
Abstract
Breast cancer is considered to be relatively sensitive to chemotherapy, and multiple combinations of cytotoxic agents are used as standard therapy. Chemotherapy is applied empirically despite the observation that not all regimens are equally effective across the population of patients. Up to date clinical tests for predicting cancer chemotherapy response are not available, and individual markers have shown little predictive value. A number of microarray studies have demonstrated the use of genomic data, particularly gene expression signatures, as clinical prognostic factors in breast cancer. The identification of patient subpopulations most likely to respond to therapy is a central goal of recent personalized medicine. We have designed experiments to identify gene sets that will predict treatment-specific response in breast cancer. Taken together with our recent trial about the construction of a high-throughput functional screening system for chemo-sensitivity related genes, studies for drug sensitivity will provide rational strategies for establishment of the prediction system with high accuracy, and identification of ideal targets for drug intervention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chemotherapy for breast cancer
Breast cancer ranks first in the incidence among gynecological cancers. According to the 2003 vital statistics records on the Japanese population, the mortality rate from breast cancer (15.2%) is increasing. For the treatment of breast cancer, surgery and several different chemotherapy regimens are used. The development of optimal consensus treatment guidelines for breast cancer requires comprehensive analysis of the results of randomized clinical trials and the interpretation of their clinical, biological and personal relevance for individual patients. Recent recommendations (St Gallen [1, 2], NIH Consensus [3]) based on clinical tests in large-scale, randomized control trials in the United States and Europe focused on the implications of evidence for patient treatment selection. They promoted the standardization of therapies in hospitals or regions, avoiding any divergent therapies.
Breast cancer is considered to be relatively sensitive to chemotherapy compared to other solid tumors. Multiple combinations of cytotoxic drugs are used as standard therapy. The most effective combination regimens include anthracyclines (epirubicine or doxorubicin), which are topoisomerase II inhibitors. The taxanes (paclitaxel and docetaxel) and Capecitabine are a new class of antimicrotubule agents that are more effective than older drugs, such as anthracyclines, and are often used in patients with advanced breast cancer with tumor cells that are resistant to anthracyclines [4–6]. Therefore, it is hoped that the addition of paclitaxel to anthracycline-containing chemotherapy will improve treatment efficacy for a subset of patients. However, not all regimens may be equally effective for all patients. It is not yet possible to select the most effective regimen for a particular individual, because there are no clinically useful predictive markers of a patient’s response to chemotherapy. Now the question is, “Is it possible to personalize chemotherapy and select the single best regimen for an individual?”
Selection of the best treatment for patients based on the gene expression profile
The recent development of DNA microarray and related technologies provides a unique molecular portrait or signature that can be correlated with clinical behavior and drug responsiveness in breast cancer [7–11]. It is anticipated that gene expression profiling of responsive compared to non-responsive tumors may allow the definition of a pattern of clinically useful discriminatory gene expression. Chang et al. [12] reported the application of DNA microarray analysis in the identification of predictive factors of response to docetaxel in 24 breast cancer patients. They identified differential expression patterns of 92 genes correlated with docetaxel response. In leave-one-out cross-validation analysis, 10 of 11 sensitive tumors (responders) and 11 of 13 resistant tumors (non-responders) were correctly classified, with an accuracy of 88%. On the other hand, Ayers et al. [13] identified a set of key genetic markers (74 genes) that predict whether patients are likely to be cured (complete pathologic response; pCR) by a chemotherapy regimen commonly given before surgery (neoadjuvant chemotherapy). It was reported that the markers predicted, with 75% accuracy, whether chemotherapy would completely eradicate tumor cells in 24 patients with early stage breast cancer treated with neoadjuvant chemotherapy (paclitaxel followed by 5-fluorouracil, doxorubicin, and cyclophosphamide; T/FAC) [13]. These findings need to be validated in large-scale clinical trials before a test to predict patients’ response to chemotherapy could routinely be used.
