
Abstract
Meta-QTL (MQTL) analysis for drought tolerance was undertaken in bread wheat to identify consensus and robust MQTLs using 340 known QTLs from 11 earlier studies; 13 MQTLs located on 6 chromosomes (1D, 3B, 5A, 6D, 7A and 7D) were identified, with maximum of 4 MQTLs on chromosome 5A. Mean confidence intervals for MQTLs were much narrower (mean, 6.01 cM; range 2.07–19.46 cM), relative to those in original QTLs (mean, 13.6 cM; range, 1.0–119.1 cM). Two MQTLs, namely MQTL4 and MQTL12, were major MQTLs with potential for use in marker-assisting breeding. As many as 228 candidate genes (CGs) were also identified using 6 of the 13 MQTLs. In-silico expression analysis of these 228 CGs allowed identification of 14 important CGs, with + 3 to − 8 fold change in expression under drought (relative to normal conditions) in a tolerant cv. named TAM107. These CGs encoded proteins belonging to the following families: NAD-dependent epimerase/dehydratase, protein kinase, NAD(P)-binding domain protein, heat shock protein 70 (Hsp70), glycosyltransferase 2-like, etc. Important MQTLs and CGs identified in the present study should prove useful for future molecular breeding and for the study of molecular basis of drought tolerance in cereals in general and wheat in particular.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Wheat is an important staple food crop world-wide with a history of remarkable success in improvement of productivity and production. The global bread wheat production increased steadily from 537.5 mt in 1995/96 (https://apps.fas.usda.gov/psdonline_legacy/circulars/grain-wheat.pdf) to ~ 759 mt in 2018–2019 (https://www.uswheat.org/wheatletter/first-look-at-2019-20-by-usda-sees-another-record-world-wheat-crop/). It occupies 17% of crop acreage globally, feeding about 2.5 billion people (40%) in ~ 90 countries and provides 20% of total food calories and protein for human nutrition (Gupta et al. 2008). India is the third largest producer of wheat (after USA and China) in the world with an estimated production of about 106.21 mt during 2019–2020 (Economic Times, India, Jan 17 2020). According to estimates of FAO, world would require around 840 mt of wheat by 2050 (~ 24% increase over 30 years). This demand excludes the requirement of animal feed and adverse impacts of climate change on wheat production.
Drought affects 42% of global wheat area and can reduce grain yield by 58–92% (Kosina et al. 2007; Farooq et al. 2014). Therefore, drought stress continues to be an important challenge to agricultural researchers and plant breeders. It has also been estimated that drought alone causes more annual loss in crop yield than all pathogens combined together and contributes to annual global loss of $30 billion (Gupta et al. 2020a, b). It is also estimated that by the year 2050, the demand for water will double and the supply of fresh water will reduce by 50% due to climate change (Gupta et al. 2020a, b). Estimates also indicate that by 2025, as many as 1.8 billion people will face absolute water shortage and 65% of the world’s population will live under water-stressed environments (Nezhadahmadi et al. 2013). Thus, in order to meet the projected demand of wheat to feed the ever-increasing human population, we need to mitigate the adverse effects of drought on productivity and production of wheat. For the sustainable development of wheat varieties with high genetic yield potential that are also tolerant to drought stress, we need to identify QTLs for drought tolerance and also the candidate genes underlying the QTLs. As many as > 1200 QTLs have already been reported for different drought responsive traits in wheat (Gupta et al. 2020a, b), but the discovery of major robust QTLs with closely associated markers with a high potential for molecular breeding still remains a challenge.
Meta-QTL (MQTL) analysis has been used to identify the consensus and robust QTLs through the use of information reported in multiple studies for the reliability of their location and effect across different genetic backgrounds and environments, as well as to refine QTL positions on a consensus map (Goffinet and Gerber 2000). MQTL analysis is generally conducted using the software Biomercator V4.2, which is based on the algorithm developed by Goffinet and Gerber (2000) in which a maximum likelihood function is used to determine the following: (1) the number of MQTLs given a set of input QTLs on a common genetic map; (2) the consensus positions of detected MQTLs based on the variance in positions of input QTLs, and (3) 95% confidence interval (CI) for each MQTL based on the variance in marker intervals of input QTL (Arcade et al. 2004). More recently, whole genome sequences (pseudomolecules) have also been utilized for meta-QTL analysis (Xu et al. 2019).
MQTL analysis requires two necessary conditions, namely a consensus map with high-density markers (Hong et al. 2010; Shirasawa et al. 2013) and a large number of independent QTLs for the same trait identified from different genetic backgrounds and environments (Goffinet and Gerber 2000). Consensus QTLs obtained from meta-analysis of a number of QTLs related to a target trait at a 95% confidence interval (CI) are called MQTLs. In the past, MQTL analysis has been successfully carried out for a variety of traits in a number of crops including the following: (1) maize (Chardon et al. 2004; Wang et al. 2006; Coque et al. 2008; Truntzler et al. 2010; Hao et al. 2010; Li et al. 2011; Chen et al. 2017; Guo et al. 2018; Zhao et al. 2018), (2) rice (Ballini et al. 2008; Norton et al. 2008; Khowaja et al. 2009; Islam et al. 2019; Khahani et al. 2020), (3) cotton (Rong et al. 2007), (4) rapeseed (Shi et al. 2009), (5) potato (Danan et al. 2011), (6) cocoa (Lanaud et al. 2009), (7) soybean (Guo et al. 2006; Sun et al. 2012) and (8) apricot (Marandel et al. 2009).
