
Abstract
As a member of tumor suppressor p53 family, p63, a gene encoding versatile protein variant, has been documented to correlate with cancer formation and progression, though it is rarely mutated in cancer patients. However, it has long been controversial on whether p63 is an oncogene or a tumor suppressor. Here, we comprehensively reviewed reports on roles of p63 in development, tumorigenesis and tumor progression. According to data from molecular cell biology, genetic models and clinic research, we conclude that p63 may act as either an oncogene or a tumor suppressor gene in different scenarios: TA isoforms of p63 gene are generally tumor-suppressive through repressing cell proliferation, survival and metastasis; ΔN isoforms, however, may initiate tumorigenesis via promoting cell proliferation and survival, but inhibit tumor metastasis and progression; effects of p63 on tumor formation and progression depend on the context of the whole p53 family, and either amplification or loss of p63 gene locus can break the balance to cause tumorigenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
p63 gene and its protein products
p63, also known as TP63 (tumor protein 63), Trp63 (transformation related protein 63), or AIS (amplified in squamous cell carcinoma), is a gene highly homologous to tumor suppressor p53. It locates on the distal long arm of human chromosome 3, 3q27 [1]. p63 gene possesses 2 promoters and 16 exons (Fig. 1a). The first promoter drives transcription of TAp63, starting at the first exon, while the second promoter triggers transcription of ΔNp63 isotypes, which starts at exon 3′ [2].

Schematic presentation of p63 gene (a) and protein isoforms (b). TAD transactivation domain, TAD2 additional transactivation domain, DBD DNA-binding domain, OD oligomerization domain, SAM sterile alpha motif, TID trans-inhibitory domain. All aliases of each isoforms are listed following the formal terms
Primary transcript of either TA or ΔN isotype p63 undergoes alternative splicing. Consequently, p63 gene can generate at least 10 different protein isoforms, namely TAp63α/β/γ/δ/ε and ΔNp63α/β/γ/δ/ε (Fig. 1b). TA isoforms of p63 protein contain a longer N-terminal transactivation domain (TAD), while ΔNp63s possess a shorter region at the N-termini, which composed 14 amino acid residues and encoded by exon 3′. All isoforms share a common DNA-binding domain (DBD) and a common oligomerization domain (OD) at the middle part. α and β isoforms of p63 (p63α and p63β) contain an additional transactivation domain (TAD2) next to OD. Additionally, p63α possesses a unique sterile alpha motif (SAM) and a trans-inhibitory domain (TID) at the C-terminus, which are involved in protein–protein interaction and activity modulation [2, 3].
Activities of p63 proteins: transactivators or trans-inhibitors?
As transcription factors belonging to p53 family, p63 proteins can recognize and bind to the canonical p53 response elements (p53-REs), two or more tandem repeats of RRRCWWGYYY, in the promoter regions of various genes. Owing to the difference in the sequence of DBD, p63 and p53 exhibit different preferences for binding sequences in target promoters. Additionally, distinctions of domains else than DBDs between different p63 isoforms may lead to nuance of DNA-binding preferences [4, 5]. p63 can also positively regulate expression of genes such as Skp2 via binding to intron regions [6].
Downstream targets of p63 proteins are involved in a variety of essential biological processes. As mentioned above, p63 and other members of p53 family share some common downstream targets, such as p21Waf1/Cip1, Puma, and Bax, enabling them to orchestrate to regulate cell cycle [7]. Like p53, TA isoforms of p63 can undoubtedly activate these downstream genes and consequently lead to cell cycle arrest, cell senescence and cell apoptosis. On the contrary, ΔNp63 proteins inhibit transcription of these genes, causing enhanced proliferation and cell survival. This effect of ΔNp63s is due to the lack of an intact TAD, and they can antagonize the transactivity of other p53 family proteins by means of forming inhibitory complexes with them or competitively binding to the p53-REs. Hence TAp63s enhance [3, 8,9,10], while ΔNp63s repress [3, 11], transcription of p21Waf1/Cip1, which can inhibit Cyclin E/Cdk2 to mediate cell senescence and to restrain cell proliferation [12]. ΔNp63, but not TAp63, can also negatively regulate p16Ink4a [13], which activates Rb via inhibition of Cyclin D/CDK4/6 [14] and facilitates the formation of senescence-associated heterochromatic foci (SAHF) to keep pro-proliferative genes in an inactive sate [15, 16].
It is traditionally accepted that TAp63s are trans-activators while ΔNp63s are trans-inhibitors [3]. However, mounting evidence demonstrates that ΔNp63s can also stimulate some downstream target genes, including Caspase-1 [17], Perp [18], K14, BPAG1 [19], MKP3 [20], Hsp70 [21]. This may be because that the N-terminal fragment composed of 14 amino acid residues functions as a TAD in the ΔNp63 proteins. Of note, some genes, such as K14 and MKP3, are transactivated by only ΔNp63s, but not TAp63s [19, 20]. And genes such as Hsp70 are reported to be up-regulated by ΔNp63α but down-regulated by TAp63γ [21]. Further study indicates that the N-terminal 68 amino acids of TAp63s may function as an extra trans-inhibitory domain. So TAp63γ demonstrates to repress transcription of Hsp70. However, TAD2 in TAp63α can eliminate this repression on Hsp70 transcription [21]. These findings suggest the roles of particular domains in different p63 isoforms in modulating their specific transactivation or trans-inhibition.
p63 in development: TAp63s or ΔNp63s are the leading actors?
Vast evidence shows that p63 gene mutation leads to ectodermal defects, including ectrodactyly, ectodermal dysplasia, and facial clefting syndrome (EEC), split hand/foot malformation syndrome (SHFM), limb–mammary syndrome (LMS), acro-dermato-ungual-lacrimal-tooth syndrome (ADULT), and ankyloblepharon-ectodermal dysplasia-clefting syndrome (AEC) [22,23,24,25,26]. These observations demonstrate the importance of p63 in development, particularly in development of ectoderm.
