
Abstract
The p53-related protein p63 has pleiotropic functions, including cell proliferation, survival, apoptosis, differentiation, senescence, and aging. The p63 gene is expressed as multiple isoforms that either contain an N-terminal p53-homologous transactivation domain (TAp63) or that lack this domain (ΔNp63). Multiple studies have demonstrated that p63 plays a crucial role in stratified epithelial development, and have shown the importance of p63 for maintaining proliferation potential, inducing differentiation, and preventing senescence. Additionally, much research focuses on the role of p63 in cancer progression. Clinical evidence suggests that p63 may play a role in inhibiting metastasis. Similarly, genetic mice models together with cell culture data strongly indicate that p63 deficiency may be a causative factor for metastatic spread. Moreover, the role of p63 in cancer metastasis has been shown to be greatly related to the ability of mutant p53 to promote cancer malignancy. However, there is still much confusion as to what the role of each specific isoform is. In this review, we highlight some of the major findings in the current literature regarding the role of specific p63 isoforms in development, tumorigenesis, and particularly in cancer metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
General Introduction
The tumor suppressor protein p53 is frequently altered in human cancers. At present, p53 is recognized as one of the most important tumor suppressor proteins, and much research focuses on reconstituting p53 function in order to effectively treat cancer patients. In simple words, p53 protects the organism from damaged and potentially life-threatening cells. Upon DNA damage or other cellular stresses, such as oxidative stress, hypoxia, or carcinogen exposure, p53 levels accumulate and become activated. Then, p53 may direct a variety of responses, including metabolic homeostasis, antioxidant defense, DNA repair, cell cycle arrest, senescence, and apoptosis, depending on the severity of the damage. The specific response depends on whether the damage can be repaired or if it is too severe and requires death of the cell in order to maintain tissue homeostasis (for reviews see [1, 2]).
Today, after more than 30 years of research on p53, the story has become more complex, albeit even more fascinating, with the discovery in the late 1990’s of two p53 family members, p63 [3] and p73 [4]. These three proteins, encoded by the TP53, TP63, and TP73 genes (Trp53, Trp63, and Trp73 in mice, respectively), are transcription factors that bind directly to DNA as tetramers, interact with other transcription factors and the transcription machinery, and together control the expression of thousands of genes involved in all aspects of life. From early on, it has been clear that p63 and p73 are involved in a broad spectrum of biological activities, including cell proliferation, survival, apoptosis, development, differentiation, senescence, and aging. In particular, p63 has emerged as a critical player in embryonic development, epithelial stem cell maintenance, and differentiation. In cancer biology, p63 has been shown to be involved in all aspects of tumorigenesis and cancer progression (for reviews see [5, 6]). From clinical correlations to mechanistic studies, accumulating evidence shows the importance of p63 in preventing cancer metastasis. In this review, we discuss the main functions of p63 in normal and cancer physiology, with an added emphasis on the mechanistic role of p63 as a metastasis suppressor.
The p53 Family
The p53 family of proteins is evolutionarily conserved in animals from worms to humans. Invertebrate organisms like C. elegans and D. melanogaster express a p53 homologue (Cep-1 in C. elegans and Dmp53 in D. melanogaster) that resembles p63 more than p53. Gene duplication gave rise to a new p53-like protein in cartilaginous fish, while bony fish and other vertebrates express all three family members. Phylogenetic and functional analyses suggest that p63 is the founding member of the family, followed by p73, and finally by p53 [7].
The full-length p53 protein contains five distinct domains, namely two N-terminal transactivation domains (TAD-I and TAD-II), a proline-rich domain (PRD), a DNA binding domain (DBD), an oligomerization domain (OD), and a C-terminal regulatory domain (CTD) that contains a nuclear localization signal [8]. Although initially believed to be transcribed as a single isoform, it is now clear that p53 is transcribed as multiple isoforms that differ in their amino and carboxy termini. Indeed, twelve different TP53 transcripts encode proteins with different functions and patterns of expression, and capable of affecting each other’s expression and activity (for reviews see [9, 10]). Similarly, p63 and p73 isoforms either contain a p53-homologous transactivation domain (TAp63 and TAp73) or lack this domain (ΔNp63 and ΔNp73), and alternative splicing generates different C-termini, for a total of at least ten p63 and over thirty p73 isoforms.
Whereas p53-null mice develop normally and are viable, disrupting p63 or p73 in mice severely affects proper development. p73-null mice suffer of hydrocephalia and hippocampal dysgenesis, are much smaller than their wild type counterparts, and 50 % to 75 % of the pups die before 4 weeks of age, mainly due to gastrointestinal hemorrhage, followed by intracranial bleeding. Those mice that survive, however, are not prone to tumorigenesis [11]. Also, unlike p53, p73 is rarely mutated in human cancers [12], suggesting that p73 cannot substitute for p53. However, TAp73 accumulates upon genotoxic stress, and it can up-regulate p21 and pro-apoptotic proteins such as PUMA, BAX and NOXA [13], indicating that p73 does play an important role in human cancer. Indeed, TAp73 is a tumor suppressor, as was demonstrated by specific TAp73 knockout in mice. In this study, seventy-three percent of the TAp73−/− mice and thirty percent of the TAp73+/− mice developed spontaneous tumors, where 66 % of the tumors from heterozygous mice displayed loss of heterozygosity [14]. On the other hand, ΔNp73 isoforms have oncogenic functions, and are able to inhibit p53 or TAp73 [15].
Even more strikingly, p63-null mice exhibit severe defects in all ectoderm-derived tissues and early post-natal lethality [7, 16]. We will discuss the role of p63 in development and cancer in more detail below.
p63
Since the cloning of the human p63 gene, it has been clear that TAp63 and ΔNp63 isoforms perform different functions. TAp63 isoforms possess strong transactivation activity on p53-responsive promoters [3], whereas ΔNp63 proteins are able to outcompete p53 for binding to p53-responsive promoters and repress gene expression [17]. Thus, ΔNp63 isoforms were initially described as simple dominant-negative proteins with the ability to inhibit TAp63 and p53 activity. However, further research has demonstrated that ΔNp63 isoforms are bona fide transcription factors with functions ranging well beyond modulation of other p53 family members [18–20]. Currently, much research focuses on dissecting the specific contributions of each class of isoforms to the functions of p63. Although there is still no definitive evidence regarding the mechanistic role of each isoform, several lines of evidence indicate that TAp63 isoforms can induce apoptosis and senescence, ΔNp63 proteins can promote cell survival and proliferation, and both kinds of isoforms are involved in cancer formation and progression. Indeed, both TAp63 and ΔNp63 have been described as metastasis inhibitors, albeit via different mechanisms. Moreover, the ratio between the two major classes of isoforms, as well as their interactions with other p53 family members, plays an important role in cancer formation and progression. Clearly, TAp63 and ΔNp63 have distinct and overlapping functions in normal and cancer tissues. Here, we discuss functions known to be specific to TAp63 versus ΔNp63. Whenever possible, we make a distinction between isoforms with different C-termini, but unfortunately the functional differences between these isoforms are still largely unknown.