MQTL analysis in wheat has also been successfully utilized to detect definitive QTL regions not only for drought tolerance (20 MQTLs for 502 independent QTLs from an earlier study; Acuña-Galindo et al. 2015), but also for several other individual traits including the following: (1) pre-harvest sprouting tolerance (Tyagi and Gupta 2012), (2) ear emergence (Hanocq et al. 2007; Griffiths et al. 2009), (3) resistance against Fusarium head blight (Häberle et al. 2009; Loffler et al. 2009; Liu et al. 2009), (4) plant height (Griffiths et al. 2012), (5) grain dietary fiber content (Quraishi et al. 2010), (6) seed size and seed shape (Gegas et al. 2010), (7) yield contributing traits (Zhang et al. 2010; Quraishi et al. 2017), (8) resistance to leaf rust (Soriano and Royo 2015) etc. Since large number of QTLs for drought tolerance have been reported after the last report of MQTL analysis for drought tolerance in wheat, the present study involving MQTL analysis was conducted (based on QTL studies conducted during 2015–2020) to supplement the list of MQTLs and candidate genes reported in the earlier MQTL study for drought tolerance (Acuña-Galindo et al. 2015).
Materials and methods
In the present study, meta-QTL (MQTL) analysis and identification of underlying candidate genes was undertaken utilizing the widely known five steps, which are briefly described:
Bibliographic review and collection of QTL information
A thorough bibliographic review was carried out and QTL information for drought tolerance in wheat was collected from 14 independent studies. This included information on chromosome location, most closely linked marker(s), QTL position, log of odds (LOD) value, confidence intervals (CIs) and R2 values. Details for individual studies used in the present study are summarized in Table 1. The MQTL analysis was conducted using BioMercator v4.2 (Arcade et al. 2004).
Construction of consensus map
A consensus genetic map was developed using the following two dense composite maps: (1) a map containing 1235 SSR loci with an average distance of 2.2 cM between two adjacent markers (Somers et al. 2004) and (2) the composite map with 4506 markers available at Grain Gene 2.0 (https://wheat.pw.usda.gov/GG3/). All markers from the framework maps used in individual QTL studies were projected on these reference maps (applying a weighted least square method) to construct a consensus genetic map, following procedure outlined in BioMercator v4.2 (www.genoplante.org) (Arcade et al. 2004). The two maps were first integrated to provide a pre-consensus map, which was then used for developing a consensus map which carried maximum number of markers and was suitable for QTL projection to facilitate meta-QTL analysis.
QTL projection
The projection of individual QTLs on consensus map was based on their LOD score, phenotypic variation explained (PVE), confidence interval and QTL position. The software, BioMercator v4.2 (Arcade et al. 2004) was used for projection utilizing positions of markers that were common in more than one study, and following the rule that the markers flanking the interval of the original QTL should correspond to the interval on the consensus chromosome map. Therefore, finally, out of the 14 studies, information from only 11 studies was used by the software for QTL projection. The remaining three studies were dropped by the software mainly due to missing essential information mentioned above.
Wherever the genetic position of a marker was not known, the markers closest to the flanking markers from the reference map were used to project QTL on the consensus map. The 95% confidence intervals (CIs) of original QTLs on their original maps were estimated using the following formula provided by Darvasi and Soller (1997): CI = 530/NR2, where, N is the population size and R2 is the proportion of the phenotypic variance explained (PVE) by individual QTL. The QTLs which could not be mapped onto the consensus map and those mapped at locations outside the consensus map were discarded.
Analysis for meta-QTL
Meta-QTL (MQTL) analysis was performed on the QTL clusters on each chromosome using BioMercator v 4.2 (Goffinet and Gerber 2000). This software allows us to find out the number (n) of MQTL, which is usually less than the number of total projected QTLs. This is done by selecting one of the five models available in the software, one model for each possibility. A specific model is selected on the basis of minimum value of Akaike Information Criterion (AIC); the lower the AIC value, the more appropriate is the model. The aim of AIC is to estimate the mean log-likelihood (MELL) for the number of real positions xi of the n QTLs (Sakamoto et al. 1986). The AIC value is computed using the following formula: AIC = − 2 × L(θ[k], X[k], X) + 2 × k., where L(θ[k], X[k], X) is the log-likelihood of the observed vector X (QTL position), k is the actual number of parameters (1, 2, 3, 4…n), and X0 the actual value of the positions of n QTLs. K is an unbiased estimator of MELL. We have chosen the model with the minimum AIC value, as recommended.
Identification of candidate genes underlying the MQTL region and expression analysis
The candidate genes (CGs) are the genes localized within a MQTL region that is identified based on the positions of markers flanking the CI of the MQTL (or the marker closest to the flanking markers). For this purpose, nucleotide sequences of markers were retrieved from GrainGenes database (https://wheat.pw.usda.gov/GG3) and used for a search of genes using wheat reference genomic sequence available in EnsemblPlants database (version 082214v1, http://plants.ensembl.org/index.html) using BLAST (maximum E-value = 1E−100, minimum 95% identity of the sequence). Gene models found within the physical regions corresponding to the QTL intervals were retrieved using BioMart of EnsemblPlant. In silico expression analysis for the validation of identified CGs was conducted with WheatExp database using the conserved domain sequences (CDS) of each individual candidate gene. Heat maps for expression data were prepared using ClustVis (https://biit.cs.ut.ee/clustvis/; Metsalu and Vilo 2015).