Besides mutation of p63, transversion or deletion of human chromosome 7, band q21.3–q22.1, which contains SLC25A13, DSS1, DLX5 and DLX6 genes, can also lead to limb malformation [26,27,28,29,30,31]. Studies have shown that p63 can bind to a cis-acting element in this segment, thereby regulating the expression of DLX5 and DLX6 [32, 33]. And simultaneous deletion of DLX5 and DLX6 has also been shown to result in limb defects [34,35,36]. Therefore, it is likely that p63 regulates limb development by controlling the transcription of DLX5 and DLX6.
TAp63 proteins are barely detectable in somatic cells, but they express at a relatively higher level in oocytes, where they play key roles in quality control through turning on genes responsible for cell apoptosis upon genotoxic stress [37,38,39]. It was also reported that TAp63s are the first isoforms expressed during mouse embryogenesis and are pivotal to initiation of epithelial stratification program and inhibition of terminal differentiation [40].
ΔNp63s are predominant isoforms encoded by p63 gene in tissues and organs, especially epithelial basal layer in embryonic ectoderm and ectoderm-derived tissues or organs [41,42,43]. During development, expression of ΔNp63s is stimulated by BMP2, BMP7 and FGF10 [44]. ΔNp63s can counteract TAp63s and promote maturation of embryonic epidermis [40]. Gerry Melino group found that ΔNp63s transactivate genes characteristic of epidermal basal layer and thymus, such as K14, FGFR2 and Jag2, while TAp63s transcribe genes characteristic of the superbasal layer, including Ets-1, K1, transglutaminases, and involucrin [19, 45, 46].
Consistent with evidence from human genetics, data from mouse model revealed that either pan-p63 or ΔNp63-specific knockout mice demonstrate dysplasia of limbs and epidermis. They are both deficient in ectodermal cells, leading to a lack of squamous epithelium and its derivatives, including breast, lacrimal gland and salivary glands [24, 47]. Using a transgenic mouse model, Rizzo et al. revealed that ΔNp63 overexpression results in atopic dermatitis via increasing many cytokines and chemokines, including IL-33 and IL-31 [48]. TAp63-specific knockout mice age prematurely and develop blisters, skin ulcerations, senescence of hair follicle-associated dermal and epidermal cells, and decreased hair morphogenesis, likely owing to a defect in maintenance of adult skin stem cells [49]. The similarity between pan-p63 knockout mice and ΔNp63-specific knockout mice indicates that ΔNp63s are the predominant isoforms of p63 gene regulating skin development [40].
p63 in tumor formation and progression: an oncogene or a tumor suppressor gene?
-
1.
Amplification or loss of p63 locus causes cancers?
Although p63 gene was originally cloned as a homologue of p53 and p73, which were both well known as tumor suppressors, it is rarely mutated in tumors [3, 50]. Therefore, the link between p63 and tumorigenesis remained obscure until it was reported that p63 gene is frequently amplified in primary cell lines derived from some squamous cell carcinoma (SCC): David Sidransky group employed fluorescent in situ hybridization (FISH) analysis and detected frequent amplification of p63 locus in primary lung SCC (LSCC) and head/neck SCC (HNSCC). Moreover, amplification of the p63 locus was accompanied by RNA and protein overexpression of its gene products ΔNp63α and ΔNp63ε, whose ectopic expression in Rat 1a endowed these fibroblast cells with characteristics of malignancies, significantly enhancing their colony growth and xenograft tumor formation. They also found that most LSCC with ΔNp63α overexpression simultaneously harbored p53 mutation [51]. Another investigation in non-small cell lung cancers demonstrated a similar observation: Pierre Massion et al. found that copy number of p63 gene and protein level of ΔNp63α were significantly increased in 88% of squamous carcinomas, 42% of large cell carcinomas and adenocarcinomas of lung [1]. These results suggest that p63 may function as an oncogene, whose amplification may lead to SCC likely in combination with p53 dysfunction.
However, evidence from mouse model revealed that loss of a p63 allele increases tumor predisposition and deteriorates tumor phenotype under the background of p53 or p73 heterozygosity. To investigate whether p53 family members genetically interact each other in tumor formation, Elsa Flores et al. intercrossed mice with heterozygote of p53, p73 or p63. They found that, compared with p53+/− mice, p53+/−; p63+/− mice spontaneously developed squamous cell carcinomas at a strikingly higher frequency. These tumors were found in multiple tissues including larynx, pharynx, cervix, and esophagus, and were more metastatic than those in p53+/− mice. On the other hand, compared to p73+/− mice, p73+/−; p63+/− mice demonstrated higher predisposition to mammary adenocarcinoma, salivary adenoma, squamous cell carcinoma, osteosarcoma, transitional cell carcinoma and rhabdomyosarcoma. Their study also revealed that tumors from p63+/− mice underwent loss of heterozygosity (LOH), which is one of the hallmarks of tumor suppressor gene inactivation [52]. Consistent with Elsa Flores’ data, analysis of tumorigenesis conducted by Alea Mills group indicated that heterozygosity of p63 significantly enhanced sarcoma development in p53-deficient mice [9]. Other independent groups reported that loss of p63 expression is associated with tumor progression and poor prognosis in human bladder carcinomas [53,54,55]. These investigations indicate that p63 gene has tumor-suppressive activities and loss of p63 locus may contribute to tumorigenesis.
-
2.
ΔNp63s: oncoproteins or tumor suppressor proteins?