Structure
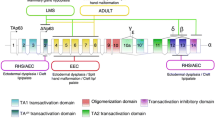
The human p63 protein is encoded by the TP63 gene, located in chromosome 3q27-28. It was discovered based on sequence homology to TP53 and TP73, primarily over the DNA-binding (DBD), oligomerization (OD), and transactivation (TA) domains [3]. TAp63 proteins contain an N-terminal TA domain that is 22 % homologous to that of p53, while ΔNp63 isoforms are transcribed from an alternative promoter within the third intron and thus bear no resemblance to the p53 TAD. Instead, the fourth exon, denominated exon 3’, encoding the N-terminus in ΔNp63 proteins, is spliced out in TAp63 transcripts. The 14 unique N-terminal amino acid residues in ΔNp63 isoforms have been shown to possess transactivation activity [18, 20], thus making ΔNp63 proteins bona fide transcription factors. Additional alternative splicing yields five different C-temini, α, β, γ, δ, and ε, for a total of ten different isoforms (Fig. 1) [21]. The DBD and OD are shared by all isoforms and are 60 % and 37 % homologous to those of p53, respectively [3, 7]. The p63α and p63β isoforms contain an additional TA domain (TA2) encoded in exons 11 and 12, which may be responsible for mediating ΔNp63α and ΔNp63β transactivation activity [19]. Further, TAp63α and ΔNp63α contain two more C-terminal domains, namely a Sterile Alpha Motif (SAM) and a Post-Inhibitory Domain (PID). The SAM domain consists of five tightly packed alpha helices, and it is important for protein-protein interactions, presumably with other SAM domain-containing proteins [22]. The PID binds to the TA domain of TAp63 isoforms, thus inhibiting gene transactivation and regulating TAp63 activity [23].

The p63 gene and protein structure. a The p63 gene (TP63) is encoded by sixteen exons and can be expressed from two different transcriptional start sites. Alternative splicing generates five different C-termini for a total of ten isoforms. b The p63 proteins contain six defined structural domains: An N-terminal transactivation domain (TA), a DNA binding domain (DBD), an oligomerization domain (OD), a second transactivation domain (TA2), a sterile alpha motif (SAM), and a post-inhibitory domain (PID). The TA domain, DBD, and OD are 22 %, 60 %, and 37 % homologous to the respective p53 domains
The p63 OD is 60 % homologous to the p73 OD. While the p53 OD contains one α-helix, the crystal structure of the p73 OD shows that this domain is stabilized by an additional C-terminal α-helix (H2) that is evolutionarily conserved in vertebrate p73 and p63 [24, 25]. It has been shown that p63 and p73 interact with each other but not with p53 in vivo [26, 27], even though previous in vitro studies using an OD lacking H2 indicated only weak interactions between p63 and p73 [28]. Newer studies using an OD including H2 have shown that p63 and p73 preferentially form heterotetramers of homodimers (a p63 dimer bound to a p73 dimer) in vitro, and confirmed that p53 does not form heterooligomers with p63 and/or p73, most likely due to the dissimilarity between the p53 and the p63/p73 OD [24, 25].
The presence or absence of the different domains in p63 isoforms affects not only their transactivation capacity, but also their stability. TAp63γ has the greatest transactivation potential on a p53-responsive promoter [3], as is expected from its TA domain and lack of a PID. However, TAp63 isoforms have a much shorter half-life than ΔNp63 isoforms and are quickly degraded when expressed exogenously [29].
Regulation
Expression Patterns
TAp63 and ΔNp63 exhibit rather different patterns of expression restricted to only a few physiological tissues. During adulthood, ΔNp63 isoforms (predominantly ΔNp63α) are expressed abundantly in basal epithelial cells, such as the basal layer of the skin, the myoepithelial cells of the breast [30], the basal cells of the prostate [31], and thymic epithelial cells [32]. During mouse development, ΔNp63α transcripts can be detected in the simple ectoderm at embryonic day 11 (E11), prior to stratification, and by E17 ΔNp63α can be strongly detected in the basal layer, but not in more superficial layers [33]. On the other hand, TAp63 isoforms are expressed at significant levels only in the female germline [34, 35], but cannot be detected in basal epithelial cells by either immunostaining or western blotting techniques. TAp63 transcript expression in epithelial cells can be detected by RT-PCR, but it is ten to several hundred folds lower than ΔNp63 expression [27, 36–38]. Given their different patterns of expression, TAp63 and ΔNp63 are regulated differently at the transcriptional, post-transcriptional, and post-translational levels.
Transcriptional Regulation
ΔNp63α expression can be induced in isolated dental epithelia from E11-E14 mice by beads releasing BMP2, BMP7, and FGF10 [33]. Also during development, ΔNp63α expression in the mouse lamboidal junction, a developmental feature during facial morphogenesis, has been shown to be under the control of Wnt9b signaling via an intronic Lef-Tcf binding element on the mouse Trp63 gene locus that is highly conserved in mammals [39]. Of note, BMP and Wnt signaling are crucial for normal epidermal and ectodermal appendage development [40, 41].
A number of signaling pathways that regulate p63 expression have been described using tissue culture methods. For example, activation of the PI3-K signaling cascade by EGF up-regulates ΔNp63α expression at the mRNA and protein levels, C/EBPα expression up-regulates ΔNp63α promoter reporter activity, and exogenous Snail and Slug down-regulate ΔNp63α expression and protein levels [42, 43]. Similarly, TAp63 expression can be induced in certain cancer cell lines. DNA damage caused by doxorubicin increases affinity of the c-Jun transcription factor to the TAp63 promoter and induces TAp63α, but not ΔNp63α expression [44, 45], and exogenous RelA expression up-regulates TAp63 promoter activation, which can be inhibited by the IκBα super-repressor [46].
Post-Transcriptional Regulation
Post-transcriptional regulation by micro RNAs (miRNA) is an emergent field of research that has proven to be of great importance to all biochemical processes. The miRNA miR-203 represses ΔNp63 isoforms during skin development. This miRNA targets a sequence on the 3′-untranslated region that is conserved in at least humans, mice, rats, dogs, and chicken. In the adult epidermis, miR-203 is expressed in the suprabasal layers, where it represses epidermal stem cell proliferation and clonogenic capacity, and basal epithelial gene expression by directly targeting ΔNp63 transcripts [47, 48]. Other miRNAs that target p63 include miR-92 and miR-21. miR-92 is thought to contribute to myeloid cell proliferation by inhibiting ΔNp63β translation and releasing myeloid cells from ΔNp63β repression of G2 cell cycle progression [49]. miR-21, on the other hand, targets TAp63 in glioblastoma cells and thus acts in an oncogenic manner [50].
