
Abstract
Antibody drug conjugates (ADCs) are promising cancer therapeutics with minimal toxicity as compared to small cytotoxic molecules alone and have shown the evidence to overcome resistance against tumor and prevent relapse of cancer. The ADC has a potential to change the paradigm of cancer chemotherapeutic treatment. At present, 13 ADCs have been approved by USFDA for the treatment of various types of solid tumor and haematological malignancies. This review covers the three structural components of an ADC—antibody, linker, and cytotoxic payload—along with their respective structure, chemistry, mechanism of action, and influence on the activity of ADCs. It covers comprehensive insight on structural role of linker towards efficacy, stability & toxicity of ADCs, different types of linkers & various conjugation techniques. A brief overview of various analytical techniques used for the qualitative and quantitative analysis of ADC is summarized. The current challenges of ADCs, such as heterogeneity, bystander effect, protein aggregation, inefficient internalization or poor penetration into tumor cells, narrow therapeutic index, emergence of resistance, etc., are outlined along with recent advances and future opportunities for the development of more promising next-generation ADCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a diseased condition where normal body cells transform into uncontrolled proliferating cells mainly due to mutation in certain genes. Most of the chemotherapeutic drugs employed in cancer treatment are non-specific in nature and precipitate off target toxicities, mostly on the fast-growing epithelial cells of hair and gastrointestinal tract. Thus, there is a continuous need for novel anticancer agents which can selectively target cancer cells and cause minimal side effects to normal cells. Monoclonal antibodies (mAbs) and immune-based therapeutics have already attracted attention due to their great potential for target specificity, multiple mechanisms of action and broad therapeutic index. The surface antigens present on cancer cells, the only distinguishing feature from normal cells, are identified as the best therapeutic target for monoclonal antibodies. Classification of cancer based on the surface antigen can help to employ a particular mAb treatment (Waldum et al. 2008). mAbs specifically target the cancer antigen on cell surface and execute their actions by the two most prominent mechanisms; ADCC (antibody-dependent cell cytotoxicity) and CDC (complement dependent cytotoxicity). Other minor mechanisms involved in cancer cell destruction are promoting apoptosis, blocking cell signalling pathways and inhibiting cell signalling (Zhou et al. 2014a). There are over 50 different mAbs in clinical use currently. Despite their remarkable success, they cannot be claimed to give a complete cure of the disease as, over a period of time, cancer cells show heterogeneity, intrinsic resistance and/or acquired resistance (Zugazagoitia et al. 2016).
Antibody-drug conjugates (ADCs) are the emerging and promising next generation therapeutics after mAbs and are highly potent drugs that have emerged after the classic blend of immunotherapy and chemotherapy. Compared to the use of mAbs alone for cancer treatment, ADCs offer a synergistic effect due to the conjugation of a mAb with a cytotoxic drug (Khongorzul et al. 2020; Sheyi et al. 2022).
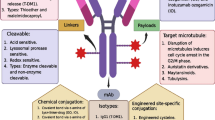
Unlike mAbs, ADC is a tri-component system that comprises monoclonal antibody, connecting linker and cytotoxic drug molecule, also known as payload (Joubert et al. 2020). Figure 1 shows the schematic representations of ADC with characteristics of each of its components. The necessity for development of ADC is to target cancer cells with fewer antigen expression which cannot be accomplished by mAbs alone. They are a promising therapy for targeted delivery of cytotoxic drugs at the cancer site. Currently, the development of ADC has reached up to 3rd generation drugs (Vankemmelbeke and Durrant 2016). 1st generation drugs have shown limited advantages compared to the parent drug due to insufficient potency of the payload and linker instability, however they have shown efficacy. 2nd generation heterogenous ADCs containing potent drugs with narrow therapeutic index have shown toxicity and found less effective due to stochastic conjugation approaches and limited penetration into tumor tissue. The 3rd generation ADCs overcame the previous problems due to the development of site-specific conjugation and stable linkers. Table 1 represents the detailed characteristics and limitations and comparison of ADCs belong to each, 1st, 2nd and 3rd generation (Vankemmelbeke and Durrant 2016; Sau et al. 2017; Zou 2023) At present, total 13 ADCs have been approved by USFDA (PEG Biopharma 2020; U.S.F.D.A Podcast 2022) and more than 100 ADCs are currently being evaluated in approximately 160 active clinical trials for the treatment of various types of haematological malignancies and solid tumors (Fu et al. 2022a, b). Table 2 represents the USFDA approved ADCs along their respective details of payload, linker, conjugation technique, DAR (Drug antibody ratio), indication etc. (Fatima and Khare 2022; Tarantino et al. 2022). The pharmacokinetic and pharmacodynamic actions of ADCs always depend on DAR which generally ranges from 0 to 8 when linker is joined through lysine base and it ranges from 2 to 4 when linker is conjugated through cysteine (Abdollahpour-Alitappeh et al. 2019). Various structural components of ADC need to selected very carefully (Tang et al. 2019) and are discussed in detail in subsequent sections.

Schematic representation of antibody-drug conjugate
Structural components of ADCs
Monoclonal antibody
Monoclonal antibody, also called as immunoglobulin, are the key component of ADC that provides selectivity to the treatment. Murine antibodies were the first mAb derived from mice using hybridoma technology (Carter and Senter 2008). However, due to their shorter life span, they proved inefficient to provide desired therapeutic response. To overcome the rejection of antibody treatment, murine antibody was later replaced by chimerized and humanized mAbs which also secure high cell target specificity (Carter and Senter 2008). These antibodies act as a good delivery system for pharmaceutical cytotoxic agent conjugated to it and hence the chances of off target toxicity were minimized (Majidi et al. 2009).