Prediction of the therapeutic response to preoperative chemotherapy in breast cancer based on gene expression profiling
We are also carrying out analysis of gene expression with DNA microarrays in order to be able to select the best treatment for patients based on the gene expression profiles of their tumors. In order to identify the gene set for the prediction of therapeutic response, gene expression analyses were performed using DNA microarray and quantitative reverse-transcription PCR (Q-RT-PCR) (Fig. 1). DNA microarray is an excellent experimental tool to analyze gene expression comprehensively. While individual microarray studies can be highly informative, there are inconsistencies between various microarray platforms, making it difficult to compare independently obtained data addressing the same biological problem [14, 15]. Also, there are some problems about dynamic range, accuracy, and universal use. On the other hand, Q-RT-PCR is widely recognized to be the gold standard method for quantifying gene expression. However, studies using RT-PCR technology as a discovery tool have historically been limited to relatively small gene sets compared to other gene expression platforms such as microarrays. For development of the prediction system with high accuracy, verification by a method that we can analyze more precisely is required. Therefore, our strategy is that we first performed gene expression profiling of 21,000 genes by DNA microarray for selection of candidate genes. Then, differentially expressed genes were selected between the drug resistant group and the drug sensitive group based on DNA microarray data. Next, expression of selected candidate genes were quantified by Q-RT-PCR for confirming the array data and increasing the reliability. Finally, we narrowed the candidate genes down to establish a prediction system based on RT-PCR data.

Gene expression analysis by microarrays and quantitative RT-PCR. Patients are divided into two groups, responder (sensitive) and non-responder (resistant), based on clinical and pathological response. Differentially expressed genes were statistically selected based on microarray data between responder and non-responder group. Expression of selected genes was quantified in all cases by real-time RT-PCR. Selection of the candidate genes for discriminating between non-responder and responder was based on the RT-PCR data for establishment of a prediction system
The scheme of identifying genetic markers for neoadjuvant chemotherapy is shown in Fig. 2. Specimens were obtained by core needle biopsy before treatment begins, and pure populations of tumor cells were collected by Laser Captured Microdissection (LCM). After RNA extraction, gene expression analyses were performed using microarray or real-time RT-PCR. In contrast, patients receive the treatment of chemotherapy, and clinical response is evaluated at completion of treatment. Furthermore, surgical specimens were examined to determine the pathological response to chemotherapy. All clinical and genomic data are entered into an integrated database and analyzed for identification of predictive factors. We have identified several sets of genes that predict patients’ response to several different types of neoadjuvant chemotherapy such as paclitaxel, epirubicine and docetaxel. Before clinical application, independent validation of the prediction system is required, and we have started a validation study for prediction of the therapreutic response to preoperative paclitaxel on new cases. These kinds of findings bring oncologists one step closer to being able to select the most effective regimen for a particular individual.

Study of identifying genetic markers for neoadjuvant chemotherapy. We have combined the laser captured microdissection (LCM) method with the microarray technologies. Tumor cells were selectively collected by LCM to exclude most of the stromal tissues to analyze cancer cells and assessed gene expression analyses. Clinical and pathological responses are evaluated at completion of treatment. Differentially expressed genes were selected for discriminating between non-responder and responder
Functional screening system for chemo-sensitivity related genes using transfection cell array
DNA microarray analysis is a revolutionary experimental tool for analyzing gene expression comprehensively, and gene expression profiling has created new possibilities for the molecular characterization of cancer. A number of microarray studies have reported a lot of candidate genes for prediction of therapeutic response or clinical outcome. However, there are not many clinically useful systems showing that it is highly precise, because it is not easy to identify true target genes by mathematical (statistical) algorithms. Functional analyses for their characterization of candidate genes are required.
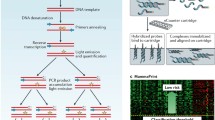
Functional validation is usually accomplished in molecular and cell based assays on a gene-by-gene basis, creating a bottle-neck effect for the characterization of the huge numbers of targets arising from genomics surveys such as microarray. A recently described, cell-based microarray system, the transfection cell array, paved a way for the high-throughput gene analysis in the field of functional genomics [14–19]. Transfection cell array (TFA) enable the high-throughput transfection of a large number of gene constructs and the detection of phenotypic changes in living cells by the different gene constructs (Fig. 3). We have designed the experiment for the construction of a high-throughput functional screening system for identification of chemo-sensitivity related genes using TFA.

High-throughput functional screening using transfection array (TFA). TFA enables the high-throughput transfection of a large number of gene constructs and the detection of phenotypic changes in living cells by the different gene constructs. Nucleic acid such as siRNA are printed together with transfection reagent on a glass slide. Cells are seeded directly onto the slide for reverse-transfection. After cell culturing, various detection assays can be performed. A process can be divided into three steps, as array preparation, reverse transfection and detection assay
In this study, candidate genes were chosen among microarray data of the paclitaxel trial with the above-described statistical selection for construction of the system. siRNA for selected candidate genes were spotted onto slides for preparation of transfection arrays and breast cancer cells were seeded onto arrays and transfected siRNA for knockdown of each candidate gene. After exposure with several concentrations of paclitaxel, cell viability was measured. To choose the chemo-sensitivity related genes on the basis of functional analysis, we selected the gene which affected sensitivity of the paclitaxel by knockdown. Then we constructed the functional gene network (pathway) that comprises known interactions derived from the database. Furthermore, we selected additional genes related to the pathway from each chemo-sensitivity related gene. Further TFA analysis is performed to confirm the significance of the constructed functional pathway.