Since p63 can encode two classes of protein isoforms, TAp63s and ΔNp63s, which were traditionally assumed to possess contrary functions in transcription regulation, the researchers tried to identify which isoform(s) is/are the prime culprit(s) in SCC tumorigenesis. The aforesaid investigations carried out by two independent groups revealed that ΔNp63α is the predominant isoform overexpressed in different cancer types, particularly squamous carcinomas, and ectopic overexpression of ΔNp63 isoforms in cultured cells can increase soft agar growth and tumor size in mice [1, 51]. And overexpression of ΔNp63α occurs in more than 80% of SCCs arising from head/neck [56, 57], lung [1], esophagus [58], and cervix [59], as well as some cases of basal breast carcinoma [60, 61]. In keeping with these observations, we found that ΔNp63α can promote cell proliferation and tumor formation [62], as well as prevent cancer cells from apoptosis upon genotoxic stress [63, 64]. According to these findings, p63 seems to exert its oncogenic functions via expressing ΔNp63α and other ΔNp63 proteins.
In the cases of ΔNp63s promoting tumorigenesis, ΔNp63s may antagonize p53 and TAp73 transactivities, consequently increasing transcription of the abovementioned genes involved in cell cycle arrest and apoptosis. As a result, cell proliferation and cell survival are enhanced, while cell senescence and cell apoptosis are inhibited [63, 65,66,67]. It was also documented that ΔNp63α may up-regulate transcription of Hsp70, which is a stress response protein and a determinant of cell death and cell transformation, to prime HNSCC [21], as well as that ΔNp63α can target transcription of chromatin remodeler Lsh to bypass oncogene-induced senescence (OIS) and drive tumorigenesis in vivo [68]. It was also reported that ΔNp63α positively regulates cell matrix adhesion molecules, including integrins α6, β1 and β4, as well as matrix protein Laminin-γ2. Ablation of ΔNp63α can cause a significant down-regulation of these proteins, resulting in cell death by anoikis, which can be perfectly rescued by restoring expression of ΔNp63α. This mechanism may interpret roles of ΔNp63α in tumorigenesis and tumor cell survival from another perspective [69]. On the other hand, Dennis McCance group found that in primary human foreskin keratinocytes expressing HPV16 E6/E7 genes, expression of pan p63 promotes cell migration, extracellular matrix (ECM) remodelling and cell invasion via inducing Src–FAK complex expression/activation [70]. In one of our recent work, we found that metformin, a drug for type II diabetes, promoted WWP1-mediated proteasomal degradation of ΔNp63α, resulting in disruption of cell matrix adhesion and subsequent apoptosis in human squamous carcinoma cells [71]. In addition, data from our group and other labs revealed that ΔNp63α activates c-Myc via several mechanisms, likely promoting cell cycle progression and tumorigenesis [72, 73].
As mentioned above, loss of p63 expression or locus is reported to associate with tumorigenesis or progression, particularly metastasis, in a variety of cancers. Among these cancer types, ΔNp63s are the predominant p63 isoforms in some of their normal tissues, such as breast [74,75,76], urothelium [53, 77], prostate [78, 79], and cervix [80]. These observations indicate that ΔNp63s may possess anti-metastasis activities. Further research using an intravenous injection assay proved that ectopic expression of ΔNp63α significantly inhibited metastasis of malignant spindle carcinoma D3S2 cells to the lungs [81].
During the initiation of metastasis for cancer progression, epithelial cells have to lose their cell polarity and cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells. This process is termed epithelial–mesenchymal transition (EMT). A reverse process termed mesenchymal–epithelial transition (MET) is believed to participate in the establishment and stabilization of distant metastases by allowing cancerous cells to regain epithelial properties and integrate into distant organs [82]. Investigations on the molecular mechanism demonstrated that ΔNp63s inhibit cell metastasis via regulating genes involved in cell adhesion, motility and migration. As has been noted previously, positive regulation of molecules involved in cell matrix adhesion may contribute to tumor repressive activities of ΔNp63s [69]. Additionally, this mechanism may also prevent tumor cells from detaching the matrix. Cell–cell adhesion is another aspect of epithelial properties, in which the transmembrane protein Perp is an important factor to maintain proper desmosome structure and function. Perp is a direct downstream target of various p53-family proteins including ΔNp63s [18]. Logically, ΔNp63s may inhibit EMT via facilitating cell matrix and cell–cell adhesion. On the other hand, ablation or reduced expression of ΔNp63s were reported to result in up-regulation of other proteins involved in cell adhesion, motility and migration, such as N-cadherin, L1 cell adhesion molecule (L1CAM), Periostin, and Wnt-5a [83]. It was also documented that ΔNp63α inhibits cell invasion via up-regulating inhibitor of differentiation-3 (Id-3), which can down-regulate expression of matrix metallopeptidase 2 (MMP2) to prevent from cleaving components of the extracellular matrix [84]. We recently reported that activation of oncogenic phosphatidylinositol 3 kinase (PI3K), Ras, and Her2 signaling can down-regulate ΔNp63α in cancer development via activating Akt, which in turn phosphorates Foxo3a to prevent it from binding to p63 promoter region. This decrease in ΔNp63α leads to down-regulation of its downstream targets including E-cadherin, Desmoplakin and Par3, resulting in enhanced cell motility and tumor metastasis [85]. In some of our other studies, we identified the metastasis suppressor CD82 (cluster of differentiation 82) and mitogen-activated protein kinase phosphatase 3 (MKP3) as direct ΔNp63α transcriptional targets, and found that ΔNp63α up-regulates CD82 or MKP3 to inhibit caner metastasis [20, 86]. It was also reported that ΔNp63s are involved in regulation of crucial players of EMT such as Snails and TGF-β [87,88,89], as well as p53-, particularly mutant p53-, mediated regulation of metastasis [81, 90, 91]. It remains unclear whether p63 is involved in MET process during the colonization and formation of a metastatic nodule [92].
-
3.