Post-Translational Regulation
Genotoxic damage and extrinsic signals may affect p63 protein levels via post-translational regulation. For example, ultraviolet exposure induces ΔNp63α phosphorylation and subsequent proteasome-dependent protein degradation [51, 52]. This phosphorylation has been shown to be at least partially mediated by the stress-activated MAP kinase p38 [53]. Similarly, extrinsic signals, such as TNF-α or chemotherapeutic agents, can target ΔNp63α for degradation. TNF-α has been shown to induce ΔNp63α degradation in head and neck squamous cell carcinoma (HNSCC). Cells by activating the IKK pathway and leading to ΔNp63α phosphorylation by IKKβ and subsequent degradation via the ubiquitin-proteasome pathway [54]. Similarly, cisplatin treatment of HNSCC cells leads to ΔNp63α phosphorylation and nuclear export mediated by 14-3-3σ (also known as Stratifin) for subsequent proteasomal degradation mediated by RACK1 [55]. Likewise, doxorubicin treatment of different cell lines results in ΔNp63α phosphorylation by HIPK2, which targets ΔNp63α for proteasomal degradation [56].
ΔNp63α ubiquitination has been shown to be mediated by the HECT-containing NEDD4-like ubiquitin protein ligase Itch (also known as AIP4) [57, 58]. In one study, Itch was shown to interact with p63α isoforms via a PY motif found in the SAM domain of TAp63α and ΔNp63α, although it shows greater affinity for ΔNp63α than TAp63α [57]. On the other hand, another study showed that Itch could interact with p63γ isoforms, which lack the SAM domain. In this study, the authors conclude that the region encompassing amino acid residues 15–26 in ΔNp63 and 109–120 in TAp63 are critical for p63-Itch interaction [58].
Unlike ΔNp63 isoforms, TAp63 is only expressed at higher levels in oocytes, where it can direct apoptosis. In contrast to p53, which is found in low levels in unstressed cells and is stabilized upon stress, TAp63 is already expressed in high quantity, suggesting that its apoptotic activity must be controlled separately from its protein levels. Indeed, it has been shown that TAp63α is kept in an inactive, dimeric form in unstressed cells, and that genotoxic stress leads to activation by phosphorylation and subsequent tetramerization. In this model, The PID of one TAp63α molecule interacts with the TA domain of another, forming a closed dimeric conformation. This interaction is inhibited upon phosphorylation, thus opening up the dimer to allow tetramerization and increasing DNA binding affinity [59].
Development
Human Genetics
Heterozygous germline mutations on p63 are strongly associated with human autosomal dominant developmental diseases. Clinical studies have shown that mutations clustering on the p63 DBD strongly correlate with Ectrodactyly Ectodermal dysplasia-Clefting syndrome (EEC) [60]. EEC is characterized by ectrodactyly (the absence of one or more central digits in hands and/or feet), ectodermal dysplasia (defects on ectodermal appendages such as hair, sweat glands, teeth and nails), and cleft lip with or without cleft palate. Ankyloblepharon-Ectodermal dysplasia Clefting syndrome (AEC) differs from EEC in that it does not have a limb malformation component. Instead, AEC patients present ankyloblepharon, a partial or complete fusion of the eyelids. AEC mutations have been found within the SAM domain of p63α isoforms [61]. Frameshift mutations on these isoforms that yield truncated C-termini lacking the PID and/or part of the SAM domain are associated with Limb-Mammary Syndrome (LMS). LMS patients suffer ectrodactyly, mammary gland and nipple hypoplasia, and cleft palate, but no epidermal defects [62, 63]. A single missense mutation (R298Q) in the p63 DBD has been shown to associate with Acro-Dermato-Ungual-Lacrimal-Tooth (ADULT) syndrome, a disease similar to EEC but without facial clefting [64]. Finally, at least 10 % of Split-Hand/Foot Malformation (SHFM) cases are believed to be caused by single missense mutations in p63 [62, 63].
Differentiation and Stem Cell Maintenance
Homozygous disruption of p63 in mice results in early post-natal lethality due to a complete lack of all stratified epithelia and ectodermal appendages, including hair, mammary glands and teeth, in addition to severe craniofacial defects and limb truncation [7, 16]. While the two original studies of a p63 knockout mouse described the same gross phenotypes, Dennis Roop’s group did not detect differentiated stratified epithelial cells, and thus concluded that p63 is required for differentiation [16]. On the other hand, Frank McKeon’s group detected small patches of differentiated skin and concluded that p63 is important for proliferation potential [7].
In support of the proliferation potential hypothesis, p63 disruption was shown to not affect commitment to differentiation and differentiation of thymic epithelial cells, as evidenced by expression of differentiation markers and the ability to mediate T-cell maturation. Of note, in addition to the phenotypes described earlier, p63 loss leads to severe thymic hypoplasia. In this study, small patches of skin in the p63-null mice were able to differentiate, albeit in a catastrophically discontinuous fashion, as shown by expression of differentiation markers. Both thymic and epidermal stem cells lacking p63, however, succumbed to proliferative rundown, as evidenced by much decreased clonogenic potential [65]. These results are supported by studies showing that miR-203, a miRNA that targets ΔNp63 isoforms and that has an expression pattern opposite to ΔNp63 in the skin during development, represses epidermal stem cell proliferation and clonogenic capacity [47, 48]. Taken together, these studies conclude that p63 is essential for maintaining the proliferative potential of stratified epithelial stem cells in the skin and thymus.
Interestingly, a more recent study challenges this notion and concludes that p63 is required for commitment to an epithelial phenotype, and that in the absence of p63, the ectoderm fails to initiate embryonic stratification. The authors first looked at the corneal epithelia, which commits at E16.5, but does not significantly regenerate or stratify until 2 weeks after birth (P14). They found that at E18.5, corneal epithelial cells from p63-null mice still expressed ectodermal markers not found in epithelial cells, but did not express corneal differentiation markers [66]. This study highlights the importance of p63 in epithelial differentiation during embryonic development independently of proliferative defects. Notably, the seemingly opposite conclusions found by the previous studies are not necessarily mutually exclusive, and they may reflect different roles for p63 at different stages of development or adulthood, and in different epithelial compartments.