Structurally, mAbs are glycoprotein molecules, with IgG1, IgG2, and IgG4 antibodies most often employed for ADC design. For an mAb to be ideal for ADC, it should possess target specificity, high antigen-binding affinity, minimal cross-reactivity, minimal heterogeneity, low immunogenicity, and a long circulation time in plasma. The binding affinity of the antibody should be in the range of 0.1−1 nM (Goli et al. 2018).The antibody is divided into different regions or fragments that possess distinct functions. The Fab fragment mediates antigen recognition by binding to antigen. The other region of antibody has one constant fragment (Fc) which interacts with immune system through its binding to neonatal Fc receptor (FcRn). Engineering of this Fc domain can change the property of antibody which results into diverse immune effector functions like activation of complement system, migration of effector cell to the tumor site, induction of cell-mediated cytotoxicity (CH2 fragment) and antibody dependent cell-mediated phagocytosis (ADCP) (Chau et al. 2019). The antibody possesses disulfide bond in heavy and light chain. Cysteine residues of hinge region are the potential site for conjugation. The number of disulfide bonds of IgG in hinge region varies in each isotype, for example; IgG1 has 2, IgG2 has 4, IgG3 has 11 and IgG4 has 2 inter chain disulfide bonds. The antibody structural integrity is maintained by disulfide bonds and gets destabilized when they are subjected to alkylation and reduction processes during its conjugation to cytotoxic payload or drug. Even on reduction of disulfide bond of IgG1 isotype of hinge region, there is loss of ADCC activity (Adair et al. 2012). The IgG1 antibody is typically used due to its ability to induce both ADCC and CDC when it binds to FcRn. Other isotypes of IgG have several drawbacks. For example, IgG2 has a higher propensity for dimerization in vivo than IgG1, while IgG3 has a broader hinge region that is prone to proteolysis and IgG4 has a Fab region in the CH3 domain that forms a monovalent bond with other functional groups, resulting in diminished clinical efficacy (Carter and Senter 2008).
Linker
Linkers are the most important part of ADCs, connecting the mAbs to cytotoxic payloads and determining the overall stability, pharmacokinetic, and pharmacodynamic parameters of an ADC. Linker stability is the most critical criteria for an ADC to maintain its concentration in the blood plasma, as premature breaking of the linker can lead to an early release of the cytotoxic drug in the systemic circulation, resulting in adverse effects. To achieve targeted delivery with minimal off-target release of the cytotoxic drug, a proper linker molecule must be developed. The linker must also be liable enough for cleavage upon the action of proteolytic enzymes after internalization into the tumor cells (Firer and Gellerman 2012; Sheyi et al. 2022). Furthermore, the hydrophobicity of the linker is also an important factor, as hydrophobic payloads coupled with hydrophobic linkers can lead to aggregation of the ADC molecules (Buecheler et al. 2018). Therefore, for such payloads, hydrophilic linkers containing polyethylene glycol (PEG) groups (Tedeschini et al. 2021), pyrophosphate diester groups (Kern et al. 2016) or negatively charged sulfonate groups (McCombs and Owen 2015) must be required .
The efficacy of ADCs also depends on the number of drug molecules loaded on the antibody. An antibody can bear 2, 4, 6 or 8 drug molecules. The DAR is the most important consideration to obtain an appropriate ADC action and therefore optimization of DAR is necessary. If it is too less, it leads to decrease in efficacy and if too many drug molecules are attached to antibody, it causes pharmacokinetic instability and toxicity. Scientific research community is trying to put efforts to link larger drug molecules to antibody to design homogeneous ADC. Overall, aim during the development of ADC is to obtain the DAR close to 4 (Feng et al. 2014).
The chemistry of linking drugs to the exposed lysine, cysteine or engineered site of antibody creates an additional level of complexity to the design of ADCs. Generally, one disulfide bond is present between heavy and light chain and 2 disulfide bonds are present between heavy chains. Almost 8 cysteine residues are involved in this disulfide bond and are common target for linking the drug molecules by mild reduction of disulfide bond. Reduction exposes the sulphur group, where a linker molecule carrying the cytotoxic payload, reacts to form a bond with antibody (Liu-Shin et al. 2018). Based on the payload release mechanism, linker molecules can be classified as: cleavable and non-cleavable linkers (Jain et al. 2015). Table 3 represents the advantages and disadvantages of various linker types.
Cleavable linkers
Cleavable linkers have a chemical trigger in their structure which is cleaved to release the cytotoxic drug in an unmodified form. Their cleavage is based on selective physiological differences between extracellular and intracellular environments such as pH, redox potential, the presence of protease/glucuronidase enzymes, and the concentration of glutathione. Around 80–85% of the marketed ADCs contain a cleavable linker, which can be further divided into pH sensitive linkers (acid-cleavable linkers), protease sensitive linkers (peptide linkers), and glutathione sensitive or reducible linkers (disulfide linkers) (Jain et al. 2015).
pH sensitive linker
This type of linkers can release the payload based on the nonspecific pH sensing mechanism. Hydrazones are the most widely used pH sensitive linker that possesses half-life of 4.4 h at pH 5 and 183 h at pH 7, which means, they are readily cleavable at the acidic environment of the cellular compartments like in lysosomes with the help of lysosomal enzymes (Savoy et al. 2021). Thus, they are susceptible to cleavage only at specifically low pH (4–6) and not at systemic pH of blood. Potential demerit of such type of linker is the non-enzymatic release of drug in systemic circulation which makes it less preferred for its use. Figure 2 represents the conjugation of Doxorubicin with hydrazone linker. These type of linker is used in ADCs like Gemtuzumab Ozogamacin and Inotuzumab Ozogamicin (Garbaccio 2014).

ADC conjugated to payload (Doxorubicin) through pH sensitive hydrazone linker which gets cleaved in the acidic environment between pH 4–5
The acid labile linker acetyl butyrate was preferred to conjugate Calicheamicin to IgG4 in Inotuzumab Ozogamicin and was proved to be pharmacodynamically efficacious in CD22 positive lymphoblastic leukemia (DiJoseph et al. 2005; Dahl et al. 2016; Savoy et al. 2021).