TAp63α: simply as a tumor suppressor?
Since a great deal of evidence demonstrates that TAp63s transactivate various genes to promote cell cycle arrest and cell apoptosis [8, 38, 93, 94], it is almost indisputably accepted that TAp63 isoforms have tumor-suppressive activity [95]. Using a TAp63-specific knockout mouse model, Alea Mills group found that TAp63 deficiency compromises Ras-induced senescence, enhances proliferation and promotes tumorigenesis in the context of p53 deficiency. Exogenous expression of TAp63s, including TAp63α, TAp63β and TAp63γ, can induce cell senescence in cultured cells and inhibit tumor formation upon xenograft implantation in nude mice [9]. Elsa Flores groups employed another conditional knockout mouse model and found that TAp63s suppress cancer metastasis through up-regulating miR-130b and Dicer [96]. TAp63s can directly bind to and transactivate the promoter of endoribonuclease Dicer, which was responsible for processing of mcroRNAs and involved in cancer metastasis [97, 98]. Deficiency of TAp63s leads to down-regulation of Dicer and a decrease in spontaneous development of highly metastatic tumors. Processing of various metastasis-related microRNAs, including miR-10b, miR-200b, miR-200c, miR-34a and miR-130b, is deficient in TAp63−/− mice. TAp63s can also directly bind to and transactivate miR-130b promoter. Most importantly, restore of either Dicer or miR-130b partially rescues invasive phenotype in TAp63−/− MEFs, while simultaneous restore of both molecules perfectly reverses invasion induced by TAp63 deficiency [96]. In addition, TAp63s can also potently activate transcription of Perp to inhibit metastasis [18].
Though TA isoforms of p63 are once assumed to induce cell senescence, which contributes to tumor-suppressive activities of TAp63s [9], TAp63-specific conditional knockout mice employed by Elsa Flores group demonstrate enhanced cell senescence [49]. This is likely due to genomic instability and increased DNA damage resulted from TAp63 deficiency. And this senescence induced by TAp63 deficiency occurs not only in dermal precursor cells [49], but also in osteosarcomas and rhabdomyosarcomas in TAp63−/−; p53+/− mice [96]. This observation is in conflict with abovementioned data from Alea Mills group that TAp63s induce cell senescence [9].
Additionally, despite the widespread concept of TAp63s as tumor suppressors, Roberta Malaguarnera et al. reported that TAp63α protein is in a high percentage of thyroid carcinomas, but not in normal thyroid cells or benign thyroid adenomas [99]. In these thyroid cancer cells, the tumor-suppressive activities of TAp63α are absent, because either endogenous or exogenous TAp63α fails to transactivate its downstream genes. On the contrary, TAp63α seems to antagonize effects of p53 on its target genes, cell viability and foci formation in these cells. Moreover, transactivity of p53 in thyroid cancer cells can be strikingly elevated by TAp63α silencing. These oncogenic effects of TAp63α likely depend on its C-terminus, which contains a unique sterile alpha motif (SAM) and a trans-inhibitory domain (TID), since neither TAp63β nor TAp63γ are still able to induce the target genes and to exert tumor-restraining effects in thyroid cancer cells [99]. Another investigation demonstrated that mRNA levels of TAp63s, but not ΔNp63s, are higher in high-grade follicular lymphomas compared to non-neoplastic lymphocytes. This overexpression of p63 is independent of gene amplification [100]. Whether and how up-regulated TAp63s are involved in formation and progression of lymphomas in these cases requires further investigation.
Concluding remarks: a double dealer depending on context
p63 gene can encode two groups of proteins, namely TA and ΔN isoforms [2]. Increasing evidence demonstrates that transactivities of p63 proteins are much complicated: besides their trans-repressive activities [3, 11, 13], ΔNp63s also possess transactivities for some downstream genes [19, 21]; TAp63s are generally transactivators [3, 9, 96], but they may lose their transactivities even act as trans-inhibitors in certain scenarios [21, 99]. Evidence from human genetics and mouse model reveals that p63 gene is essential for organism development [19, 24, 40, 41, 47, 49]: TAp63s are expressed at a relatively high level in oocytes and during early stage of embryogenesis to maintain certain progenitor cells, particularly of skin stem cells; expression of ΔNp63s gathers along with the differentiation of ectoderm layer to promote its maturation and stratification of epithelium.
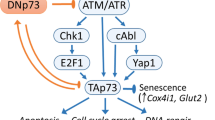
TA and ΔN isoforms of p63 regulate tumor formation, growth and metastasis via multiple mechanisms (Fig. 2). TAp63s are generally assumed to function as tumor suppressor proteins, because they can transactivate a batch of genes to induce cell cycle arrest and cell apoptosis [8, 38, 93, 94]. Evidence from mouse models also reveals that TAp63s repress tumorigenesis and metastasis [9, 96]. Tumor-suppressive activity of TAp63s seems to depend on genetic background or cell type: TAp63 deficiency induces cell senescence in normal epidermal cells and epithelium-derived cancer cells [49, 96], but increases proliferation and enhances Ras-mediated oncogenesis in the context of p53 deficiency in vivo [9]; in some thyroid carcinomas, TAp63α loses its tumor-suppressive activity and even exhibits oncogenic effects via antagonizing p53 transactivity [99]. ΔNp63s also have dual effects on cancers: in various types of squamous cell carcinomas, they are overexpressed as a consequence of p63 gene amplification and function as oncoproteins to initiate tumor formation [1, 51]. Oncogenic effects of ΔNp63s may be due to their activities on cell proliferation and cell survival [63, 65,66,67,68,69]. On the other hand, ΔNp63s can also transactivate a vast body of genes involved in EMT [18, 20, 65, 69, 82, 84]. This may account for the observation that ΔNp63s can suppress metastasis during the progress of some cancer types [81, 85], though some evidence indicates that p63, including ΔNp63 isoforms, can drive cell invasion under some circumstances [70].