Genetic complementation studies support a greater role for ΔNp63 than for TAp63 in stratified epithelial development. Restoration of ΔNp63α expression on the basal epidermal layer results in significant basal layer formation, suggesting that ΔNp63α expression in this system can partly revert the proliferative defects of the p63-null epidermis. On the other hand, TAp63α reintroduction has no discernible effect on skin development. Notably, double expression of ΔNp63α and TAp63α rescues the knockout phenotype to a greater extent, inducing expression of differentiation markers and a greater degree of re-epithelialization, but still cannot fully recover proper embryonic development [38, 67]. Likewise, thymus development in p63-null mice is partially rescued by re-introduction of ΔNp63α, but not TAp63α, into thymic epithelial cells. In this study, p63-null thymi exhibited no apparent defects in differentiation, but displayed marked deficiencies in size, structure, and number of γδ epithelial cells. ΔNp63α significantly rescued these defects mainly by up-regulating FGF Receptor 2 and the Notch ligand JAG2 [32].
ΔNp63α expression in the skin is highest in the proliferative, non-terminally differentiated basal layer, and it is absent in the differentiated suprabasal layers, suggesting that ΔNp63α expression is negatively correlated with differentiation. Indeed, ΔNp63α inhibits differentiation by directly down-regulating the Notch ligand JAG2 and the Notch effector HES-1 [68, 69]. While in most cell types Notch signaling promotes stem cell potential, in keratinocytes Notch signaling induces differentiation. However, p63 ablation impairs keratinocyte differentiation [36]. These seemingly contradictory observations may be reconciled in that ΔNp63α is important for the induction of differentiation, and in its absence keratinocytes fail to commit to differentiation, but terminal differentiation requires ΔNp63α down-regulation in order to suppress stem cell features. Accordingly, Notch activation upon differentiation induction also down-regulates ΔNp63α, serving as a negative feedback that allows for terminal differentiation [68].
TAp63−/− mice develop blisters and ulcerated wounds in the skin, in addition to exhibiting impaired wound healing and accelerated aging. However, TAp63−/− mice develop fully differentiated stratified epithelia, demonstrating that TAp63 isoforms are dispensable for epithelial development. Interestingly, TAp63 deletion in the epidermal compartment in mice does not affect skin development or wound healing, does not result in ulcerations of the skin, and does not affect proliferation, indicating that these defects are not due to the absence of TAp63 in the basal layer of the skin. Instead, TAp63 loss affects dermal skin precursors, thus affecting adult skin maintenance (see Senescence and Aging below) [70].
Cell Cycle
The role of p63 in cell cycle regulation is cell type and context dependent. Exogenous TAp63γ expressed in erythroleukemia cells accumulates in response to genotoxic stress and can up-regulate p21 expression, thus halting cell cycle progression [71], while exogenous ΔNp63α directly binds to the p21 promoter to inhibit reporter expression [17, 54]. ΔNp63α is thought to be required for G1 progression in keratinocytes, since ablation of endogenous p63 expression in human primary keratinocytes increases p21 expression and leads to reduced proliferation and G1 arrest [72, 73]. Accordingly, reduced endogenous ΔNp63α in mouse primary keratinocytes results in decreased Cyclin D1, CDK4, and CDK2 expression. Interestingly, miR-34a and miR-34c expression is elevated in mouse skin with abrogated p63 expression, and inhibition of these miRNAs prevents Cyclin D1 and CDK4 down-regulation and rescues the proliferation defects elicited by p63 deficiency [74].
Senescence and Aging
p63 is also involved in cellular senescence and aging. Germline disruption of p63 expression results in a dramatic increase of senescence-associated β-Galactosidase (SA β-Gal) staining in whole-mount E17.5 mouse embryos, which is phenocopied by somatic disruption of p63 expression in basal epithelial cells using a Cre-Lox system driven by a K5 promoter. Strikingly, disrupting p63 expression by activating Cre in eight-month old mice results in an aging phenotype, characterized by alopecia and curvature of the spine (an age-related phenotype), in addition to skin defects and weight loss [75]. Importantly, abrogating p63 expression using a K5 promoter affects primarily the ΔNp63 isoforms. As mentioned previously, disrupting TAp63 expression in the basal epithelial compartment does not recapitulate the skin defects found in the adult TAp63−/− mice. Rather, dermal precursor cells without TAp63 expression hyperproliferate and succumb to senescence, thus leading to the observed phenotypes in the adult epidermis. Additionally, absence of TAp63 in dermal precursor cells results in genomic instability and increased DNA damage. These observations indicate that TAp63 restrains dermal precursor cell proliferation, and that its deficiency brings about senescence triggered by increased DNA damage. Accordingly, TAp63-null mice age prematurely, as evidence from increased curvature of the spine and shorter lifespan [70]. Nonetheless, exogenous expression of TAp63 isoforms in cell culture leads to increased cellular senescence independently of p53, as evidenced from increased SA β-Gal staining and decreased proliferation. Moreover, TAp63 is required for Ras-mediated oncogene-induced senescence (OIS). Ras expression in wild type MEFs leads to cell senescence, which is abrogated in TAp63-deficient MEFs [76]. These observations highlight the importance of both ΔNp63 and TAp63 in cell senescence, and demonstrate that positive or negative modulation of either p63 isoform may result in senescence by different molecular mechanisms.
Role in Cancer
While it is now clear that ΔNp63 isoforms promote survival and TAp63 isoforms induce cell death, the role of p63 in cancer is not dictated just by the specific actions of each individual isoform, but by the interactions of all p53 family members, including mutant p53. Genetic studies in mice have provided conflicting results, as one group reported that Trp63+/− mice (C57BL/6 × 129/SvJae) develop spontaneous tumors [77], while another group reported that Trp63 heterozygosity in mice (C57BL/6J × 129S5) does not result in spontaneous tumor formation [78]. Nevertheless, the scientific community has reached some consensus in that ΔNp63 may promote tumorigenesis, TAp63 is a tumor suppressor in the female germline, and p63 may be a metastasis suppressor. The specific molecular mechanisms, however, are still being deciphered, and it may take another 10 years before we have a clearer picture regarding the role of p63 in cancer.
ΔNp63 Isoforms Exhibit Oncogenic Features
In contrast to p53, the TP63 gene is rarely mutated in human cancers [12, 79, 80]. Rather, ΔNp63α is often over-expressed in low-grade squamous cell carcinoma (SCC), generally due to chromosomal amplification [12, 81–83]. Indeed, clinical studies have found ΔNp63α over-expression ranging from 85 % to 100 % of all SCC cases, including SCC of the head and neck [12, 84], esophagus [85], lung [83], and cervix [86], in addition to a subset of basal breast carcinomas [87, 88].