Peptide linker
It is the most stable linker among the cleavable linkers. The peptide molecules are cleaved only by proteases which are intracellular constituents. Most commonly used dipeptide linker molecule, Valine-Citrulline (Val-Cit), undergoes cleavage upon the action of lysosomal protease enzyme, Cathepsin B, under the acidic condition (Sheyi et al. 2022). The valine-citrulline linker is used to conjugate mono methyl auristatin E (MMAE) to anti-CD79b antibody as shown in Fig. 3 which is used in hodgkin’s lymphoma (Dornan et al. 2009; Singh and Erickson 2009). These types of linkers are cleaved by intracellular proteases to release cytotoxic payload and they provide a selective action by cleaving the dipeptide bond. Moreover, the Val-Cit linker is proved to be around 100 times stable than hydrazone linker in the human plasma. In addition to that, they have shown minimum in vivo toxicity and maximum in vitro specificity, as compared to their hydrazone conjugates (Doronina et al. 2003). Another most commonly used dipeptide linker is Valine-Alanine (Val-Ala) (Sheyi et al. 2022). One of ADCs in clinic containing this linker is Loncastuximab tesirine which also contains a pegylated spacer to balance the lipophilicity of the payload from PDB dimer family, SG3199 (Lee 2021). Thus, Val-Ala linker is superior over Val-Cit in terms of accommodating lipophilic payload without much aggregation. Lastly, Phenylalanine-Lysine (Phe-Lys) linker is also considered as a potential linker, although is less stable as compared to the Val-Cit linker (Doronina et al. 2003).

ADC conjugated to payload (MMAE) through peptide (valine-citrulline) linker: Amide bond between valine-citrulline linker and p-amino benzyl alcohol (PABC) is cleaved by lysosomal enzyme and resultant moiety undergoes spontaneous self-immolation of the PABC portion through 1,6-elimination process to release the free payload, MMAE into the tumor cell
Disulfide linker
The third most common cleavable linker is the disulfide linker, which has a higher stability in systemic circulation due to its selective cleavage by the high intracellular concentration of glutathione. Concentrations of reduced glutathione are up to 1000-fold higher in tumor cells’ cytoplasm compared to normal cells. After the degradation and internalization of the disulfide bridge, the drug is released in the lysosome. Research has suggested that most of the disulfide-linked drugs are initially released intact by proteolytic degradation of the antibody in the lysosomes, then liberated as active metabolites through reducing agents such as glutathione or through disulfide exchange (Zhang et al. 2021). For example, Cantuzumab Ravtansine/IMGN-242 has the cytotoxic maytansinoids (DM4) conjugated to the huC242 antibody via a reducible disulfide linker, SPDB (N-succinimidyl 4-(2-pyridyldithio) butanoate), as shown in Fig. 4.The only challenging limitation of disulfide linker containing ADCs is the bystander effect (a process where cytotoxic drug gets released after being internalisation into the target cell into an extracellular fluid and then moves into a cell which may or may not lack ADC specific antigen).

ADC conjugated to payload (DM4) through disulfide linker: Disulphide bond is reduced by high concentration of reduced glutathione (GSH) within tumor cell and payload gets released as active metabolite
β-glucuronide linker
According to the published report, lysosomal β-glucuronidase is overexpressed in some tumor types (Albin et al. 1993; Graaf et al. 2002) which can selectively release cytotoxic payload through cleavage in a tumor cell unlike many hydrazone- and disulfide-based linkers which release the drug outside the targeted tissue. Amine group containing cytotoxic agents are delivered with the help of β-glucuronide linker. It is a type of enzymatically cleavable linker which is highly hydrophilic in nature and so can be used to deliver hydrophobic drugs. It possesses greater stability and solubility in plasma. These characteristics of linker provide ADCs which are well tolerated at high doses as well as are majorly active in vitro and in vivo. β-glucosidase acts on the glycosidic bond to release the drug. This enzyme has high intracellular concentration and lesser extracellular concentration which increases the systemic stability of ADC and reduces the off-target toxicity. Because of its hydrophilicity, it can also prevent the aggregation of ADCs containing highly hydrophobic drugs. Drugs like Auristatins bind to the payload through this type of linker (Bargh et al. 2019). Additionally, to expand the utilization of this linker toward phenol group containing drugs like SN38, duocarmycin, psymberin etc., incorporation of N,N’-dimethylethylene diamine self-immolative spacer was introduced with the linker as shown in Fig. 5. Release of drug takes place by the exposure of β-glucuronidase with the drug linker. The resulting ADCs were therapeutically selective and active against CD70 positive breast and lung cancer (Jeffrey et al. 2010).

ADC conjugated to payload (SN38) through β-glucuronide linker: Release mechanism involves initial de-glucuronidation using enzyme β-glucuronidase followed by 1,6-elimination, decarboxylation, and lastly cyclization of the dimethylethylene diamine (DMED, self-immolative spacer) carbamate to liberate free payload, SN38
Non-cleavable linker
Non-cleavable linkers do not contain any chemical triggers in their structure, and they form part of the payload along with an amino acid appendage. They only release the drug after the degradation of the monoclonal antibody within the lysosome after its internalization. The conjugation of the antibody to this linker is typically based on a lysine or cysteine amino acid pair. This type of linker is the most stable linker used in ADCs due to its increased plasma stability. By limiting the amount of free drug in the extracellular domain, non-cleavable linkers reduce the adverse effects of ADCs, since they are non-breakable in nature (Polson et al. 2009; Govindan et al. 2016). Examples of non-cleavable linkers include thioether and maleimidocaproyl (MC). As presented in Fig. 6 the non-cleavable linker, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC), when linked with a cytotoxic agent, maytansine DM1, in a clinically useful ADC, Kadcyla, shows less toxicity and good efficacy as compared to cleavable disulfide linker (Lu et al. 2016). The active metabolite, Lys-SMC-DM1, generated after catabolism has poor permeability and so it cannot exert Bystander effect. Similar is observed with MC linked with monomethyl aurustatin F (MMAF). When conjugated to non-cleavable linker, drugs like dolastatin was found to be required in minimum effective concentration for tubulin inhibition (Gianolio et al. 2012). Conjugation of non-cleavable linker, amino-PEG6 with transglutaminase isopeptide, forms an amide bond which is stable enough to carry auristatin analogues to the target site. These observations can prove the non-cleavable linkers better than cleavable linkers for the site-specific delivery of payload (Dorywalska et al. 2015). The only drawback related to the use of non-cleavable ADCs is that, several drugs possessing charged amino acids are unable to penetrate the membrane, which makes them less efficient to combat the surrounding tumor cells (Kovtun and Goldmacher 2007).