Roles of p63 in tumor formation, growth and metastasis. TAp63s mainly act as tumor suppressors by inhibiting cell proliferation, survival and tumor metastasis, and occasionally exert oncogenic activity via repressing p53 trans-activity. ΔNp63s promote formation and growth of some epithelium-derived tumors through enhanced cell proliferation and survival. On the other hand, ΔNp63s repress tumor metastasis via inhibition of EMT. A full arrow means a positive regulation, while a blunt arrow represents an inhibition
Since ΔN and TA isoforms of p63 can, respectively, promote or inhibit tumor initiation, effects of p63 gene on tumorigenesis are delicate. In normal somatic cells, oncogenic ΔNp63s are balanced with tumor-suppressive TAp63s, TAp73s and p53. Under certain scenarios, particularly in the absence of sufficient functional p53 or p73, either amplification or loss of p63 gene locus may exacerbate this subtle imbalance, leading to predisposition to cancer.
Owing to dual roles of p63s in cancer formation and progression, future investigations are required to elucidate how p63 isoforms are regulated at either transcriptional or post-transcriptional levels, and how they are regulated to exert their oncogenic or tumor-suppressive activities in different cell types, as well as how they switch their different activities at different stage of tumor development. With these questions addressed, it would be possible to explore some small molecule drugs targeting specific p63 isoforms, their regulators or downstream genes for cancer therapy according to different scenarios.
References
Massion PP, Taflan PM, Jamshedur Rahman SM, Yildiz P, Shyr Y, Edgerton ME, Westfall MD, Roberts JR, Pietenpol JA, Carbone DP, Gonzalez AL (2003) Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res 63(21):7113–7121
Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, D’Erchia AM (2009) Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res 37(18):6092–6104
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F (1998) p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2(3):305–316. doi:10.1016/S1097-2765(00)80275-0
Osada M, Park HL, Nagakawa Y, Yamashita K, Fomenkov A, Kim MS, Wu G, Nomoto S, Trink B, Sidransky D (2005) Differential recognition of response elements determines target gene specificity for p53 and p63. Mol Cell Biol 25(14):6077–6089
Perez CA, Ott J, Mays DJ, Pietenpol JA (2007) p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene 26(52):7363–7370
McDade SS, Patel D, McCance DJ (2011) p63 maintains keratinocyte proliferative capacity through regulation of Skp2–p130 levels. J Cell Sci 124(Pt 10):1635–1643
Murray-Zmijewski F, Lane DP, Bourdon JC (2006) p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 13(6):962–972
Osada M, Ohba M, Kawahara C, Ishioka C, Kanamaru R, Katoh I, Ikawa Y, Nimura Y, Nakagawara A, Obinata M, Ikawa S (1998) Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat Med 4(7):839–843
Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, Vogel H, Mills AA (2009) TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol 11(12):1451–1457. doi:10.1038/ncb1988
Lo Iacono M, Di Costanzo A, Calogero RA, Mansueto G, Saviozzi S, Crispi S, Pollice A, La Mantia G, Calabro V (2006) The Hay Wells syndrome-derived TAp63alphaQ540L mutant has impaired transcriptional and cell growth regulatory activity. Cell Cycle 5(1):78–87
Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA (2003) The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 23(7):2264–2276
Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120(4):513–522. doi:10.1016/j.cell.2005.02.003
Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA (2005) p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev 19(17):1986–1999
Serrano M, Hannon GJ, Beach D (1993) A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366(6456):704–707
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113(6):703–716
Takahashi A, Ohtani N, Hara E (2007) Irreversibility of cellular senescence: dual roles of p16INK4a/Rb-pathway in cell cycle control. Cell Div 2:10
Celardo I, Grespi F, Antonov A, Bernassola F, Garabadgiu AV, Melino G, Amelio I (2013) Caspase-1 is a novel target of p63 in tumor suppression. Cell Death Dis 4:e645
Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, Mills AA, Attardi LD (2005) Perp is a p63-regulated gene essential for epithelial integrity. Cell 120(6):843–856. doi:10.1016/j.cell.2005.01.008
Candi E, Rufini A, Terrinoni A, Dinsdale D, Ranalli M, Paradisi A, De Laurenzi V, Spagnoli LG, Catani MV, Ramadan S, Knight RA, Melino G (2006) Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ 13(6):1037–1047
Bergholz J, Zhang Y, Wu J, Meng L, Walsh EM, Rai A, Sherman MY, Xiao ZX (2013) DeltaNp63alpha regulates Erk signaling via MKP3 to inhibit cancer metastasis. Oncogene 33(2):212–224. doi:10.1038/onc.2012.564
Wu G, Osada M, Guo Z, Fomenkov A, Begum S, Zhao M, Upadhyay S, Xing M, Wu F, Moon C, Westra WH, Koch WM, Mantovani R, Califano JA, Ratovitski E, Sidransky D, Trink B (2005) DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res 65(3):758–766
van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG (2001) p63 Gene mutations in EEC syndrome, limb-mammary syndrome, and isolated split hand–split foot malformation suggest a genotype–phenotype correlation. Am J Hum Genet 69(3):481–492