Results
Bibliography search for QTLs for drought tolerance
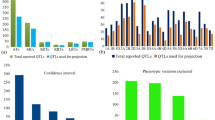
Out of 14 studies on QTL analysis for drought tolerance in wheat, listed in Table 1, in the present study only 11 studies could be used for MQTL analysis. Each study involved. a separate mapping population (total 11 populations), leading to detection of as many as ~ 340 QTLs. These 340 QTLs were distributed on all the 21 chromosomes belonging to 7 homoeologous groups (1–7) and 3 sub-genomes (A, B and D). Significant differences were observed for the number of QTLs not only among all the seven homoeologous groups, but also among individual chromosomes within a homoeologous group (Fig. 1). Each QTL is characterized by its map position [most likely position and confidence interval (CI) around this position], LOD value and the proportion of PVE (estimated through R2 value). Whenever, the required information about position and R2 value for the QTLs was not available from a particular study, the most likely position of QTLs was determined as the middle point between the two flanking markers, and the R2 value of closest flanking marker was taken as the R2 value of the QTL.

Distribution of individual QTLs for drought tolerance on 7 homoeologous groups and the A, B and D sub-genomes of wheat
Construction of a consensus map and QTL projection
In bread wheat, a number of framework genetic maps are available, one each for an individual mapping population used for QTL interval mapping in a particular study. However, the number of common markers among the individual maps that were used in the present study were not adequate for construction of a consensus map and for the reliable projection of QTL positions. Therefore, for developing a consensus map, a pre-consensus map was first generated from two important available maps (see “Materials and methods” section for details). The pre-consensus map so developed was used for developing a consensus map using the framework maps. Following criteria were used for construction of consensus map using framework maps from 11 studies: (1) A chromosome in a framework map having no more than one common marker in the corresponding chromosome of the pre-consensus map was excluded. (2) Inversion of marker order was filtered out by discarding inconsistent loci with the exception of very closely linked markers. If two or more markers in a map are available in inverted orientation relative to pre-consensus map, then one of the two closest markers available in inverted order and separated by a distance of < 1 cM, was dropped to retain a maximum number of common markers. (3) When all the common markers were in reverse order with respect to the pre-consensus map, we used inverted genetic map for projection.
Meta-QTL analysis
Only 86 QTLs from the 340 QTLs that were identified from bibliographic search were initially selected for meta-QTL analysis, since the remaining 254 QTLs did not have common flanking markers with the markers in the consensus map. Out of these 86 QTLs, only 42 QTLs could be projected on to the consensus map based on the lowest AIC values. Out of these 42 QTLs, maximum number of QTLs was available on chromosome 5A, followed by QTLs on chromosomes 7A, 1D, 3B, 6D and 7D in that order. Remaining 44 QTLs were discarded by the software probably due to high AIC value as mentioned earlier. Using these 42 original QTLs, 13 MQTLs (Table 2; Fig. 2) were identified on six different chromosomes, namely 1D, 3B, 5A, 6D, 7A and 7D (for details, see Table 3). The confidence intervals (CI) for MQTLs were narrow (mean: 6.01 cM; range 2.07–19.46 cM) relative to those for the original QTLs (mean: 13.6 cM; range 1.0–119.1 cM; for details see Table 2). Further, it was observed that 10 of the 13 MQTLs, each controlled more than one trait indicating their pleiotropic nature. Out of these 13 MQTLs, following two MQTLs were important: (1) MQTL12, which is the major MQTL located on chromosome 7D with R2 (PVE %) of 23.40%, and a confidence interval of 3.18 cM. This MQTL also had CI that was reduced by 50% (3.18 cM against original 6.10 cM). (2) MQTL4, which comprised maximum number of original QTLs (10), each controlling a separate drought responsive trait (Table 2).

Distribution and location of 13 MQTLs on 6 different chromosomes in bread wheat. The traits for the original QTLs associated with each MQTL are indicated in parenthesis against each MQTL. DTM: Days to maturity; DTA days to anthesis, DTH days to heading, HI harvest index, SPKL culm length, SKW single kernel weight, WSC water soluble carbohydrate, PH plant height, GY grain yield, FLL flag leaf length, FLA flag leaf area, dsSPDM drought susceptibility index spike dry matter, TDR total dry matter, CID carbon isotope discrimination, CT canopy temperature, OP osmotic potential, GP germination percentage, CHL chlorophyll content, LR leaf rolling, TW test weight, GFD grain filling duration, LL leaf length
Candidate genes (CGs) identified underlying the MQTLs region
In the present study, 228 drought responsive CGs were initially identified, which belonged to the following 6 MQTLs: MQTL2 (34), MQTL4 (30), MQTL5 (31), MQTL7 (20), MQTL8 (19) MQTL13 (94). No candidate genes (CGs) could be identified for the remaining 7 MQTLs, since these MQTLs were flanked by markers like AFLP/RFLP with no sequences data available for BLAST search.