Tissue culture experiments using HNSCC cells derived from primary tumors have shown that ΔNp63α is required for cell survival. Indeed, p63 ablation results in apoptosis, evidenced by increased cleaved PARP levels, increased Annexin V staining, and increased PUMA and NOXA expression [27, 72]. Interestingly, ΔNp63α over-expression is not correlated with wild type p53 status in SCC, indicating that ΔNp63α does not act to inhibit p53-mediated apoptosis in these settings [12, 82]. Instead, ΔNp63α has been shown to inhibit TAp73-mediated apoptosis. In this model, ΔNp63α either competes with TAp73 for binding to promoters of pro-apoptotic proteins to inhibit their transcription, or it directly binds to TAp73 to inhibit its function [27, 72]. Similar effects have been observed in a relatively small subset of invasive breast carcinoma primary cell samples that express ΔNp63α. In these cells, cisplatin treatment leads to c-Abl-dependent TAp73 phosphorylation on Tyr99, resulting in ΔNp63α-TAp73 complex dissociation and TAp73 activation, TAp73-dependent transcription of apoptotic molecules, and ultimately apoptosis [89]. Transcriptional repression of pro-apoptotic molecules by ΔNp63α in SCC has been shown to be mediated in association with Histone Deacetylase 1 (HDAC1) and HDAC2, hence suggesting new therapeutic avenues for treating SCC with HDAC inhibitors [90]. Alternatively, ΔNp63α may also contribute to tumorigenesis by yet other mechanisms. For example, ΔNp63α has been shown to up-regulate Heat-Shock Protein 70 (Hsp70) expression, and the expression of both proteins is significantly correlated in primary HNSCC biopsy samples [91]. Since the chaperone protein Hsp70 displays proliferative and anti-apoptotic characteristics, this mechanism may also contribute to the oncogenicity of ΔNp63α.
In another study, ΔNp63α was shown to inhibit oncogene-induced senescence in keratinocytes when co-expressed with Ras. It has been shown that concomitant ΔNp63α and Ras expression cooperate to induce tumor formation in mouse xenografts, while either Ras or ΔNp63α over-expression alone is not enough for supporting tumor growth, thus indicating that ΔNp63α may also act as an oncogene by potentiating the oncogenic features of Ras [92].
TAp63 is A Tumor Suppressor
In contrast to the role of ΔNp63α in promoting cell proliferation and survival, TAp63α promotes cell cycle arrest, senescence, and apoptosis. TAp63 is required for protecting the female germline from genotoxic stress during meiotic arrest. Indeed, TAp63α is highly expressed in oocytes, but not in testis or other tissues. Upon exposure to ionizing radiation, oocytes die within 5 days. TAp63-null mouse oocytes are resistant to death induced by radiation, while p53-null oocytes still succumb to apoptosis triggered by ionizing radiation, indicating that TAp63α, but not p53, is important for mediating these effects [35]. In fact, further research has shown that DNA damage induced by cisplatin treatment leads to activation of c-Abl, which phosphorylates and activates TAp63 to direct oocyte cell death. Wild type TAp63α, but not a phosphorylation-resistant mutant TAp63α-Y149F, can induce PUMA and NOXA gene expression when expressed exogenously in p53-null human lung adenocarcinoma H1299 cells [34]. Moreover, exogeneous expression of either TAp63α, TAp63β, or TAp63γ inhibits tumor formation upon xenograft implantation in nude mice [76], demonstrating that TAp63 acts to suppress tumorigenesis in vivo.
Intriguingly, TAp63 isoforms have been found to be over-expressed in high-grade follicular lymphoma, but the biological significance of these observations is yet to be elucidated [93].
Role in Metastasis
Metastasis results from the acquisition of migratory and invasive abilities as a result of not only accumulated mutations and changes in gene expression, but also as a response to stromal cell stimuli. One of the most dramatic changes involves the loss of epithelial markers and the gain of mesenchymal traits through a process termed epithelial to mesenchymal transition (EMT). Cells that become metastatic are able to locally invade the underlying mesenchyme, intravasate into blood or lymph vessels, travel in circulation, and then finally extravasate into a distal tissue for colonization and the formation of a metastatic nodule resembling the primary tumor.
Clinical Studies
Notwithstanding the oncogenic properties of ΔNp63α, numerous studies over the past few years suggest that reduced p63 expression is associated with cancer progression. Analyses of biopsy samples have shown that reduced p63 expression is associated with progression in a variety of cancers. In cervical carcinomas, loss of p63 expression is associated with undifferentiated morphology [86, 94]. Further, immunohistochemistry combined with RT-PCR analysis have shown that ΔNp63α expression is progressively reduced at advanced stages of breast [30, 95, 96], prostate [31], urothelial [97, 98], and bladder cancers [99], and lost in the majority of invasive cancers and metastatic nodes. Moreover, gene expression profiling studies of prostate cancer [100], melanoma [101], and esophageal SCC [102] reveal a clear correlation between reduced p63 expression and cancer progression.
Mouse Genetics Studies
Genetic studies show that p63 haploinsufficiency in p53+/− mice markedly increases metastatic frequency. While p53+/− mice develop spontaneous tumors with a relatively low incidence of metastasis, tumors from p53+/−; p63+/− compound heterozygous mice show a much more aggressive behavior, with a metastatic frequency of 50 %, compared to 5 % in p53+/− mice [77].
There is evidence demonstrating that both TAp63 and ΔNp63 isoforms are involved in inhibiting metastasis, although likely via different molecular mechanisms. TAp63−/− and TAp63+/− mice develop spontaneous tumors that are highly metastatic [103]. TAp63−/− MEFs show increased invasion in matrigel, which can be reverted by expression of Dicer and miR-130b, both of which are direct targets of TAp63. These observations suggest that TAp63 inhibits metastasis by inducing Dicer and miR-130b [103].
Cell Culture Studies
Knockdown of TAp63 in human lung carcinoma p53-null H1299 cells, which express mainly TAp63α and TAp63β as detected by PCR, leads to increased cell invasion, thus further supporting a role for TAp63 in inhibiting metastasis [104]. ΔNp63 isoforms are also thought to be important contributors to inhibiting metastasis. Exogenous ΔNp63α expression in malignant spindle carcinoma D3S2 cells markedly decreases metastatic frequency to the lungs upon intravenous injection into recipient mice [105]. Accordingly, reduced ΔNp63α expression in SCC, keratinocyte, and urothelial cell lines up-regulates genes involved in cell motility and induces cell migration and invasion, while enforced ΔNp63α expression inhibits these traits [36, 43, 106–108].
Cell Adhesion
Another important signaling axis under control of p63 involves cell adhesion. Ablation of all p63 isoforms in mammary epithelial MCF-10A cells causes a major down-regulation of cell-to-matrix adhesive molecules, particularly of integrins β1, β4 and α6, and of the matrix protein Laminin-γ2, resulting in death by anoikis, which can be completely prevented by expression of an shRNA-insensitive ΔNp63α, but not by TAp63γ. Similarly, reintroduction of integrin β4 into a p63-deficient background can partially protect from anoikis, highlighting the importance of cell-matrix adhesion for epithelial tissue integrity and cell survival [37].