Recent advances in our knowledge of cancer biology have led to the development of therapies targeting specific signaling pathways. Molecular targeting promises to improve ability to predict who will respond by assessing the state of these pathways in patients. We hope that these functional analyses will provide rational strategies for establishment of the prediction system with high accuracy and identification of ideal targets for drug intervention.
References
Rabaglio M, Aebi S, Castiglione-Gertsch M. Controversies of adjuvant endocrine treatment for breast cancer and recommendations of the 2007 St Gallen conference. Lancet Oncol. 2007;8:940–9.
Cinieri S, Orlando L, Fedele P, Cusmai A, D’Amico M, Rizzo P, Chetri MC. Adjuvant strategies in breast cancer: new prospectives, questions and reflections at the end of 2007 St Gallen international expert consensus conference. Ann Oncol. 2007;18(Suppl):63–5.
Eifel P, Axelson JA, Costa J, Crowley J, Curran WJ Jr, Deshler A, Fulton S, Hendricks CB, Kemeny M, Kornblith AB, Louis TA, Markman M, Mayer R, Roter D. National institutes of health consensus development conference statement: adjuvant therapy for breast cancer, November 1–3, 2000. J Natl Cancer Inst. 2001;93:979–89.
Fisher B, Bryant J, Wolmark N, Mamounas E, Brown A, Fisher ER, Wickerham DL, Begovic M, DeCillis A, Robidoux A, Margolese RG, Cruz AB Jr, Hoehn JL, Lees AW, Dimitrov NV, Bear HD. Effect of preoperative chemotherapy on the outcome of women with operable breast cancer. J Clin Oncol. 1998;16:2672–85.
Rivera E, Holmes FA, Frye D, Valero V, Theriault RL, Booser D, Walters R, Buzdar AU, Dhingra K, Fraschini G, Hortobagyi GN. Phase II study of paclitaxel in patients with metastatic breast carcinoma refractory to standard chemotherapy. Cancer. 2000;89:2195–201.
Wolmark N, Wang J, Mamounas E, Bryant J, Fisher B. Preoperative chemotherapy in patients with operable breast cancer: nine-year results from national surgical adjuvant breast and bowel project B-18. J Natl Cancer Inst Monogr. 2001;30:96–102.
Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC, Lashkari D, Shalon D, Brown PO, Botstein D. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci USA. 1999;96:9212–7.
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–52.
Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, Wilfond B, Borg A, Trent J. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–48.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–74.
van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer Nature. 2002;415:530–6.
Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, O’Connell P. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–9.
Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, Lecocke M, Metivier J, Booser D, Ibrahim N, Valero V, Royce M, Arun B, Whitman G, Ross J, Sneige N, Hortobagyi GN, Pusztai L. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–93.
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, Brown PO, Børresen-Dale AL, Botstein D. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100(14):8418–23.
Kruse JJ, Stewart FA. Gene expression arrays as a tool to unravel mechanisms of normal tissue radiation injury and prediction of response. World J Gastroenterol. 2007;13(19):2669–74.
Ziauddin J, Sabatini DM. Microarrays of cells expressing defined cDNAs. Nature. 2001;411:107–10.
Silva JM, Mizuno H, Brady A, Lucito R, Hannon GJ. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc Natl Acad Sci USA. 2004;101:6548–52.
Vanhecke D, Janitz M. High-throughput gene silencing using cell arrays. Oncogene. 2004;23:8353–8.
Castel D, Debily MA, Pitaval A. Gidrol X.Cell microarray for functional exploration of genomes. Methods Mol Biol. 2007;381:3753–84.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is based on a presentation delivered at Presidential Symposium 1, “Breast cancer: individualized diagnosis for tailored treatment,” held on 29 June 2007 at the 15th Annual Meeting of the Japanese Breast Cancer Society in Yokohama.
About this article
Cite this article
Nagasaki, K., Miki, Y. Molecular prediction of the therapeutic response to neoadjuvant chemotherapy in breast cancer. Breast Cancer 15, 117–120 (2008). https://doi.org/10.1007/s12282-008-0031-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-008-0031-6