ADC conjugated to payload (DM1) through non-cleavable (MCC) linker: Release mechanism involves complete lysosomal proteolytic degradation of the antibody after internalization into the cell which releases the active metabolite of payload, lysine-MCC-DM1
Payload
Majority of payloads used in ADCs are highly cytotoxic agents and primarily involve two targets: one is cell cycle independent (targets DNA) and other is cell cycle dependent (microtubule inhibition and G2/M arrest) (Mack et al. 2014; Kostova et al. 2021) The selection criteria for payload involve stability, solubility and conjugation (Tang et al. 2019). Following conjugation, it is crucial for the payload to remain stable in the systemic circulation before reaching to the target site. Alkene-, epoxide- and disulfide-containing drugs may be transformed or reduced by the cellular enzymes, while acid labile drugs may degrade in the lysosome before reaching the cytosolic target. An efficient payload must be soluble enough for allowing the antibody to conjugate in aqueous buffers, as greater concentrations of organic solvent may lead to denaturing of the antibody scaffold. The microtubule polymerization inhibitors include etoposide, vinblastine, vincristine, taxol, auristatin analogues and maytansinoids. By inhibiting tubulin formation, the cell cycle gets arrested and eventually cell undergoes apoptosis. Another class of drugs involved in ADCs that targets DNA of cancer cells includes camptothecin, duocarmycin, pyrrolobenzodiazepine and calicheamicin (Deonarain et al. 2015). Figure 7 shows the structures of various payloads used in ADCs (Goundry and Parker 2022). and described here.

Structures of various payloads used in ADCs
Calicheamicin
It is an antitumor antibiotic extracted from Micromonospora eichnospora calichensis. It belongs to family enedyiene. The prominent mechanism of action of calcheamicin is binding to DNA and cleaving the ribose sugar from it. During this, it searches for the nucleotide sequence TCCT in 5’-3’ sequence of DNA through its iodine group in aryl tetrasaccharide moiety. Upon finding the particular sequence, it prevents transcription factor from binding to it (Perez et al. 2014; Robak and Robak 2014). The two ADCs that contain derivative of calicheamicin are gemtuzumab ozogamicin and inotuzumab ozogamicin which contain humanized monoclonal IgG4 antibody that are directed against CD33 and CD22 respectively which ultimately bring about regression in tumor (Ricart and Tolcher 2007).
Doxorubicin
It is an anticancer antibiotic extracted from Streptomyces peucetius. It has central anthracycline nucleus. The primary mechanism of action of doxorubicin is to intercalate the DNA base pair and to stop the process of ligation –religation carried out by topoisomerase II. Doxorubicin is conjugated to milatuzumab which targets tumor cell through CD74 antigen (Widdison and Chari 2013).
Maytansinoids
Maytansinoids are derived from the African shrub, Maytenus ovatus, and are 19-membered macrolides attached to a chlorinated benzene, with a C-3 side chain (N-acyl-N-methyl-l-alanyl ester) that has been found to have biological activity. The primary mechanism of action of maytansinoids is to inhibit microtubule polymerization and arrest the cell cycle between G2 and M phases. Clinically, maytansinoids are used in a type of antibody-drug conjugate (ADC) called trastuzumab emtansine, which targets a HER2 antigen (Widdison and Chari 2013; Feng et al. 2014). The thiolated group present in S-methyl-DM1 and S-methyl-DM4 is used in a Michael addition reaction to form a disulfide bond (Tan 2015).
Dolastatin 10
Dolastatin 10, mitotic spindle inhibitor, is one of the extremely potent cytotoxic natural pentapeptide which is obtained from the intestine of marine sea hare, cyanobacteria species, Dolabella auricularia. Structurally it is a linear pentamer that comprises of 4 amino acids at N-terminal and 1 amino acid at C-terminal. Both, N-terminus and C-terminus are preferable to undergo modification. The N terminal-α,α dimethyl substitution gives better in vitro and in vivo efficacy. The efficiency of dolavaline, a structural analogue of dolastatin 10, is observed by replacing the N-terminal N,N-dimethyl alanine with N,N-dimethyl glycine (Maderna et al. 2014). The drug binds close to anti-cancer alkaloids, vincristine and colchicine binding β subunits of tubulin and is more potent than other tubulin binding drugs. Because, it expresses anti-tubulin activity at maximum therapeutic dose, a targeted delivery system is needed to avoid any toxicity (Gaya and Rustin 2005). When dolastatin is used in combination with brentuximab vedotin, it promotes dendritic cell and T-cell maturation to increase immune response against tumor (Müller et al. 2014).
Auristatins
Auristatins are synthetic derivatives of dolastatin 10. They disrupt the microtubule formation and induce apoptosis of cancerous cells (Feng et al. 2014). The derivatives of auristatin, mono methyl auristatins E (MMAE) and mono methyl auristatin F (MMAF) are much potent than vinblastine. However, they are susceptible to resistance too as they are sensitive to efflux pump and p-glycoprotein (Goldmacher and Kovtun 2011). The MMAF carries a negatively charged C terminal phenylalanine residue rather than carboxyl group which is the reduced form and has less cell permeability. To overcome this problem, use of non-cleavable linker is recommended which makes it internalised (Gerber et al. 2013; Dugal-Tessier et al. 2017). Researchers have found that increasing the hydrophilicity decreases the aggregation of ADCs and also reduces the bystander effect of auristatin analogues (Mendelsohn et al. 2017).
Amatoxins
Amatoxins are the class of naturally occurring toxins, containing 9 structurally related toxins, and obtained from basidiomycetes mushrooms belonging to genus Amanita. The most prominent members are alpha and beta-Amanitin. This payload introduced the new mechanism of anticancer action by inhibition of RNA Polymerase II through binding to cleft Rpb1 and Rpb2. Thus, it prevents the initiation and elongation state of transcription. They are chemically resistant to heat, acid and enzymatic degradation. Moreover, amatoxins possess high water solubility which makes them stable in plasma and their clearance gets increased. Due to their hydrophilicity, they do not penetrate through the cell via passive diffusion.
Amatoxins have shown ideal conjugation property as they are able to retain the payload characteristics even after joining through linker and thereby reduce the chances of off target toxicity. The linker is attached to the -OH (tryptophan) or -NH2 (asparagine) group of amanitin for better stability. Amanitin, in its conjugated form, is highly active against multi-drug resistant tumor cells. Its action against both, dormant and active tumor cells, reduces the chance of cancer to relapse (Pahl et al. 2018; Zhao et al. 2020).