Duijf PH, Vanmolkot KR, Propping P, Friedl W, Krieger E, McKeon F, Dotsch V, Brunner HG, van Bokhoven H (2002) Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum Mol Genet 11(7):799–804
Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398(6729):714–718. doi:10.1038/19539
Berdon-Zapata V, Granillo-Alvarez M, Valdes-Flores M, Garcia-Ortiz JE, Kofman-Alfaro S, Zenteno JC (2004) p63 gene analysis in Mexican patients with syndromic and non-syndromic ectrodactyly. J Orthop Res 22(1):1–5. doi:10.1016/S0736-0266(03)00166-9
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P (2000) Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 67(1):59–66. doi:10.1086/302972
Scherer SW, Poorkaj P, Allen T, Kim J, Geshuri D, Nunes M, Soder S, Stephens K, Pagon RA, Patton MA et al (1994) Fine mapping of the autosomal dominant split hand/split foot locus on chromosome 7, band q21.3-q22.1. Am J Hum Genet 55(1):12–20
Duijf PH, van Bokhoven H, Brunner HG (2003) Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 12(Spec No 1):R51–R60
Faiyaz-Ul-Haque M, Zaidi SH, King LM, Haque S, Patel M, Ahmad M, Siddique T, Ahmad W, Tsui LC, Cohn DH (2005) Fine mapping of the X-linked split-hand/split-foot malformation (SHFM2) locus to a 5.1-Mb region on Xq26.3 and analysis of candidate genes. Clin Genet 67(1):93–97. doi:10.1111/j.1399-0004.2004.00369.x
Del Campo M, Jones MC, Veraksa AN, Curry CJ, Jones KL, Mascarello JT, Ali-Kahn-Catts Z, Drumheller T, McGinnis W (1999) Monodactylous limbs and abnormal genitalia are associated with hemizygosity for the human 2q31 region that includes the HOXD cluster. Am J Hum Genet 65(1):104–110. doi:10.1086/302467
Ugur SA, Tolun A (2008) Homozygous WNT10b mutation and complex inheritance in split-hand/foot malformation. Hum Mol Genet 17(17):2644–2653. doi:10.1093/hmg/ddn164
Lo Iacono N, Mantero S, Chiarelli A, Garcia E, Mills AA, Morasso MI, Costanzo A, Levi G, Guerrini L, Merlo GR (2008) Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 135(7):1377–1388. doi:10.1242/dev.011759
Kouwenhoven EN, van Heeringen SJ, Tena JJ, Oti M, Dutilh BE, Alonso ME, de la Calle-Mustienes E, Smeenk L, Rinne T, Parsaulian L, Bolat E, Jurgelenaite R, Huynen MA, Hoischen A, Veltman JA, Brunner HG, Roscioli T, Oates E, Wilson M, Manzanares M, Gomez-Skarmeta JL, Stunnenberg HG, Lohrum M, van Bokhoven H, Zhou H (2010) Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet 6(8):e1001065. doi:10.1371/journal.pgen.1001065
Simeone A, Acampora D, Pannese M, D’Esposito M, Stornaiuolo A, Gulisano M, Mallamaci A, Kastury K, Druck T, Huebner K et al (1994) Cloning and characterization of two members of the vertebrate Dlx gene family. Proc Natl Acad Sci USA 91(6):2250–2254
Robledo RF, Rajan L, Li X, Lufkin T (2002) The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev 16(9):1089–1101. doi:10.1101/gad.988402
Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G (1999) Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development 126(17):3795–3809
Deutsch GB, Zielonka EM, Coutandin D, Weber TA, Schafer B, Hannewald J, Luh LM, Durst FG, Ibrahim M, Hoffmann J, Niesen FH, Senturk A, Kunkel H, Brutschy B, Schleiff E, Knapp S, Acker-Palmer A, Grez M, McKeon F, Dotsch V (2011) DNA damage in oocytes induces a switch of the quality control factor TAp63alpha from dimer to tetramer. Cell 144(4):566–576. doi:10.1016/j.cell.2011.01.013
Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, Mattei M, Candi E, De Felici M, Melino G, Cesareni G (2009) Inhibition of the c-Abl–TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med 15(10):1179–1185. doi:10.1038/nm.2033
Deutsch GB, Zielonka EM, Coutandin D, Dotsch V (2011) Quality control in oocytes: domain–domain interactions regulate the activity of p63. Cell Cycle 10(12):1884–1885
Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR (2004) p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 18(2):126–131
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A (1999) p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398(6729):708–713. doi:10.1038/19531
Kouwenhoven EN, Oti M, Niehues H, van Heeringen SJ, Schalkwijk J, Stunnenberg HG, van Bokhoven H, Zhou H (2015) Transcription factor p63 bookmarks and regulates dynamic enhancers during epidermal differentiation. EMBO Rep 16(7):863–878
Chakravarti D, Su X, Cho MS, Bui NH, Coarfa C, Venkatanarayan A, Benham AL, Flores Gonzalez RE, Alana J, Xiao W, Leung ML, Vin H, Chan IL, Aquino A, Muller N, Wang H, Cooney AJ, Parker-Thornburg J, Tsai KY, Gunaratne PH, Flores ER (2014) Induced multipotency in adult keratinocytes through down-regulation of DeltaNp63 or DGCR8. Proc Natl Acad Sci USA 111(5):E572–E581
Laurikkala J, Mikkola ML, James M, Tummers M, Mills AA, Thesleff I (2006) p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development 133(8):1553–1563
Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, Melino G, Aberdam D (2011) DeltaNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ 18(5):887–896. doi:10.1038/cdd.2010.159
Candi E, Rufini A, Terrinoni A, Giamboi-Miraglia A, Lena AM, Mantovani R, Knight R, Melino G (2007) DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc Natl Acad Sci USA 104(29):11999–12004. doi:10.1073/pnas.0703458104
Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, Sinha S (2012) DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development 139(4):772–782