Expression analysis of CGs under drought
In the database, expression data was available for only 42 of 228 CGs, and that too for a solitary drought tolerant wheat genotype TAM107. This expression data was utilized for in silico expression analysis of the CGs, and allowed us to prepare heat maps. Only 14 CGs exhibited significant changes (FC = + 3 to − 8) in expression due to drought stress (1 h and 6 h drought stress at seedling stage in tolerant cv. TAM107); for the remaining 28 CGs, although expression data was available, no significant change in expression due to drought was observed, as evident from the heat maps presented in Fig. 3. These 14 CGs showing significant changes in expression due to drought belonged to only 5 of the 6 MQTLs (Table 3); for the remaining one MQTL (MQTL7), although 20 CGs were available, but expression data was available for only three genes, and this was not sufficient for expression analysis. The 14 CGs encoded a variety of proteins and under drought stress, 11 of these CGs showed down-regulation whereas the remaining three genes showed up-regulation (for details see Table 3). These CGs can be validated through a study of their expression in seedlings of drought sensitive and drought tolerant wheat cultivars grown under moisture stress.

Heat maps showing differential expression (in silico) of important proteins encoded by candidate genes underlying the following 5 MQTLs: a MQTL2, b MQTL4, c MQTL5, d MQTL8, e MQTL13. The scale shown on the left of each heatmap represents is the fold changes ranging from + 3 to − 8 which differ in different heatmaps. Proteins which showed siginificant FC in expression are marked by an asterisk (*)
Discussion
Meta-QTL analysis combines results from multiple QTL studies, permitting refinement of QTL locations and identification of closely associated molecular markers for the traits of interest. In the present study, 13 MQTLs for drought tolerance were identified in wheat. For this purpose, after thorough bibliographic search, 11 studies reporting 340 QTLs were selected. However, all the 340 QTLs could not be utilised by the software, mostly due to the following two reasons: (1) unavailability of complete information about the QTL (lack of common markers in the consensus map and those flanking the QTL) and (2) high AIC value.
A meta-QTL study for drought and heat tolerance in wheat was also conducted earlier (Acuña-Galindo et al. 2015). In this earlier study, information from 30 studies (till 2015) reporting 854 QTLs (including 502 QTLs for drought tolerance) was utilized. The present study was thus a supplement to this earlier study, and largely reports MQTLs based on QTLs reported during 2015–2020. The earlier study reported 20 MQTLs that were specific to drought stress and 43 MQTLs that were common for both heat and drought, MQTL analysis for drought tolerance have also been undertaken in other cereals like rice (Courtois et al. 2009; Khowaja et al. 2009), maize (Liu et al. 2019; Hao et al. 2010) and barley (Li et al. 2013). These MQTLs for drought tolerance reported in rice, maize and barley along with MQTL reported in two studies (including the present study) conducted in wheat may be used in future studies to identify ortho-MQTLs and the candidate genes (CGs) for drought tolerance in cereals, as earlier done for nitrogen-use efficiency (NUE) in wheat (Quraishi et al. 2011) and for yield-related traits in rice (Khahani et al. 2020).
Drought is a complex quantitative trait involving numerous physiological processes controlled by a large number of genes, such as early flowering, plant height and osmotic adjustment (Cattivelli et al. 2008; Shabala and Pottosin 2014; Gupta et al. 2020a, b). Therefore, it is always a challenge for plant breeders to collect data on phenotype with precision and then select drought-tolerant genotypes (Hu and Xiong 2014; Tuberosa 2012). In the present study, QTLs for a fairly large number of drought responsive (> 40) traits were utilized. A critical analysis would suggest that, some individual MQTLs were based each on QTLs for only one specific trait/parameter, while others were based on QTLs for more than one traits/parameters (Table 2).
Each of the 13 MQTLs that were identified during the present study had a relatively narrow confidence interval (CI), thus providing markers that are more closely associated with the corresponding MQTL. Some of the important features of 13 MQTLs that are relevant for their use in MAS include the following: (1) Stable MQTLs under different environments: MQTL4 is based on 3 QTLs for days to maturity (DTM) from three different studies (Peleg et al. 2009; Pinto et al. 2010; Gahlaut et al. 2017) involving three different environments. Similarly, MQTL10 is based on two QTLs for DTM from two out of three above studies. Therefore, MQTL4 and MQTL10 were also stable for the trait DTM. (2) More than one MQTLs for the same trait: MQTL1, MQTL4, MQTL6 and MQTL10 were each based on QTLs for DTA and MQTL1, MQTL4, MQTL5, MQTL6 and MQTL10 were based each on QTL for DTM; see Table 2). These MQTLs, each based on more than one trait seem to be more robust. (3) Pleiotropic MQTL: nine out of 13 MQTLs each controlled more than one trait. For instance, MQTL4 was derived from 10 QTLs each for different traits and MQTL 10 was based on 7 original QTLs each for a different trait. Interestingly, four QTLs were common among the QTLs, on which these two MQTLs are based. Similarly, there were seven other pleiotropic QTLs (for details see Table 2). These QTLs may, therefore, each represent a complex locus, each controlling more than one drought responsive traits. The above three categories of MQTLs should prove useful not only for MAS, but also for searching CGs and possible map-based cloning.