Pseudomonas exotoxin-A/PE38
Pseudomonas exotoxins A (PE), 638 amino acid proprotein, is the most toxic and immunogenic virulence factor of the Gram-negative bacterial species, Pseudomonas aeruginosa. After processing, it is converted into single chain polypeptide, 613 amino acid, mature toxin. The active form this PE (enzymatically active C-terminal domain having 405–613 amino acids) performs the ADP-ribosylation activity, inactivates elongation factor-2 (EF-2) and thereby inhibit the protein synthesis of the host cells which led to cell death (Wolf and Elsässer-Beile 2009). PE38 (38 kDa) is a truncated form of PE with lesser immunogenicity which lacks the receptor binding domain and a coupling site is created for cross-linking to suitable targeting agents. It is better tolerated in in-vivo experiments due to the breaking of intramolecular disulfide bond (Wu and Zhu 2021). Full or truncated PEs are used for the construction of immunotoxins (ITs) which can be used in targeted cancer therapy. Many scFv ITs containing PE38 are under clinical trials which targets antigens like CD22, mesothelin, CD25 etc. (Havaei et al. 2021). It has been observed that, in case of treatment with PE-based Its, around 2–5 cycles of treatment are needed to get remarkable clinical response including some complete remissions (Mazor et al. 2016). The approved ADC, Lumoxit, contains PE38 as payload and used for the treatment of hairy cell leukaemia.
Camptothecin derivative, SN38
Camptothecin is the natural alkaloid having highly lipophilic pentacyclic structure due to which its clinical application for the treatment of cancer was unsuccessful. Its hydrophilic prodrug, irinotecan is approved for the treatment of metastatic colorectal cancer. It has also shown clinical activity against cervical, ovarian, gastric, lung cancer and malignant lymphoma. SN38 (7-ethyl-10-hydroxycamptothecin) is the active metabolite of irinotecan which is a moderately cytotoxic drug with 1–6 nM IC50 against several human cancer cell lines. Its serum levels were observed to be about 100 times lower than the corresponding irinotecan levels. It acts via inhibition of nuclear enzyme, topoisomerase I which in turn introduces double-stranded DNA breaks while cells are in the S-phase (Hageman and Morozowich 2007; Ramesh et al. 2010; Govindan et al. 2019). SN-38 is highly hydrophobic with very limited number of available coupling sites in its structure. Through some unique conjugation method, the lactone ring of the SN-38 molecule is stabilized and the molecule is protected from glucuronidation which could end up into the development of FDA approved ADC, Trodelvy (Sacituzumab govitecan), for the treatment of TNBC (Goldenberg and Sharkey 2019).
Pyrrolobenzodiazepine (PBD) dimer, tesirine
Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs) are a family of antitumor antibiotics that exert the cytotoxicity through binding and selective alkylation in the minor groove of DNA. The N2 of DNA base, guanine, forms a covalent bond with the electrophilic N10/C11 imine of the PBD and ultimately damage the DNA of cancer cells. Dimers of PBDs are the highly potent synthetic compounds made from two individual PBD molecules linked through flexible propyldioxy tether (Gauzy-Lazo et al. 2020; Hartley 2021). Tesirine (SG3249) is one of the representatives of the PBD family which was developed by Seattle Genetics from SGD-1910 or talirine in order to decrease the hydrophobicity while maintaining potency. They introduced discrete PEG8 spacer to enhance the solubility (Tiberghien et al. 2016).
Conjugation chemistry of ADCs
The clinical success of ADC not only depends on potency of the payload and linker stability but also equally on bioconjugation technique used to attach the payload. To ensure the good pharmacokinetic profile of ADCs, precise and site-specific conjugation between payload and antibody is necessary (Tsuchikama and An 2018). First and second-generation ADCs were designed using conventional non-specific or stochastic methods of conjugation that use endogenous amino acids; cysteine and lysine which led to heterogenous mixture of ADCs having positional isomerism and ultimately results in a variety of DARs. In contrast, third generation ADCs, Enhertu®, Trodelvy® and other approved ADCs after 2020 are mostly designed through site specific conjugation to natural or non-natural amino acids which produce homogeneous ADCs with well-defined DAR. Primarily, this site-specific engineered conjugation technique restricts the distribution of drug within the antibody (Drenkard et al. 2007). Table 4 represents the widely explored conjugation techniques and reactions for the design of ADCs.
Stochastic (non-specific) conjugation techniques
Stochastic conjugation techniques use surface lysine or interchain cysteine residues as drug attachment sites on the mAb.
Lysine amide conjugation
Lysine (Lys) was the first amino acid residue employed in antibody conjugation. Generally, lysine residues are more in number in the antibody structure. There are around 85 Lys residues on the IgG1 antibody of which more than 40 are modifiable with accessible hydrophilic amino groups located on the surface of antibody. So, they become a preferable site for payload attachment. Amide coupling takes place when nucleophilic amine functional group of Lys residue reacts with the electrophilic activated carboxylic acid group of payloads. Lysine residues are usually modified by conventional electrophilic reagents like heterobifunctional NHS (N-hydroxy succinimide) ester, sulfonyl chloride, iminoboronates, isocyanates and isothiocyanates, diazonium salts, etc. (Dennler et al. 2015; Deonarain et al. 2015). This technique was used to produce clinically approved ADCs like Besponsa, Mylotarg, and Kadcyla. Drugs like dolastatin 15, α-amanitin were linked directly with the antibody through lysine conjugation. However, this is not widely preferred technique as it fails to provide homogeneous ADCs. For the given DAR of ADC that should range from 2 to 4, up to 106 different isomers of ADCs are generated by this conjugation method. These heterogeneous ADCs may differ in their DAR and conjugation-site. ADCs with very high DAR can cause toxicity due to high potency while unconjugated mAbs compete with drug-loaded species for the binding with antigen on target cells. This led to antibody aggregation. Also, each ADC in a mixture can have different stability and clearance rate and therefore it gives variable pharmacokinetic properties. For example, Methotrexate conjugation via lysine residue of Fab fragment of IgG antibody is efficacious, but gives lesser bioavailability at tumor site because of short half-life of Fab fragment (Kulkarni et al. 1985). To overcome the existing limitations of lysine conjugation, it is very essential to achieve the site-selectivity for which many site and residue-specific modifications on lysine by chemical reagents have been tried and published by many researchers (Sang et al. 2020; Haque et al. 2021). It’s very challenging task to selectively modify certain lysine residues only and thus represents the future challenges for the development of next generation ADCs. However, through recent advancement of site-specific conjugation technique, it has become possible to modify the specific lysine residues in the Fc region, a constant domain, of various antibodies. Yamada, K. et al. reported a new regiodivergent conjugation technology, AJICAP™, through which they functionalized selective lysine residues in the mAbs by simply changing a peptide sequence. They successfully introduced thiol functional groups on Lys246 and Lys248 residues using Fc affinity peptide reagents without antibody engineering and payload was then connected to this new thiol groups. The resulting ADC showed promising in vivo efficacy. (Yamada et al. 2019; Fujii et al. 2023) Recently in 2022, Dingdong Yuan et al. published a facile way of site-selective IgG functionalization where they selectively acetylated Lys248 in the Fc region of heavy chain of IgG antibody using a peptide-guided, proximity-driven group transfer reaction. They synthesized bispecific antibody complex (bsAbC) containing two antigen-binding sites to each antigen which showed nanomolar effector-cell-mediated cytotoxicity in-vitro (Yuan et al. 2022).