Rizzo JM, Oyelakin A, Min S, Smalley K, Bard J, Luo W, Nyquist J, Guttman-Yassky E, Yoshida T, De Benedetto A, Beck LA, Sinha S, Romano RA (2016) DeltaNp63 regulates IL-33 and IL-31 signaling in atopic dermatitis. Cell Death Differ 23(6):1073–1085
Su X, Paris M, Gi YJ, Tsai KY, Cho MS, Lin YL, Biernaskie JA, Sinha S, Prives C, Pevny LH, Miller FD, Flores ER (2009) TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell 5(1):64–75
Sunahara M, Shishikura T, Takahashi M, Todo S, Yamamoto N, Kimura H, Kato S, Ishioka C, Ikawa S, Ikawa Y, Nakagawara A (1999) Mutational analysis of p51A/TAp63gamma, a p53 homolog, in non-small cell lung cancer and breast cancer. Oncogene 18(25):3761–3765
Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE, Ratovitski EA, Jen J, Sidransky D (2000) AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 97(10):5462–5467
Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, Yang A, McKeon F, Jacks T (2005) Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7(4):363–373. doi:10.1016/j.ccr.2005.02.019
Koga F, Kawakami S, Fujii Y, Saito K, Ohtsuka Y, Iwai A, Ando N, Takizawa T, Kageyama Y, Kihara K (2003) Impaired p63 expression associates with poor prognosis and uroplakin III expression in invasive urothelial carcinoma of the bladder. Clin Cancer Res 9(15):5501–5507
Park BJ, Lee SJ, Kim JI, Lee SJ, Lee CH, Chang SG, Park JH, Chi SG (2000) Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer Res 60(13):3370–3374
Urist MJ, Di Como CJ, Lu ML, Charytonowicz E, Verbel D, Crum CP, Ince TA, McKeon FD, Cordon-Cardo C (2002) Loss of p63 expression is associated with tumor progression in bladder cancer. Am J Pathol 161(4):1199–1206
Weber A, Bellmann U, Bootz F, Wittekind C, Tannapfel A (2002) Expression of p53 and its homologues in primary and recurrent squamous cell carcinomas of the head and neck. Int J Cancer 99(1):22–28. doi:10.1002/ijc.10296
Sniezek JC, Matheny KE, Westfall MD, Pietenpol JA (2004) Dominant negative p63 isoform expression in head and neck squamous cell carcinoma. Laryngoscope 114(12):2063–2072
Hu H, Xia SH, Li AD, Xu X, Cai Y, Han YL, Wei F, Chen BS, Huang XP, Han YS, Zhang JW, Zhang X, Wu M, Wang MR (2002) Elevated expression of p63 protein in human esophageal squamous cell carcinomas. Int J Cancer 102(6):580–583. doi:10.1002/ijc.10739
Wang TY, Chen BF, Yang YC, Chen H, Wang Y, Cviko A, Quade BJ, Sun D, Yang A, McKeon FD, Crum CP (2001) Histologic and immunophenotypic classification of cervical carcinomas by expression of the p53 homologue p63: a study of 250 cases. Hum Pathol 32(5):479–486
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Matos I, Dufloth R, Alvarenga M, Zeferino LC, Schmitt F (2005) p63, cytokeratin 5, and P-cadherin: three molecular markers to distinguish basal phenotype in breast carcinomas. Virchows Arch 447(4):688–694
Li C, Chang DL, Yang Z, Qi J, Liu R, He H, Li D, Xiao ZX (2013) Pin1 modulates p63alpha protein stability in regulation of cell survival, proliferation and tumor formation. Cell Death Dis 4:e943
Li X, Chen J, Yi Y, Li C, Zhang Y (2012) DNA damage down-regulates DeltaNp63alpha and induces apoptosis independent of wild type p53. Biochem Biophys Res Commun 423(2):338–343. doi:10.1016/j.bbrc.2012.05.126
Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, Ratovitski E, Sidransky D (2011) DeltaNp63alpha confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res 71(3):1167–1176. doi:10.1158/0008-5472.CAN-10-1481
DeYoung MP, Johannessen CM, Leong CO, Faquin W, Rocco JW, Ellisen LW (2006) Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res 66(19):9362–9368. doi:10.1158/0008-5472.CAN-06-1619
Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW (2006) p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 9(1):45–56. doi:10.1016/j.ccr.2005.12.013
Ramsey MR, He L, Forster N, Ory B, Ellisen LW (2011) Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res 71(13):4373–4379
Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, Muthuswamy SK, Mills AA (2011) DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 8(2):164–176. doi:10.1016/j.stem.2010.12.009
Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, Brugge JS, Ellisen LW (2006) p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol 8(6):551–561. doi:10.1038/ncb1420
Srivastava K, Pickard A, McDade S, McCance DJ (2017) p63 drives invasion in keratinocytes expressing HPV16 E6/E7 genes through regulation of Src-FAK signalling. Oncotarget 8(10):16202–16219
Yi Y, Chen D, Ao J, Sun S, Wu M, Li X, Bergholz J, Zhang Y, Xiao ZX (2017) Metformin promotes AMP-activated protein kinase-independent suppression of deltaNp63alpha protein expression and inhibits cancer cell viability. J Biol Chem 292(13):5253–5261
Wu N, Rollin J, Masse I, Lamartine J, Gidrol X (2012) p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J Biol Chem 287(8):5627–5638. doi:10.1074/jbc.M111.328120
Han A, Li J, Li Y, Wang Y, Bergholz J, Zhang Y, Li C, Xiao ZhX (2016) p63alpha modulates c-Myc activity via direct interaction and regulation of MM1 protein stability. Oncotarget 7(28):44277–44287
Barbareschi M, Pecciarini L, Cangi MG, Macri E, Rizzo A, Viale G, Doglioni C (2001) p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol 25(8):1054–1060
Wang X, Mori I, Tang W, Nakamura M, Nakamura Y, Sato M, Sakurai T, Kakudo K (2002) p63 expression in normal, hyperplastic and malignant breast tissues. Breast Cancer 9(3):216–219