The above 13 MQTLs were also compared with the 20 MQTLs for drought tolerance that were reported earlier by Acuña-Galindo et al. (2015). Some MQTLs identified in the present study had markers located close to the markers flanking the MQTLs reported in this earlier study. Therefore, these markers can be considered as important for MAS. Two such MQTLs identified in the present study include MQTL2 and MQTL12; MQTL2 (chromosome 1D) was flanked by two SSR markers, Xcfd65 and Xbarc169, which are respectively located at a distance of 3 cM and 2 cM from the closest markers for MQTL9 reported by Acuña-Galindo et al. (2015), so that MQTL2 of the present study may correspond to MQTL9 of this earlier study. Similarly, MQTL12 (chromosome 7D) of the present study may correspond to MQTL64 of the earlier study. Since both these MQTLs are located close to the markers Xbarc5 and Xbarc154, these markers may be important for MAS, while breeding wheat cultivars for drought tolerance. Notwithstanding these two seemingly common MQTLs in two studies, 11 MQTLs identified during the present study seem to be novel, thus adding to the list of known MQTLs for drought tolerance.
The present study also supports the view that drought tolerance is affected by the cumulative effect of several QTLs for different drought responsive traits. Therefore, a breeder will have to select appropriate MQTLs for MAS to improve drought tolerance. For this purpose, a breeder may select one or more MQTLs (for drought with or without one or more other associated traits) that comprise a large number of original QTLs, each having a narrow CI. Thus, a few MQTLs out of the 13 MQTLs identified in the present study may prove to be important for the development of drought tolerant wheat varieties using MAS. We propose two of the 13 MQTLs to be relatively more important based on their specific features. These MQTL include the following: (1) MQTL12 which is the only major effect QTL with R2/PVE % of 23.4% with a narrow CI of 3.18 cM and controlling three important traits including spike length, days to heading and grain filling duration, and (2) MQTL4 (CI = 3.1 cM and average R2/PVE = 6.13%), which is based on 10 QTLs for 10 different drought responsive traits. These two MQTLs may be used for marker-aided introgression of independent QTLs and MQTLs in any adaptive genetic background for development of drought tolerant wheat cultivars.
CGs underlying MQTLs were also identified using wheat genomic sequences available at EnsemblPlants. Out of 228 CGs identified by us; 11 CGs were also identified in two earlier studies; three of these 11 CGs involved in drought and heat stress in wheat, encoded FAD binding domain, glycosyl hydrolase and F-box (Acuña-Galindo et al. 2015). The remaining eight CGs involving tolerance to drought stress in grasses were identified by (Swamy et al. 2011); these genes encoded proteins for ABC transporter, cytP50, zinc finger, protein kinase, glutaredoxin, F-Box, aquaporin, and NAC transcription factor.
Fourteen other CGs, encoding important proteins, were identified on the basis of significant change in gene expression (FC + 3 to − 8); these genes can be subjected to further studies for an understanding of the molecular mechanism of drought tolerance as well as development of markers for breeding drought tolerant wheat cultivars (for further details, see Table 3 and Fig. 3). Interestingly some of these CGs encodintg NAC transcription factors, Zn finger protein, homeobox domain proteins, protein containing kinase domain and HSP70, were also earlier reported to play a role during drought stress response in cereals like rice and wheat (Min et al. 2013; Fang et al. 2014; Wu et al. 2015; Li et al. 2020). For instance, gene encoding NAC A/B superfamily protein showed down-regulation due to drought tolerance. A similar gene encoding NAC protein was earlier shown to be a potential target of a miRNA in rice where its down-regulation was shown to enhance drought tolerance (Fang et al. 2014). Similarly, downregulation of the CG encoding a kinase domain containing protein was observed in the present study. The down-regulation of a leucine-rich repeat (LRR)-RLK gene, namely LP2 (Leaf Panicle2) was also reported earlier under drought stress in rice (Wu et al. 2015); the expression of LP2 is regulated by a zink finger transcription factor DROUGHT AND SALT TOLERANCE (DST). In contrast, genes encoding HSP70, Zn finger containing proteins and homeobox domain containing proteins showed up-regulation due to drought stress, indicating their role as positive regulators of drought tolerance. The role of Hsp70 in drought tolerance was reviewed earlier (for details, see Cho and Choi 2009). Similarly, the overexpression of wheat genes encoding a Zn finger protein (TaZnFP) and homeobox domain containing proteins in transgenic Arabidopsis showed enhanced drought tolerance (Min et al. 2013; Li et al. 2020).
Conclusion
Thirteen key genomic regions (in the form of MQTLs) associated with agronomic performance and drought responsive traits were identified during the present study; 11 of these MQTLs were novel and two were perhaps common to those identified earlier by Acuña-Galindo et al. (2015) Two of the 13 MQTLs regions were also recommended for use in MAS for improvement of drought tolerance in wheat. Candidate genes were also identified from six of the 13 MQTL regions. These candidate genes mainly encoded proteins for ABC transporter, F box domain, cytP50, zinc finger, protein kinase, glutaredoxin etc. Some of these proteins were also shown to be involved in drought tolerance in wheat and other cereals.