The transmembrane protein Perp facilitates cell-cell adhesions by maintaining proper desmosome structure and function. This protein, which had been previously shown to be a p53 target, is also a direct p63 target in skin development. Perp expression is absent in p63-null mice embryos. Likewise, p63 ablation in mouse keratinocytes down-regulates Perp expression. Moreover, exogenous TAp63α, TAp63γ, ΔNp63α, ΔNp63γ, and p53 are able to transactivate Perp reporter expression, and endogenous p63 binds to a putative p53/p63 binding site in intron 1 on the Perp gene. Of note, even though ΔNp63 isoforms are expressed at much higher level than TAp63 isoforms, TAp63α and TAp63γ are able to transactivate Perp expression several folds higher than ΔNp63α and ΔNp63γ, as is expected from their greater transactivation potential. Interestingly, Perp-null mice develop dramatically blistering skin and die shortly after birth, resembling some of the phenotypes found in the p63-null mice [109].
Cell Migration and Invasion
Many of the molecular mechanisms about the role of p63 in cell migration and invasion described to date involve the identification of specific genes known to influence cell motility. For example, ΔNp63α ablation in SCC cell lines was shown to up-regulate N-cadherin, L1 adhesion molecule, Periostin, and Wnt-5A, all of which are involved in cell motility [36]. N-cadherin, a mesenchymal cell-cell adhesion molecule involved in epithelial to messenchymal transition (EMT), has also been shown to increase expression upon ΔNp63α knockdown in urothelial carcinoma cell lines [106]. However, other groups have reported that expression of both N-cadherin and E-cadherin, an epithelial cell-cell adhesion molecule normally lost through EMT, is not modulated by ΔNp63α [43, 107]. This discrepancy may reflect cell-context differences, but also suggest that the gain in cell motility upon reduced ΔNp63α is independent and different from classical EMT. ΔNp63α has also been shown to affect cell invasion by inducing Inhibitor of Differentiation-3 (Id-3), which may inhibit Ets-1 mediated transcription of MMP2, thereby inhibiting cell invasion [107]. Finally, we have found that ΔNp63α, but not ΔNp63γ or TAp63 isoforms, modulates Erk2 signaling to inhibit mammary cell migration, invasion, and metastasis (Fig. 2a; unpublished).

The p63 protein inhibits metastasis via multiple mechanisms. a ΔNp63α has been shown to inhibit metastasis by a variety of molecular mechanisms, including negative regulation of Erk2 signaling, positive regulation of Id-3 signaling, and by maintaining cell adhesion in epithelial cells. b Under certain cellular conditions, TGFβ induces a trimeric complex between mutant p53 (mtp53), Smad2, and p63 that inactivates p63 activity. In this context, p63 is thought to inhibit metastasis by up-regulation of Sharp-1 and Cyclin G2 expression. c Mutant p53 may also inhibit p63 from promoting integrin recycling to the plasma membrane. Integrin recycling enhances PI3-K/Akt signaling, which induces cell motility and cancer metastasis. d TAp63 isoforms have been shown to inhibit metastasis by up-regulating DICER and miR-130b. The studies described above regarding mtp53 inhibition of p63 attribute anti-metastatic properties primarily to TAp63 isoforms, as these are the isoforms that are predominantly expressed in the systems used in these studies
The Role of Mutant p53
More recently, accumulating evidence indicates that mutant p53 (mtp53) may induce cancer progression and metastasis by inhibiting p63. In one study, Transforming Growth Factor β (TGFβ) was shown to induce a trimeric complex between p63, mtp53, and Smad2, leading to inhibition of p63 and increased cell invasion and migration. The authors identified Sharp-1 and Cyclin G2 as two clinically relevant p63 targets responsible for inhibiting cell migration and invasion (Fig. 2b) [105]. In another study, p63 was shown to inhibit RCP-mediated integrin α5β1 recycling to the plasma membrane via an unknown mechanism, thus down-regulating downstream signaling from these complexes to Akt and thereby inhibiting cell migration and invasion (Fig. 2c). The authors also show that mtp53 inhibits p63 transcriptional activity and induces cell invasion to levels comparable to p63 ablation in H1299 cells. Notably, p63 knockdown does not further enhance cell invasion elicited by mtp53 expression. The authors attribute these effects mostly to TAp63, since H1299 cells express mainly TAp63 isoforms and TAp63α over-expression reverts cell invasion induced by mtp53 back to control levels in this system (Fig. 2d) [104]. This negative modulation of Akt signaling is likely to be cell context-dependent, as ΔNp63α has been shown to inhibit PTEN expression in keratinocytes, hence acting as a positive modulator of Akt signaling [110]. Of note, TAp63 isoforms in H1299 cells are detectable by PCR, but not by western blotting [104], while normal epithelial cells and carcinomas express ΔNp63 isoforms, although relatively small levels of TAp63 expression may be sufficient for inhibiting metastasis. Since there is no significant correlation between ΔNp63 over-expression and p53 mutation in SCC, but there is a correlation between reduced ΔNp63 expression and cancer progression, mtp53 may not be sufficient to inhibit ΔNp63 from preventing metastasis in these cases. Alternatively, mtp53 may promote metastasis via additional mechanisms independent of p63 [111]. Thus, reduced ΔNp63 expression may cooperate with mtp53 to promote metastasis (Fig. 3).

p63 is a metastasis inhibitor, while mutant p53 promotes malignancy. Mutant p53 (mtp53) may promote metastasis by inhibiting p63. Moreover, mtp53 may enhance metastatic spread via other mechanisms, and p63 can inhibit mtp53 from inducing metastasis. Thus, loss of p63 expression may cooperate with mtp53 to promote metastasis
Conclusions
The TP63 gene and all its protein isoforms serve pleiotropic functions encompassing nearly all aspects of cellular life. The scientific community recognized early the importance of discerning between different p63 isoforms, thus facilitating the study of functions specific to each class of protein. Still, we are only at the tip of the iceberg. Genetic studies in mice have shed much light on the roles of TAp63 versus ΔNp63 isoforms in development and adult epithelial homeostasis. ΔNp63 isoforms are required for commitment to stratification, whereas TAp63 isoforms are dispensable for development. Both classes of isoforms are required for maintaining stratified epithelial integrity in the adult, and their individual absence results in cellular senescence and premature aging. It is interesting that different cellular compartments use TAp63 or ΔNp63 to maintain homeostasis by seemingly opposite mechanisms. While TAp63 isoforms inhibit dermal precursors hyperproliferation that may lead to senescence, ΔNp63 proteins maintain the proliferative potential of basal skin stem cells. TAp63 and ΔNp63 maintain stratified epithelial integrity by working together, but separated into different compartments, as it would be expected from the ability of ΔNp63 to inhibit TAp63.
