies of experimental colitis where IL-1 [47] or IL-18 [48] are blocked result in attenuated disease. Although it may be difficult to reconcile the different functions of the inflammasome pathway in IBD, several balancing factors are likely at play in disease pathogenesis during homeostasis versus chronic inflammation.
Inflammatory cytokine pathways
The success of antibodies directed towards TNF in the treatment of moderate to severe forms of CD and UC clearly identifies the centrality of this cytokine in IBD pathogenesis. Despite this knowledge, the mechanisms by which TNF may act in IBD have not been clearly determined, and genetic studies have not definitively implicated defects in TNF as conferring risk for these diseases. It may therefore be inferred that TNF has broad reaching roles in IBD pathogenesis or that it represents the final event in converging pathways (Fig. 1). Nevertheless, it may be important to study mechanisms of TNF activity based on what is understood from clinical practice using anti-TNF agents and from the study of the TNFΔ ARE (AU-rich element in the 3′ untranslated region that is important in TNF expression) mice that develop a spontaneous CD-like transmural ileo-colonic inflammation and ulceration with granulomas [49, 50]. For example, a TNF receptor, TNFR1, is expressed on the mesenchymal cells adjacent to IECs and may regulate mediators in parenchymal destruction, such as matrix metalloproteinases [50]. As such, this ability of the anti-TNF blockade to target innate pathways which might be shared between CD and UC may explain why TNF blockade as a therapy may succeed in not only CD but also in the more superficial inflammation of UC. Another possible activity of TNF that may establish shared mechanisms in both forms of IBD is its role in modifying epithelial apoptosis [51]. An examination of tissues in patients with CD suggests that apoptosis is increased in active disease and decreased with anti-TNF treatment. Reduced apoptosis by anti-TNF agents may at least also partially explain the clinical observation that these agents function to “heal the mucosa” [52].
Other TNF family members have been implicated in IBD, including TNF ligand-related molecule 1A (TL1A), which is encoded by TNFSF15. Interestingly, polymorphisms in this gene confer varying levels of risk in diverse ethnic populations, with Japanese and Korean populations being the most susceptible [53, 54]. TL1A is a TNF-like cytokine that is released by antigen-presenting cells and T cells and requires innate immune stimuli, such as TLR signaling. TL1A acts on T cells through the death receptor-3 (DR3) to ultimately help to induce proliferation of both Th1 and Th17 cells in an inflammatory milieu [55]. TL1A antibodies are effective in dampening colonic inflammation in DSS-treated mice and spontaneous intestinal inflammation in SAMP1/yit mice [56, 57]. Therefore, it is conceivable that blockade of TL1A may have therapeutic benefit in both forms of IBD in Asian populations.
The IL-12/IL-23 pathway is also of interest as it has been linked to CD and UC after the first GWAS detected polymorphisms in IL23R that conferred protection from disease [28]. Several drugs manufactured to block the components of this pathway are currently in clinical trials or in current practice so that an understanding of the biology in disease pathogenesis is important. IL-12 and IL-23 comprise subunit dimers-p35 for IL-12 and p19 for IL-23, as well as a p40 subunit that is common to both. These cytokines are produced by dendritic cells and function largely to induce and maintain Th1 (for IL-12) and Th17 (for IL-23) responses [58]. Blockade of IL-12 in TBS-induced colitis leads to amelioration of inflammation, thereby demonstrating the importance of this pathway in IBD [59]. The role of IL-23 has been studied in a CD-like animal model of colitis (Rag2-deficient animals treated with anti-CD40 antibody) that display an exuberant production of Th1 cytokines, such as TNF and interferon alpha (INFα) as well as IL-12p40. In one study, blocking antibodies to the various subunits of IL-12 and IL-23 were used in a systematic manner in order to understand the roles of each component of the IL-12/23 pathway in intestinal inflammation [60]. A key finding to come out of all these studies was that the p19 subunit that is unique to IL-23 is necessary for colitis in this experimental model. Furthermore, Th17 expression was found to be markedly increased and dependent on IL-23. These results from animal studies indicate that inflammatory signals derived from innate antigen-presenting cells (e.g., dendritic cells) drive the chronic inflammation seen in IBD and that Th17 via IL-23 may be an important mediator in this process. The role of the Th17 pathway in IBD has been recently reviewed [55].
Based upon these studies and the efficacy of ustekinumab (humanized monoclonal antibody directed at the IL-12/IL-23-shared p40 subunit) in patients with psoriasis [61, 62], it has been predicted that this drug would have similar efficacy in patients with CD. Although remission rates in phase II studies did not demonstrate significant differences compared with placebo through blockade of IL-12p40 [63], there are several other drugs designed to block the components of the IL-12/IL-23 pathway currently in clinical development.
One novel inflammatory cytokine pathway that may play an important role in the pathogenesis of UC is the Th2 cytokine IL-13. A chronic model of oxalozone-induced colitis, which phenotypically resembles UC, showed that lamina propria natural killer T (NKT) cells that are restricted to CD1d produce increased IL-13 that is necessary for the development of inflammation and can attenuate the colitis if blocked [64]. In human studies, stimulated lamina propria T cells from patients with UC produce increased amounts of IL-13 compared to the T cells from patients with CD that produce small amounts [65]. This cytokine was subsequently found to augment the NKT cell-mediated cytolysis of HT-29 epithelial cells after LPS stimulation. Furthermore, IL-13 directly alters the electrical resistance of these cultured epithelial monolayers, in part due to increased apoptosis and changes in tight junction proteins, such as claudins [66]. Taken together, these data suggest that IL-13 derived from NKT cells works at the level of the superficial mucosal compartment to induce and/or potentiate chronic colitis observed in patients with UC [66]. In a recent proof-of-concept clinical study on the use of INF-β1a therapy in patients with UC, a positive response to this agent correlated with decreased levels of IL-13 from lamina propria T cells compared to those observed in non-responders [67].