References
Acuña-Galindo MA, Mason RE, Subramanian NK, Hays DB (2015) Meta-analysis of wheat QTL regions associated with adaptation to drought and heat stress. Crop Sci 55:477–492
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Ayalew H, Liu H, Börner A, Kobiljski B, Liu C, Yan G (2018) Genome-wide association mapping of major root length QTLs under PEG induced water stress in wheat. Front Plant Sci 9:1759
Ballini E, Morel JB, Droc G, Price A, Courtois B, Notteghem JL, Tharreau D (2008) A genome-wide meta-analysis of rice blast resistance genes and quantitative trait loci provides new insights into partial and complete resistance. Mol Plant Microbe Interact 21:859–868
Bennett D, Izanloo A, Reynolds M, Kuchel H, Langridge P, Schnurbusch T (2012) Genetic dissection of grain yield and physical grain quality in bread wheat (Triticum aestivum L.) under water-limited environments. Theor Appl Genet 125:255–271
Cattivelli L, Rizza F, Badeck FW, Mazzucotelli E, Mastrangelo AM, Francia E, Mare C, Tondelli A, Stanca AM (2008) Drought tolerance improvement in crop plants: an integrated view from breeding to genomics. Field Crops Res 105:1–14
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A (2004) Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168:2169–2185
Chen L, An Y, Li Y-x, Li C, Shi Y, Song Y, Zhang D, Wang T, Li Y (2017) Candidate loci for yield-related traits in maize revealed by a combination of metaQTL analysis and regional association mapping. Front Plant Sci 8:2190
Cho EK, Choi YJ (2009) A nuclear-localized HSP70 confers thermoprotective activity and drought-stress tolerance on plants. Biotechnol Lett 31:597–606
Coque M, Martin A, Veyrieras JB, Hirel B, Gallais A (2008) Genetic variation for N-remobilization and postsilking N-uptake in a set of maize recombinant inbred lines. 3. QTL detection and coincidences. Theor Appl Genet 117:729–747
Courtois B, Ahmadi N, Khowaja F, Price AH, Rami JF, Frouin J, Hamelin C, Ruiz M (2009) Rice root genetic architecture: meta-analysis from a drought QTL database. Rice 2:115–128
Danan S, Veyrieras JB, Lefebvre V (2011) Construction of a potato consensus map and QTL meta-analysis offer new insights into the genetic architecture of late blight resistance and plant maturity traits. BMC Plant Biol 1:16
Darvasi A, Soller M (1997) A simple method to calculate resolving power and confidence interval of QTL map location. Behav Genet 27:125–132
El-Feki WM, Byrne PF, Reid SD, Haley SD (2018) Mapping quantitative trait loci for agronomic traits in winter wheat under different soil moisture levels. Agronomy 8:133
Fang Y, Xie K, Xiong L (2014) Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J Exp Bot 65:2119–2135
Farooq M, Hussain M, Siddique KH (2014) Drought stress in wheat during flowering and grain filling periods. Crit Rev Plant Sci 33:331–349
Gahlaut V, Jaiswal V, Tyagi BS, Singh G, Sareen S, Balyan HS, Gupta PK (2017) QTL mapping for nine drought-responsive agronomic traits in bread wheat under irrigated and rain-fed environments. PLoS ONE 12: e0182857
Gegas VC, Nazari A, Griffiths S, Simmonds J, Fish L, Orford S, Sayers L, Doonan JH, Snape JW (2010) A genetic framework for grain size and shape variation in wheat. Plant Cell 22:1042–1046
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Griffiths S, Simmonds J, Leverington M, Wang Y, Fish L, Sayers L, Alibert L, Orford S, Wingen L, Herry L, Faure S, Laurie D, Bilham L, Snape J (2009) Meta-QTL analysis of the genetic control of ear emergence in elite European winter wheat germplasm. Theor Appl Genet 119:383–395
Griffiths S, Simmonds J, Leverington M, Wang Y, Fish L, Sayers L, Alibert L, Orford S, Wingen L, Snape J (2012) Meta-QTL analysis of the genetic control of crop height in elite European winter wheat germplasm. Mol Breed 29:159–171
Guo B, Sleper DA, Lu P, Shannon JG, Nguyen HT, Arelli PR (2006) QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Sci 46:595–602
Guo J, Chen L, Li Y, Shi Y, Song Y, Zhang D, Li Y, Wang T, Yang D, Li C (2018) Meta-QTL analysis and identification of candidate genes related to root traits in maize. Euphytica 2014:223
Gupta PK, Mir RR, Mohan A, Kumar J (2008) Wheat genomics: present status and future prospects. Int J Plant Genomics 2008:896451
Gupta A, Rico-Medina A, Ana I, Caño-Delgado AI (2020a) The physiology of plant responses to drought. Science 368:266–269
Gupta PK, Balyan HS, Sharma S, Kumar R (2020b) Genetics of yield, abiotic stress tolerance and biofortification in wheat (Triticum aestivum L). Theor Appl Genet 133:1569–1602
Häberle J, Holzapfel J, Schweizer G, Hartl L (2009) A major QTL for resistance against Fusarium head blight in European winter wheat. Theor Appl Genet 119:325–332
Hanocq E, Laperche A, Jaminon O, Laine AL, Le Gouis J (2007) Most significant genome regions involved in the control of earliness traits in bread wheat, as revealed by QTL meta-analysis. Theor Appl Genet 114:569–584
Hao Z, Li X, Liu X, Xie C, Li M, Zhang D, Zhang S (2010) Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica 174:165–177