Also interesting is the great difference in expression levels. ΔNp63α is expressed at high levels in basal epithelial cells, such as myoepithelial cells and keratinocytes. Thus, the majority of the studies using these cells as a model system have attributed the functions of p63 to this particular isoform. We have demonstrated that at least in mammary epithelial cells, this is indeed the case, as reintroduction of ΔNp63α, but not ΔNp63γ or TAp63 isoforms, is able to revert cells back to a non-motile phenotype after p63 ablation (unpublished). However, as mentioned earlier, tissue culture and animal genetics studies have shown that TAp63 is a metastasis suppressor, despite low expression levels. Clearly, the effects of p63 on cancer metastasis are cell context-dependent, and more research is needed in order to understand the molecular mechanisms underlying isoform-specific effects.
The roles of p63 in tumorigenesis, cancer progression, and metastasis are still being discovered. As we search for new combinations of molecular targets for cancer therapy, the need for understanding the basic molecular mechanisms underlying cancer progression is as great as ever. The description of both TAp63 and ΔNp63 as metastasis suppressors opens new avenues for developing cancer treatments. Thus, a better understanding of the roles and interactions of the different p63 isoforms and other p53 family members will undoubtedly prove beneficial to the design of effective cancer therapies.
References
Levine AJ, Oren M (2009) The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9(10):749–758
Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137(3):413–431
Yang A et al (1998) p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2(3):305–316
Kaghad M et al (1997) Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90(4):809–819
Melino G (2011) p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ
Vanbokhoven H et al (2011) p63, a story of mice and men. J Invest Dermatol
Yang A et al (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398(6729):714–718
May P, May E (1999) Twenty years of p53 research: structural and functional aspects of the p53 protein. Oncogene 18(53):7621–7636
Khoury MP, Bourdon J-C (2011) p53 isoforms: an intracellular microprocessor? Genes Cancer 2(4):453–465
Marcel V, Hainaut P (2009) p53 isoforms - a conspiracy to kidnap p53 tumor suppressor activity? Cell Mol Life Sci 66(3):391–406
Yang A et al (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404(6773):99–103
Weber A et al (2002) Expression of p53 and its homologues in primary and recurrent squamous cell carcinomas of the head and neck. Int J Cancer 99(1):22–28
Zawacka-Pankau J et al (2010) p73 tumor suppressor protein: a close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle 9(4)
Tomasini R et al (2008) TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 22(19):2677–2691
Deyoung MP, Ellisen LW (2007) p63 and p73 in human cancer: defining the network. Oncogene 26(36):5169–5183
Mills AA et al (1999) p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398(6729):708–713
Westfall MD et al (2003) The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 23(7):2264–2276
Dohn M, Zhang S, Chen X (2001) p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 20(25):3193–3205
Ghioni P et al (2002) Complex transcriptional effects of p63 isoforms: identification of novel activation and repression domains. Mol Cell Biol 22(24):8659–8668
Helton ES, Zhu J, Chen X (2006) The unique NH2-terminally deleted (DeltaN) residues, the PXXP motif, and the PPXY motif are required for the transcriptional activity of the DeltaN variant of p63. J Biol Chem 281(5):2533–2542
Mangiulli M et al (2009) Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res 37(18):6092–6104
Thanos CD, Bowie JU (1999) p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Sci 8(8):1708–1710
Serber Z et al (2002) A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol 22(24):8601–8611
Coutandin D et al (2009) Conformational stability and activity of p73 require a second helix in the tetramerization domain. Cell Death Differ 16(12):1582–1589
Joerger A et al (2009) Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci USA
Gaiddon C et al (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21(5):1874–1887
Rocco JW et al (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9(1):45–56
Davison TS et al (1999) p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 274(26):18709–18714
Ying H et al (2005) DNA-binding and transactivation activities are essential for TAp63 protein degradation. Mol Cell Biol 25(14):6154–6164
Barbareschi M et al (2001) p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol 25(8):1054–1060
Signoretti S et al (2000) p63 is a prostate basal cell marker and is required for prostate development. Am J Pathol 157(6):1769–1775
Candi E et al (2007) DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc Natl Acad Sci USA 104(29):11999–12004
Laurikkala J et al (2006) p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development 133(8):1553–1563
Gonfloni S et al (2009) Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 15(10):1179–1185
Suh E-K et al (2006) p63 protects the female germ line during meiotic arrest. Nature 444(7119):624–628
Barbieri CE et al (2006) Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res 66(15):7589–7597
Carroll DK et al (2006) p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol 8(6):551–561
Romano R-A et al (2009) An active role of the DeltaN isoform of p63 in regulating basal keratin genes K5 and K14 and directing epidermal cell fate. PLoS One 4(5):e5623
Ferretti E et al (2011) A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell 21(4):627–641
Aberdam D et al (2007) Key role of p63 in BMP-4-induced epidermal commitment of embryonic stem cells. Cell Cycle 6(3):291–294
Mikkola ML (2007) p63 in skin appendage development. Cell Cycle 6(3):285–290
Herfs M et al (2010) Regulation of p63 isoforms by snail and slug transcription factors in human squamous cell carcinoma. Am J Pathol
Higashikawa K et al (2007) Snail-induced down-regulation of DeltaNp63alpha acquires invasive phenotype of human squamous cell carcinoma. Cancer Res 67(19):9207–9213
Petitjean A et al (2005) The expression of TA and DeltaNp63 are regulated by different mechanisms in liver cells. Oncogene 24(3):512–519
Yao J-Y, Pao C-C, Chen J-K (2010) Transcriptional activity of TAp63 promoter is regulated by c-jun. J Cell Physiol 225(3):898–904
Wu J et al (2010) TAp63 is a transcriptional target of NF-kappaB. J Cell Biochem 109(4):702–710
Lena A. et al (2008) miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ
Yi R et al (2008) A skin microRNA promotes differentiation by repressing ‘stemness’. Nature 452(7184):225–229
Manni I et al (2009) The microRNA miR-92 increases proliferation of myeloid cells and by targeting p63 modulates the abundance of its isoforms. FASEB J
Papagiannakopoulos T, Shapiro A, Kosik KS (2008) MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 68(19):8164–8172
Liefer KM et al (2000) Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res 60(15):4016–4020
Westfall MD et al (2005) Ultraviolet radiation induces phosphorylation and ubiquitin-mediated degradation of DeltaNp63alpha. Cell Cycle 4(5):710–716
Papoutsaki M et al (2005) A p38-dependent pathway regulates DeltaNp63 DNA binding to p53-dependent promoters in UV-induced apoptosis of keratinocytes. Oncogene 24(46):6970–6975