Conclusions
Our understanding of the pathogenesis of IBD has been significantly advanced by the discovery of genes and their immunologic pathways that confer risk in IBD. It has become clear that biologic pathways, such as those rooted in microbial sensing, are central to both the initiation and perpetuation of IBD. Many of these pathways are linked in a manner consistent with the complex interplay of often redundant systems that ultimately control homeostasis in the gut. In any pathway, perturbations at the level of the genes, environment (e.g., microbes), and/or altered immune responses disrupts homeostasis and ultimately determines the phenotype of the patient with IBD (Fig. 1). The most effective therapeutic option will target a molecule that represents the “final common pathway” for IBD, whatever that may be and which may vary among patients, highlighting the syndromic nature of these complex disorders.
References
Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474(7351):307–17.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9(5):313–23.
Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621.
Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14(24):2217–27.
Watanabe T, Kitani A, Strober W. NOD2 regulation of Toll-like receptor responses and the pathogenesis of Crohn’s disease. Gut. 2005;54(11):1515–8.
Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5(8):800–8.
Hedl M, Li J, Cho JH, Abraham C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci USA. 2007;104(49):19440–5.
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–4.
Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102(50):18129–34.
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–11.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8.
Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One. 2008;3(10):e3391.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Natl Rev Mol Cell Biol. 2007;8(7):519–29.
Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134(5):743–56.
Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4(4):337–49.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–31.
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171(2):513–24.
Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–78.
Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH et al. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J Clin Invest. 2001;107(5):585–93.
Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122(1):44–54.
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–21.
Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115(6):1405–13.
Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12(1):99–113.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65.
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104(34):13780–5.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40(8):955–62.
Kaser A, Ludwiczek O, Holzmann S, Moschen AR, Weiss G, Enrich B et al. Increased expression of CCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24(1):74–85.
Varona R, Cadenas V, Flores J, Martinez AC, Marquez G. CCR6 has a non-redundant role in the development of inflammatory bowel disease. Eur J Immunol. 2003;33(10):2937–46.
Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008;181(12):8391–401.
Katchar K, Kelly CP, Keates S, O’Brien MJ, Keates AC. MIP-3alpha neutralizing monoclonal antibody protects against TNBS-induced colonic injury and inflammation in mice. Am J Physiol Gastrointest Liver Physiol. 2007;292(5):G1263–71.
Sandborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC et al. Natalizumab induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2005;353(18):1912–25.
Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–8.
Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–61.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6.
Cox MA, Jackson J, Stanton M, Rojas-Triana A, Bober L, Laverty M, et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J Gastroenterol. 2009;15(44):5549–57.
Treem WR, Ahsan N, Shoup M, Hyams JS. Fecal short-chain fatty acids in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1994;18(2):159–64.
Kanauchi O, Suga T, Tochihara M, Hibi T, Naganuma M, Homma T et al. Treatment of ulcerative colitis by feeding with germinated barley foodstuff: first report of a multicenter open control trial. J Gastroenterol. 2002;37(Suppl 14):67–72.
Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707-35.
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet. 2009;41(1):71–6.
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–57.
Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol. 1995;154(5):2434–40.
Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol. 1999;163(1):143–7.
Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF Jr, Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162(11):6829–35.
Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest. 1990;86(3):972–80.
Sivakumar PV, Westrich GM, Kanaly S, Garka K, Born TL, Derry JM, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut. 2002;50(6):812–20.
Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10(3):387–98.
Armaka M, Apostolaki M, Jacques P, Kontoyiannis DL, Elewaut D, Kollias G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–7.
Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53(9):1295–302.
Rutgeerts P, Vermeire S, Van Assche G. Mucosal healing in inflammatory bowel disease: impossible ideal or therapeutic target? Gut. 2007;56(4):453–5.
Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum Mol Genet. 2005;14(22):3499–506.
Yang SK, Lim J, Chang HS, Lee I, Li Y, Liu J, et al. Association of TNFSF15 with Crohn’s disease in Koreans. Am J Gastroenterol. 2008;103(6):1437–42.
Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1756–67.
Bamias G, Mishina M, Nyce M, Ross WG, Kollias G, Rivera-Nieves J et al. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc Natl Acad Sci USA. 2006;103(22):8441–6.
Takedatsu H, Michelsen KS, Wei B, Landers CJ, Thomas LS, Dhall D, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135(2):552–67.
Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev. 2004;202:96–105.
Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182(5):1281–90.
Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25(2):309–18.
Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008;371(9625):1665–74.
Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371(9625):1675–84.
Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, et al. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology. 2008;135(4):1130–41.
Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 2002;17(5):629–38.
Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113(10):1490–7.
Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129(2):550–64.
Mannon PJ, Hornung RL, Yang Z, Yi C, Groden C, Friend J, et al. Suppression of inflammation in ulcerative colitis by interferon-beta-1a is accompanied by inhibition of IL-13 production. Gut. 2011; 60(4):449–55.
Acknowledgments
The authors acknowledge support from the U.S. National Institutes of Health and Grants DK044319, DK051362, DK053056, and DK088199 and from the Harvard Digestive Diseases Center DK034854 (RSB and MJH) and the Crohn’s and Colitis Foundation of America (MJH).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hamilton, M.J., Snapper, S.B. & Blumberg, R.S. Update on biologic pathways in inflammatory bowel disease and their therapeutic relevance. J Gastroenterol 47, 1–8 (2012). https://doi.org/10.1007/s00535-011-0521-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-011-0521-8