Hong Y, Chen X, Liang X, Liu H, Zhou G, Li S, Wen S, Holbrook CC, Guo B (2010) A SSR-based composite genetic linkage map for the cultivated peanut. (Arachis hypogaea L) genome. BMC Plant Biol 10:17
Hu H, Xiong L (2014) Genetic engineering and breeding of drought-resistant crops. Annu Rev Plant Biol 65:715–741
Islam MS, Ontoy J, Subudhi PK (2019) Meta-analysis of quantitative trait loci associated with seedling-stage salt tolerance in rice (Oryza sativa L). Plants 8:33
Khahani B, Tavakol E, Shariati V, Fornara F (2020) Genome wide screening and comparative genome analysis for meta-QTLs, ortho-MQTls and candidate genes controlling yield and yield-related traits in rice. BMC Genom 21:294
Khowaja FS, Norton GJ, Courtois B, Price AH (2009) Improved resolution in the position of drought-related QTLs in a single mapping population of rice by meta-analysis. BMC Genom 10:276
Kosina P, Reynolds M, Dixon J, Joshi A (2007) Stakeholder perception of wheat production constraints, capacity building needs, and research partnerships in developing countries. Euphytica 157:475–483
Kumar S, Sehgal SK, Kumar U, Prasad PV, Joshi AK, Gill BS (2012) Genomic characterization of drought tolerance-related traits in spring wheat. Euphytica 186:265–276
Lanaud C, Fouet O, Clément D, Boccara M, Risterucci AM, Surujdeo-Maharaj S, Legavre T, Argout X (2009) A meta-QTL analysis of disease resistance traits of Theobroma cacao L. Mol Breed 24:361–374
Li JZ, Zhang ZW, Li YL, Wang QL, Zhou YG (2011) QTL consistency and meta-analysis for grain yield components in three generations in maize. Theor Appl Genet 122:771–782
Li WT, Liu C, Liu YX, Pu ZE, Dai SF, Wang JR, Lan XJ, Zheng YL, Wei YM (2013) Meta-analysis of QTL associated with tolerance to abiotic stresses in barley. Euphytica 189:31–49
Li S, Chen N, Li F, Mei F, Wang Z, Cheng X, Kang Z, Mao H (2020) Characterization of wheat homeodomain-leucine zipper family genes and functional analysis of TaHDZ5-6A in drought tolerance in transgenic Arabidopsis. BMC Plant Biol 20:50
Liu S, Hall MD, Griffey CA, McKendry AL (2009) Meta-analysis of QTL associated with Fusarium head blight resistance in wheat. Crop Sci 49:1955–1968
Liu S, Zenda T, Wang X, Liu G, Jin H, Yang Y, Dong A, Duan H (2019) Comprehensive meta-analysis of maize QTLs associated with grain yield, flowering date and plant height under drought conditions. J Agric Sci 11:1–19
Loffler M, Schon CC, Miedaner T (2009) Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol Breed 23:473–488
Malik S, Malik TA (2015) Genetic mapping of potential QTLs associated with drought tolerance in wheat. J Anim Plant Sci 25: 1032–1040
Marandel G, Salava J, Abbott A, Candresse T, Decroocq V (2009) Quantitative trait loci meta-analysis of Plum pox virus resistance in apricot (Prunus armeniaca L.) new insights on the organization and the identification of genomic resistance factors. Mol Plant Pathol 10:347–360
Mathews KL, Malosetti M, Chapman S, McIntyre L, Reynolds M, Shorter R et al (2008) Multi-environment QTL mixed models for drought stress adaptation in wheat. Theor Appl Genet 117:1077–1091
McIntyre CL, Mathews KL, Rattey A, Chapman SC, Drenth J, Ghaderi M et al (2010) Molecular detection of genomic regions associated with grain yield and yield-related components in an elite bread wheat cross evaluated under irrigated and rainfed conditions. Theor Appl Genet 120:527–541
Metsalu T, Vilo J (2015) ClustVis: a web tool for visualising clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res 43:426–570
Min DH, Zhao Y, Huo DY, Li LC, Chen M, Xu ZS, Ma YZ (2013) Isolation and identification of a wheat gene encoding a zinc finger protein (TaZnFP) responsive to abiotic stresses. Acta Physiol Plant 35:1597–1604
Nezhadahmadi A, Zakaria HP, Golam F (2013) Drought tolerance in wheat. Sci World J 2013:610721
Norton GJ, Aitkenhead MJ, Khowaja FS, Whalley WR, Price AH (2008) A bioinformatic and transcriptomic approach to identifying positional candidate genes without fine mapping: an example using rice root-growth QTLs. Genomics 92:344–352
Onyemaobi I, Ayalew H, Liu H, Siddique KH, Yan G (2018) Identification and validation of a major chromosome region for high grain number per spike under meiotic stage water stress in wheat (Triticum aestivum L.). PLoS ONE 13:3
Peleg Z, Fahima T, Krugman T, Abbo S, Yakir D, Korol AB et al (2009) Genomic dissection of drought resistance in durum wheat × wild emmer wheat recombinant inbred line population. Plant, Cell Environ 32:758–779
Pinto RS, Reynolds MP, Mathews KL, McIntyre CL, Olivares-Villegas JJ, Chapman SC (2010) Heat and drought adaptive QTL in a wheat population designed to minimize confounding agronomic effects. Theor Appl Genet 121:1001–1021
Quraishi UM, Murat F, Abrouk M, Pont C, Confolent C, Oury FX, Ward J, Boros D, Gebruers K, Delcour JA, Courtin CM, Bedo Z, Saulnier L, Guillon F, Balzergue S, Shewry PR, Feuillet C, Charmet G, Salse J (2010) Combined meta-genomics analyses unravel candidate genes for the grain dietary fiber content in bread wheat (Triticum aestivum L.). Funct Integr Genomics 11:71–83
Quraishi UM, Abrouk M, Murat F et al (2011) Cross-genome map based dissection of a nitrogen use efficiency ortho-metaQTL in bread wheat unravels concerted cereal genome evolution. Plant J 65:745–756