Cysteine conjugation
Cysteine conjugation technique is based on the reaction between free thiol group of cysteine residues present in the antibody with the thiol reactive functional groups present in the payload. Unlike Lys conjugation which uses freely accessible hydrophilic amino groups, present on selective Lysine residues, no such free thiol groups are present on Cysteine residues for the conjugation. Instead, all cysteine residues form disulfide bonds which need to be reduced using mild reducing agents like dithiothreitol (DTT) or tris (2-carboxyethyl) phosphine (TCEP) to generate free thiol groups. Mostly, interchain disulfide bonds are selectively reduced which are least critical for the structural stability of the antibody. Depending on the reduction condition, it can generate 2, 4, 6, or 8 free thiols which can take part in conjugation reaction after getting modified by nucleophilic displacement with the thiolate group or by nucleophilic addition to the Michel acceptor. Most commonly used linker for cysteine conjugation is maleimide functionalized linker. Thus, unlike, lysine conjugation, cysteine conjugation is not a chemically straightforward process. Secondly, reduction of disulfide bonds affects the antibody structural integrity and stability adversely. Another drawback with maleimide conjugation is the instability in human serum due to the reversibility of the reaction while lysine amide conjugation is much stable. Structurally, sulfur atom is located the beta position of maleimide and Cys carboxyl thereby undergoing retro-Michael addition reaction upon oxidation of the S atom in blood serum/vessel. Attached drug (thiosuccinimide product) can be shed from the antibody via retro-Michael elimination/deconjugation reaction in presence of thiols in the serum and led to many side effects due to premature release of toxic drug. This technique was used to produce clinically approved ADC, Adcetris. In comparison to lysine, cysteine residues are less in number, so, comparatively stoichiometric homogeneous conjugates (~ 15 distinct ADCs) are produced which provide controlled DAR (ranging from 0 to 8) and heterogeneity (Nadkarni 2020).
Still, this can be improved through engineered cysteine technology. The first cysteine-based conjugation was carried out by Michael M.C. and his co-workers where they conjugated IgG1 antibody to MMAE through alkylation and reoxidization of thiol group of cysteine (Sun et al. 2005). The engineered cysteine residue involves replacement of cysteine residue with serine and resulting new antibody contains only 2, 4 and 6 accessible sites of cysteine with defined stoichiometry and pharmacokinetics (Mcdonagh et al. 2006). Moreover, this type of conjugation provides good yield and purity while operating on cytotoxic drug at very low scale concentration. One of such engineered cysteine technologies called THIOMAB can generate highly homogeneous ADCs with DAR of 2 and has shown very promising results in in-vivo studies (Junutula et al. 2008).
Cysteine rebridging (Griebenow et al. 2016) and many other cysteine oriented site-specific conjugation strategies (You et al. 2021) have been reported so far which provided advantages in terms of stability, homogeneity and controlled DAR. Despite of them; major challenges in cysteine conjugation are instability of the linker after the reaction and the heterogeneity of ADCs for which new technologies are still awaited (Sussman et al. 2018).
Site specific engineered conjugation techniques
Enzymatic conjugation
Alteration or addition of enzyme moiety has opened up a new site-specific conjugation approach to link antibody with payloads. The technique is introduced to reduce the heterogeneous conjugation of cytotoxic drug to various amino acid base pair of antibodies. Antibodies are genetically engineered at specific amino acid and such modified sites/reaction handles are identified by the enzymes which then selectively allow it to react with counterpart functional groups present on payload. For example, insertion of genetically encoded amino acid tags at specific antibody sequence which is recognized by the enzyme to perform site specific conjugation. This technique offers precise, homogeneous and defined DAR as well as maintains the structural integrity of antibody (Beerli and Grawunder 2017; van Berkel and van Delft 2018). The enzyme conjugations are listed as follows:
Using microbial transglutaminase enzyme (MTGase)
Transglutaminase is a bacterial enzyme obtained from Streptomyces mobaraensis. These enzymes catalyse isopeptide bond formation between primary amine functional group of substrate/payload and glutamine side chain at position 295 of the deglycosylated antibody under mild conditions. The resultant isopeptide formed is stable against proteases and offers ADC with DAR 2. Experimentation has shown that it can bind four payloads to IgG1–Fc region (Dennler et al. 2014; Hussain et al. 2021).
Using sortase enzyme
It is an enzyme involved in transpeptidation process during cell wall biosynthesis of gram-positive bacteria like S. Aureus. The sortase enzyme is divided into 6 subfamilies, Sortase A-F, amongst which Sortase A catalyses the transpeptidation for protein ligation. Sortase A specifically recognizes the C terminal of antibody modified with sequence LPXTG (X: any amino acid), cleaves the amide bond between glycine and threonine (T-G) and attaches an oligo-glycine containing molecule/payload to the newly generated C-terminus. The engineered antibody fragments, single chain Fv (ScFv) and Fab fragment, bind to the payload through this enzymatic technique. The conjugation technique is modified from nucleophilic transpeptidation reaction to azide-alkyne cycloaddition reaction which led to increase in the DAR (Falck and Müller 2018; Hussain et al. 2021).