Stefanou D, Batistatou A, Nonni A, Arkoumani E, Agnantis NJ (2004) p63 expression in benign and malignant breast lesions. Histol Histopathol 19(2):465–471
Koga F, Kawakami S, Kumagai J, Takizawa T, Ando N, Arai G, Kageyama Y, Kihara K (2003) Impaired delta Np63 expression associates with reduced beta-catenin and aggressive phenotypes of urothelial neoplasms. Br J Cancer 88(5):740–747
Vanaja DK, Cheville JC, Iturria SJ, Young CY (2003) Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res 63(14):3877–3882
Signoretti S, Waltregny D, Dilks J, Isaac B, Lin D, Garraway L, Yang A, Montironi R, McKeon F, Loda M (2000) p63 is a prostate basal cell marker and is required for prostate development. Am J Pathol 157(6):1769–1775
Quade BJ, Yang A, Wang Y, Sun D, Park J, Sheets EE, Cviko A, Federschneider JM, Peters R, McKeon FD, Crum CP (2001) Expression of the p53 homologue p63 in early cervical neoplasia. Gynecol Oncol 80(1):24–29
Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S (2009) A mutant-p53/smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137(1):87–98
Yang J, Weinberg RA (2008) Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14(6):818–829
Barbieri CE, Tang LJ, Brown KA, Pietenpol JA (2006) Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res 66(15):7589–7597. doi:10.1158/0008-5472.CAN-06-2020
Higashikawa K, Yoneda S, Tobiume K, Saitoh M, Taki M, Mitani Y, Shigeishi H, Ono S, Kamata N (2009) DeltaNp63alpha-dependent expression of Id-3 distinctively suppresses the invasiveness of human squamous cell carcinoma. Int J Cancer 124(12):2837–2844
Hu L, Liang S, Chen H, Lv T, Wu J, Chen D, Wu M, Sun S, Zhang H, You H, Ji H, Zhang Y, Bergholz J, Xiao ZJ (2017) DeltaNp63alpha is a common inhibitory target in oncogenic PI3K/Ras/Her2-induced cell motility and tumor metastasis. Proc Natl Acad Sci U S A 114(20):E3964–E3973
Wu J, Liang S, Bergholz J, He H, Walsh EM, Zhang Y, Xiao ZX (2014) DeltaNp63alpha activates CD82 metastasis suppressor to inhibit cancer cell invasion. Cell Death Dis 5:e1280
Melino G (2011) p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ 18(9):1487–1499. doi:10.1038/cdd.2011.81
Higashikawa K, Yoneda S, Tobiume K, Taki M, Shigeishi H, Kamata N (2007) Snail-induced down-regulation of DeltaNp63alpha acquires invasive phenotype of human squamous cell carcinoma. Cancer Res 67(19):9207–9213
Lindsay J, McDade SS, Pickard A, McCloskey KD, McCance DJ (2011) Role of DeltaNp63gamma in epithelial to mesenchymal transition. J Biol Chem 286(5):3915–3924
Muller PA, Vousden KH, Norman JC (2011) p53 and its mutants in tumor cell migration and invasion. J Cell Biol 192(2):209–218. doi:10.1083/jcb.201009059
Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139(7):1327–1341
Olsen JR, Oyan AM, Rostad K, Hellem MR, Liu J, Li L, Micklem DR, Haugen H, Lorens JB, Rotter V, Ke XS, Lin B, Kalland KH (2013) p63 attenuates epithelial to mesenchymal potential in an experimental prostate cell model. PLoS One 8(5):e62547
Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, Lena AM, Candi E, Terrinoni A, Catani MV, Oren M, Melino G, Krammer PH, Stremmel W, Muller M (2005) TAp63alpha induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J 24(13):2458–2471. doi:10.1038/sj.emboj.7600708
Wu G, Nomoto S, Hoque MO, Dracheva T, Osada M, Lee CC, Dong SM, Guo Z, Benoit N, Cohen Y, Rechthand P, Califano J, Moon CS, Ratovitski E, Jen J, Sidransky D, Trink B (2003) DeltaNp63alpha and TAp63alpha regulate transcription of genes with distinct biological functions in cancer and development. Cancer Res 63(10):2351–2357
Li C, Xiao ZX (2014) Regulation of p63 protein stability via ubiquitin-proteasome pathway. Biomed Res Int 2014:175721. doi:10.1155/2014/175721
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, Leung ML, El-Naggar A, Creighton CJ, Suraokar MB, Wistuba I, Flores ER (2010) TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467(7318):986–990. doi:10.1038/nature09459
Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T (2009) Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 23(23):2700–2704
Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, Parenti AR, Daidone MG, Bicciato S, Piccolo S (2010) A MicroRNA targeting dicer for metastasis control. Cell 141(7):1195–1207
Malaguarnera R, Mandarino A, Mazzon E, Vella V, Gangemi P, Vancheri C, Vigneri P, Aloisi A, Vigneri R, Frasca F (2005) The p53-homologue p63 may promote thyroid cancer progression. Endocr Relat Cancer 12(4):953–971. doi:10.1677/erc.1.00968
Pruneri G, Fabris S, Dell’Orto P, Biasi MO, Valentini S, Del Curto B, Laszlo D, Cattaneo L, Fasani R, Rossini L, Manzotti M, Bertolini F, Martinelli G, Neri A, Viale G (2005) The transactivating isoforms of p63 are overexpressed in high-grade follicular lymphomas independent of the occurrence of p63 gene amplification. J Pathol 206(3):337–345
Acknowledgements
This work was supported by National Natural Science Foundation of China (#31671423) and Science and Technology Department of Sichuan Province (#2016JY0152). We thank the members of Zhi-Xiong Xiao lab for stimulative discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Chen, Y., Peng, Y., Fan, S. et al. A double dealing tale of p63: an oncogene or a tumor suppressor. Cell. Mol. Life Sci. 75, 965–973 (2018). https://doi.org/10.1007/s00018-017-2666-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2666-y