Chatterjee A et al (2010) Regulation of p53 Family Member Isoform Delta}Np63{alpha by the Nuclear Factor-{kappa}B Targeting Kinase I{kappa}B Kinase {beta}. Cancer Res 70(4):1419–1429
Fomenkov A et al (2004) RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 3(10):1285–1295
Lazzari C et al (2011) HIPK2 phosphorylates ΔNp63α and promotes its degradation in response to DNA damage. Oncogene 30(48):4802–4813
Rossi M et al (2006) The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA 103(34):12753–12758
Rossi M et al (2006) Itch/AIP4 associates with and promotes p63 protein degradation. Cell Cycle 5(16):1816–1822
Deutsch GB et al (2011) DNA damage in oocytes induces a switch of the quality control factor TAp63α from dimer to tetramer. Cell 144(4):566–576
Celli J et al (1999) Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99(2):143–153
McGrath JA et al (2001) Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 10(3):221–229
Brunner HG, Hamel BCJ, van Bokhoven H (2002) P63 gene mutations and human developmental syndromes. Am J Med Genet 112(3):284–290
van Bokhoven H, McKeon F (2002) Mutations in the p53 homolog p63: allele-specific developmental syndromes in humans. Trends Mol Med 8(3):133–139
Rinne T et al (2006) Delineation of the ADULT syndrome phenotype due to arginine 298 mutations of the p63 gene. Eur J Hum Genet 14(8):904–910
Senoo M et al (2007) p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell 129(3):523–536
Shalom-Feuerstein R et al (2010) ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ
Candi E et al (2006) Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ 13(6):1037–1047
Nguyen B-C et al (2006) Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev 20(8):1028–1042
Wu G et al. ΔNp63α and tap63α regulate transcription of genes with distinct biological functions in cancer and development. Cancer Res
Su X et al (2009) TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 5(1):64–75
Katoh I et al (2000) p51A (TAp63gamma), a p53 homolog, accumulates in response to DNA damage for cell regulation. Oncogene 19(27):3126–3130
DeYoung MP et al (2006) Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res 66(19):9362–9368
Truong AB et al (2006) p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev 20(22):3185–3197
Antonini D et al (2010) Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J Invest Dermatol 130(5):1249–1257
Keyes WM et al (2005) p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev 19(17):1986–1999
Guo X et al (2009) TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol 11(12):1451–1457
Flores ER et al (2005) Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7(4):363–373
Keyes WM et al (2006) p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc Natl Acad Sci USA 103(22):8435–8440
Hagiwara K et al (1999) Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res 59(17):4165–4169
Sunahara M et al (1999) Mutational analysis of p51A/TAp63gamma, a p53 homolog, in non-small cell lung cancer and breast cancer. Oncogene 18(25):3761–3765
Björkqvist AM et al (1998) DNA gains in 3q occur frequently in squamous cell carcinoma of the lung, but not in adenocarcinoma. Genes Chromosomes Canc 22(1):79–82
Hibi K et al (2000) AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 97(10):5462–5467
Massion PP et al (2003) Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res 63(21):7113–7121
Sniezek JC et al (2004) Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope 114(12):2063–2072
Hu H et al (2002) Elevated expression of p63 protein in human esophageal squamous cell carcinomas. Int J Cancer 102(6):580–583
Wang TY et al (2001) Histologic and immunophenotypic classification of cervical carcinomas by expression of the p53 homologue p63: a study of 250 cases. Hum Pathol 32(5):479–486
Matos I et al (2005) p63, cytokeratin 5, and P-cadherin: three molecular markers to distinguish basal phenotype in breast carcinomas. Virchows Arch 447(4):688–694
Perou CM et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Leong C-O et al (2007) The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 117(5):1370–1380
Ramsey MR et al (2011) Physical Association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res 71(13):4373–4379
Wu G et al (2005) DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res 65(3):758–766
Keyes WM et al (2011) ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 8(2):164–176
Pruneri G et al (2005) The transactivating isoforms of p63 are overexpressed in high-grade follicular lymphomas independent of the occurrence of p63 gene amplification. J Pathol 206(3):337–345
Quade BJ et al (2001) Expression of the p53 homologue p63 in early cervical neoplasia. Gynecol Oncol 80(1):24–29
Stefanou D et al (2004) p63 expression in benign and malignant breast lesions. Histol Histopathol 19(2):465–471
Wang X et al (2002) p63 expression in normal, hyperplastic and malignant breast tissues. Breast Cancer 9(3):216–219
Koga F et al (2003) Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin Cancer Res 9(15):5501–5507
Koga F et al (2003) Impaired Delta Np63 expression associates with reduced beta-catenin and aggressive phenotypes of urothelial neoplasms. Br J Cancer 88(5):740–747
Urist MJ et al (2002) Loss of p63 expression is associated with tumor progression in bladder cancer. Am J Pathol 161(4):1199–1206
Vanaja DK et al (2003) Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res 63(14):3877–3882
Haqq C et al (2005) The gene expression signatures of melanoma progression. Proc Natl Acad Sci USA 102(17):6092–6097
Su H et al (2003) Gene expression analysis of esophageal squamous cell carcinoma reveals consistent molecular profiles related to a family history of upper gastrointestinal cancer. Cancer Res 63(14):3872–3876
Su X et al (2010) TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467(7318):986–990
Muller PAJ et al (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139(7):1327–1341
Adorno M et al (2009) A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137(1):87–98
Fukushima H et al (2009) Loss of DeltaNp63alpha promotes invasion of urothelial carcinomas via N-cadherin/Src homology and collagen/extracellular signal-regulated kinase pathway. Cancer Res 69(24):9263–9270
Higashikawa K et al (2009) DeltaNp63alpha-dependent expression of Id-3 distinctively suppresses the invasiveness of human squamous cell carcinoma. Int J Cancer 124(12):2837–2844
Kommagani R et al (2009) Regulation of VDR by {Delta}Np63{alpha} is associated with inhibition of cell invasion. J Cell Sci
Ihrie RA et al (2005) Perp is a p63-regulated gene essential for epithelial integrity. Cell 120(6):843–856
Leonard MK et al (2011) ΔNp63α regulates keratinocyte proliferation by controlling PTEN expression and localization. Cell Death Differ
Girardini JE et al (2011) A Pin1/Mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20(1):79–91
Acknowledgments
This work was supported by the National Key Basic Research Program (973 Program) of China (2012CB910700) and National Science Foundation of China (#31171362) to ZX. X., and United States Department of Defense Congressionally Directed Medical Research Programs grant W81XWH-10-1-0161 to J.B.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bergholz, J., Xiao, ZX. Role of p63 in Development, Tumorigenesis and Cancer Progression. Cancer Microenvironment 5, 311–322 (2012). https://doi.org/10.1007/s12307-012-0116-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-012-0116-9