Using formylglycine generating enzyme (FGE)
The formylgycine-generating enzyme binds to the specific sequence CXPXR (X: glycine, alanine, threonine or serine) in the heavy and light chain of antibody and catalyse the post translational modification, i.e., oxidation of cysteine to a formyl-glycine which can be later used for conjugation chemistry (Beerli and Grawunder 2017; Krüger et al. 2019).
Using tubulin tyrosine ligase (TTL) enzyme
Tubulin tyrosine ligase is another enzyme which catalyses addition tyrosine derivatives and produces site specific conjugation at C-terminus of a protein of interest by recognizing the 14-amino acid hydrophilic sequence (VDSVEGEGEEEGEE, Tub-tag) within the peptide region. The unique chemical handle present on the tyrosine derivatives can be functionalized through chemo-selective conjugation. Very less information regarding this enzyme has been reported in literature and clinical significance of this conjugation chemistry is yet to be explored (Gerlach et al. 2019; Hussain et al. 2021).
Using trypsiligase and subtiligase enzymes
Enzymatic modification on N-terminus of amino acids is inaccessible, however, few proteinases are engineered to conjugate payload at the specific position of peptide of antibody. Trypsiligase, a designer enzyme for both N- and C-terminal site-specific labelling of peptides and proteins, has four variants K60E/N143H/E151H/D189K with different specificity for chemo-enzymatic conjugation. Click chemistry approach can be used to produce homogeneous ADC using this enzyme (Meyer et al. 2016). Similarly, subtiligase is an engineered peptide ligase enzyme derived from Bacillus amyloliquefaciens which performs ligation reaction of an acyl-donor peptide ester to the N-terminal α-amine of the acceptor peptide, forming a native peptide bond. The enzyme has two mutated amino acids, S221C and P225A compared to parent subtilisin enzyme (Zhang et al. 2018). A proper pharmacokinetics is not established for trypsiligase and subtiligase yet (Falck and Müller 2018; Hussain et al. 2021).
Using phosphopantetheinyl transferase enzyme
4’-Phosphopantetheinyl transferases (PPTase) are site-specific conjugating enzymes that carry out posttranslational priming of acyl carrier proteins (ACP) and peptidyl carrier proteins (PCP) by attachment of 4’-phosphopantetheniyl cofactor (P-pant) with highly conserved serine residues of each domain through phosphodiester bond. They are considered as versatile biocatalyst for site specific modification of proteins due to their broad substrate tolerance. The P-pant prosthetic group serves as a flexible linker which is highly stable and irreversible. The two most commonly utilized PPTase, Bacillus subtilis Sfp and Escherichia Coli AcpS, produce dual labelled antibody. The enzyme can carry out conjugation in one-step or two-steps process, but higher DAR can be achieved with 2-step process. The enzyme can attach payload across the full length of antibody, however, the conjugation kinetics is optimal only in specific domains (CH1). The PPTase can counter both, high drug loading and drug resistance issues, in an innovative way (Yin et al. 2004; Grünewald et al. 2019).
Using spyLigase enzyme
SpyLigase is a synthetic enzyme that catalyzes the formation of isopeptide bond irreversibly between the two peptides, SpyTag and KTag. This selective peptie-peptide ligation technique can be used for the conjugation of cytotoxic payloads to antibodies. Using this enzyme-based conjugation technique, Siegmund V. et al. have designed and prepared new class of ADC based on these recognition peptide tags, SpyTag and KTag. They prepared ADCs by initially fusing SpyTag to the C-terminus of the anti-EGFR mAb, Cetuximab. SpyLigase was used to covalently link KTag with SpyTag in a site-specific manner and lastly, a cytotoxic payload, MMAE was attached to the chemically synthesized KTag via click chemistry. The obtained ADC exhibited subnanomolar IC50 values. The ligation technique also seemed advantageous for the enhancement of DAR (Fierer et al. 2014; Siegmund et al. 2016).
Using O6-alkylguanine-DNA alkyltransferase (AGT) (SNAP-Tag) enzyme
This enzyme repairs the O6-alkylguanine in double‐stranded DNA by transferring the alkyl group from the DNA to the cysteine residues of O6‐alkylguanine‐DNA alkyltransferase irreversibly. Thereby it forms S‐alkylcysteine (thioether) and AGT enzyme gets deactivated. SNAP-Tag is a conjugation technique which is based on AGT enzyme action. O6‐benzylguanine (BG) modified payloads can be conjugated to SNAP‐tag in a site‐specific manner. The nucleophilic substitution reaction carried out by the SNAP-tag has significance in the conjugation of ADC as it reduces the heterogeneous products. The enzyme acts on the single chain fragment variable (scFv) of the VH and VL domains of the antibody and produces a homogeneous conjugation of the drug to an antibody having a stoichiometric ratio of 1:1 (Fang et al. 2005). The conjugation of MMAF and anti-Her2 mAb using SNAP-Tag has significantly enhanced the cytotoxic activity against breast cancer. The only limitation with the enzyme is that to increase the DAR, is a challenge (Woitok et al. 2016).
Glycan conjugation
Glycan conjugation is an exclusively site-specific conjugation approach because almost all IgG type antibodies carry one and only N-glycan linked to Asn297 in its conserved site of Fc region. This N-glycan is a suitable payload conjugation site because it is located very far from the variable region. Glycosylation of small payload to Asn297 glycans generates homogeneous ADC. This technique provides structural benefits like the conjugation portion lies far from the antigen binding region of antibody so antigen binding is not affected by the conjugation. Second, its chemical structures are different from amino acids hence no interaction with polypeptide is possible. Carbohydrate chemistry is totally distinct from peptide chemistry. Third, 2 or 4 conjugation per antibody is possible with biantennary nature of two oligosaccharides (Qasba 2015). The two most common sugar moieties exploited are N-acetylglucosamine and mannose. Along with them, galactose and sialic acid are also widely explored carbohydrate molecules (Agarwal and Bertozzi 2015). One of the examples of glycan-based conjugation is the periodate oxidation of glycans to produce aldehydes on the antibody which can be later used for conjugating payload with hydrazide or aminooxy moiety. Using this, the ADCs can be generated that give DAR in the range of 4–6. As the glycosylation is a heterogeneous posttranslational modification, the main challenge is to generate homogeneous glycans. The generated glycans exist as mixture of oxidised fucose, galactose and sialic acid residues. Many approaches have been explored to overcome this limitation like periodate oxidation of oligosaccharides to modify the mAb (Zuberbühler et al. 2012), introduction of periodate-sensitive sialic acid and galactose residues into the native antibody N-glycan (Zhou et al. 2014b) conjugation after remodeling of the glycans of antibodies with azido-containing sialic acid etc. (Li et al. 2014) .