Quraishi UM, Pont C, Ain Q-U, Flores R, Burlot L, Alaux M, Quesneville H, Salse J (2017) Combined genomic and genetic data integration of major agronomical traits in bread wheat (Triticum aestivum L.). Front Plant Sci 8:1843
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL, Smith CW (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Sakamoto Y, Ishiguro M, Kitagawa G (1986) Akaike information criterion statistics. D. Reidel, Dordrecht, p 81
Shabala S, Pottosin I (2014) Regulation of potassium transport in plants under hostile conditions: implications for abiotic and biotic stress tolerance. Physiol Plant 151:257–279
Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, Bancroft I, Zhao J, Meng J (2009) Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics 182:851–861
Shirasawa K, Bertioli DJ, Varshney RK, Moretzsohn MC, Leal-Bertioli SC, Thudi M, Pandey MK, Rami JF, Foncéka D, Gowda MV, Qin H (2013) Integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes of Arachis and divergence of the legume genomes. DNA Res 20:173–184
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Soriano JM, Royo C (2015) Dissecting the genetic architecture of leaf rust resistance in wheat by QTL meta-analysis. Phytopathology 105:1585–1593
Sun YN, Pan JB, Shi XL, Du XY, Wu Q, Qi ZM, Jiang HW, Xin DW, Liu CY, Hu GH, Chen QS (2012) Multi-environment mapping and meta-analysis of 100-seed weight in soybean. Mol Biol Rep 39:9435–9443
Swamy BM, Vikram P, Dixit S, Ahmed HU, Kumar A (2011) Meta-analysis of grain yield QTL identified during agricultural drought in grasses showed consensus. BMC Genom 12:319
Truntzler M, Barrière Y, Sawkins MC, Lespinasse D, Betran J, Charcosset A, Moreau L (2010) Meta-analysis of QTL involved in silage quality of maize and comparison with the position of candidate genes. Theor Appl Genet 121:1465–1482
Tuberosa R (2012) Phenotyping for drought tolerance of crops in the genomics era. Front Physiol 3:347
Tyagi S, Gupta PK (2012) Meta-analysis of QTLs involved in pre-harvest sprouting tolerance and dormancy in bread wheat. Triticeae Genom Genet 3:9–24
Verma V, Foulkes MJ, Worland AJ, Sylvester-Bradley R, Caligari PDS, Snape JW (2004) Mapping quantitative trait loci for flag leaf senescence as a yield determinant in winter wheat under optimal and drought-stressed environments. Euphytica 135:255–263
Wang Y, Yao J, Zhang ZF, Zheng YL (2006) The comparative analysis based on maize integrated QTL map and meta-analysis of plant height QTLs. Chin Sci Bull 51:2219–2230
Wu F, Sheng P, Tan J, Chen X, Lu G, Ma W, Heng Y, Lin Q, Zhu S, Wang J, Wang J, Guo X, Zhang X, Lei C, Wan J (2015) Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the DROUGHT AND SALT TOLERANCE transcription factor to regulate drought sensitivity in rice. J Exp Bot 66:271–281
Xu J, Dai X, Ramasamy RK, Wang L, Zhu T, McGuire PE, Jorgensen CM, Dehghani H, Gulick PJ, Luo MC, Müller HG (2019) Aegilops tauschii genome sequence: a framework for meta-analysis of wheat QTLs. G3 Genes Genomes Genet 9:841–853
Yang D, Liu Y, Cheng H, Chang L, Chen J, Chai S, Li M (2016) Genetic dissection of flag leaf morphology in wheat (Triticum aestivum L) under diverse water regimes. BMC Genet 17:94
Zhang LY, Liu DC, Guo XL, Yang WL, Sun JZ, Wang DW, Zhang A (2010) Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. J Integr Plant Biol 52:996–1007
Zhao X, Peng Y, Zhang J, Fang P, Wu B (2018) Identification of QTLs and meta-QTLs for seven agronomic traits in multiple maize populations under well-watered and water-stressed conditions. Crop Sci 58:507–520
Zheng BS, Le Gouis J, Leflon M, Rong WY, Laperche A, Brancourt- Hulmel M (2010) Using probe genotypes to dissect QTL environment interactions for grain yield components in winter wheat. Theor Appl Genet 121:1501–1517
Acknowledgements
The work was carried out, when AK, IJ and KK held JRF positions under a research project funded by Department of Biotechnology, New Delhi, India and GS held SRF position under NASF-ICAR program of Government of India. PKG was awarded Hony Scientist position and HSB was awarded Senior Scientist position both from Indian National Science Academy (INSA). Head, Department of Genetics and Plant Breeding, CCS University, Meerut, provided the necessary infrastructure. Authors are also thankful to Dr. Sandhya Tyagi, Division of Genetics, IARI, New Delhi for providing inputs to AK for conducting MQTL analysis.
Author information
Authors and Affiliations
Contributions
PKG, HSB and PKS conceived the study and also edited and finalized the manuscript. AK conducted MQTL analysis and wrote the first draft of the MS jointly with GS; IJ and KK helped AK in preparing the files for MQTL analysis.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, A., Saripalli, G., Jan, I. et al. Meta-QTL analysis and identification of candidate genes for drought tolerance in bread wheat (Triticum aestivum L.). Physiol Mol Biol Plants 26, 1713–1725 (2020). https://doi.org/10.1007/s12298-020-00847-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12298-020-00847-6