Unnatural amino acid (UAA) conjugation
This site-specific conjugation technique involves the incorporation of structurally unique unnatural/non canonical amino acids over the surface of antibody where liker-payload conjugates in a chemoselective manner. This unnatural amino acid contains a bio-orthogonal handle that enables the site-specific attachment of a drug with defined stoichiometry in the presence of all 20 naturally occurring amino acid functional groups. This is very challenging as rearrangement of the antibody sequence is required for which new tRNA/aminoacyl tRNA synthetase pair is needed. This engineered tRNA synthetase inserts the UAA into the protein in response to an unassigned codon, mostly an amber stop codon (TAG). Extensive work has been done by Ambrx, Inc. USA in the area of developing such technologies that enhance the expression of proteins containing UAAs. p-Acetylphenylalanine (pAcPhe) and p-Azidophenylalanine (pAzPhen) are the examples of UAAs. One of the reported methods of UAA conjugation is to develop of pAcPhe through orthogonal tRNA/ aminoacyl-tRNA synthetase (aaRS) expressing cell line in which amber stop codon are substituted so antibody synthesized from them contain pAcPhe chain. This pAcPhe chain is stable and contains a keto group which donates electron to a drug containing alkoxy-amine, like MMAF (mystanoid analogue). This leads to an oxime ligated conjugate (Panowski et al. 2014; Schumacher et al. 2016). The pAcPhe is structurally related to Fab region and is conjugated with drug through hydrazone linker using click chemistry (Sochaj et al. 2015).
The second method is addition of selenocysteine (Sec) in which sulphur atom is replaced by selenium ion which proves to be more nucleophilic in nature than existing thiol group of cysteine. The selenium group is introduced into the mAb using Sec labelling technique where the UGA codon is labelled with non-Sec protein to alter the transcription action (Adumeau et al. 2016).
This site-specific technique of conjugation works best to control the exact localization and number of reactive residues to give homogenous ADCs (Kang et al. 2021). The only concern is that suitable choice of UAA is important because its incorporation may result into instability of mAb as well as it can induce antigenic response.
C-/N-terminal selective conjugation
Modification of C-/N-terminus of mAb is another site-specific conjugation approach to introduce biorthogonal motifs or affinity tags which can be further functionalised. Compared to N-terminus, C-terminal positions are distal from antigen binding sites and so have wider scope for the modifications (Walsh et al. 2021). According to a recently published study, low levels of C-terminal amidation is typically detected in biotherapeutic mAbs which does not raise any safety concerns. Based on the results, further studies to access its safety and efficacy potential in much higher concentration are needed (Shah et al. 2022). On the other hand, modification at N-terminus, although widely explored, is more challenging and needs close monitoring to ensure that it does not hinder antigen binding affinity. The more nucleophilic α-amine of the N terminus, having pKa of 6–8, is a uniquely reactive site which can be modified even under milder conditions by various methods like pH control, modification of specific amino acids; cysteine, tryptophan, serine or threonine etc. through introduction of reactive aldehyde or ketone handle for conjugation through oxime or hydrazone linkages, N-terminal transamination and oxime ligation etc. (Rosen and Francis 2017). SeriMab technology by ImmunoGen (Harris et al. 2015) is one of the successful examples of N-terminal modification. Recently, Ko, M.J. et al. have reported a method to develop a new ADC type where payload is conjugated to N-terminal of mAb through amine bonds introduced via reductive alkylation reactions (NTERM) and compared the stability, efficacy and toxicity of ADC with thiol-conjugated and the lysine-conjugated ADCs (Ko et al. 2021). During both, in-vitro and in-vivo screening, NTERM conjugated ADC was found better in terms of therapeutic window, stability and toxicity.
Analytical techniques for ADCs
Physical characterization of ADCs is done using various analytical techniques to identify the heterogeneous nature of ADC. The analysis of each component in ADC (antibody, linker and drug) needs specificity with highest resolution so that a perfect evaluation can be done (McCombs and Owen 2015). The various techniques involved in characterization of ADC are chromatography, mass spectroscopy, UV spectroscopy and other hyphenated techniques like LC-MS, RPLC-MS etc. Even bioanalytical technique like ELISA is used for analysing drug distribution in ADC (Neupane and Bergquist 2017).
An analyst faces difficulty in characterization and quantification of ADCs owing to their complex and heterogenous structure compared to the drug and antibody individually. Various comprehensive reviews are published by different authors describing various analytical methods that can be used for characterization and quantification of ADCs. Various quality attributes that required precise control are DAR, drug distribution, size variant analysis of conjugates, charge based separations, analysis of unconjugated drug, peptide mapping analysis and biophysical analysis (Wakankar et al. 2011; Wagh et al. 2018). Various techniques used for the measurement of said quality attributes are described in the following sections.
UV−visible spectroscopy
It uses electromagnetic radiation and measures the amount of radiation absorbed by drug and mAb. It is the most common technique to measure DAR when used in continuous with hydrophobic chromatography. However, photo lability of drug like calcheamicin limits its use for DAR determination. Also ADCs having different absorption maxima limit its use (Neupane and Bergquist 2017).
Owing to its simplicity and relatively cheaper instrument, UV−visible spectroscopy is a method of choice by various researchers. This technique is most widely used to measure the Drug-Antibody ratio. For determination of drug-antibody ratio, Amax values of ADC and antibody requires to be calculated followed by solving the simultaneous equation, mole of drug per mole of antibody can be calculated (Wakankar et al. 2011; Wagh et al. 2018). However, drugs like calcheamicin cannot be estimated using UV-Visible spectroscopy due to its photolability. Further, if ADCs having different absorption maxima, then it cannot be estimated using the technique (Neupane and Bergquist 2017).
Hydrophobic interaction chromatography (HIC)
HIC evaluates both, drug load distribution and average DAR. In this technique proteins are separated based on their relative hydrophobicity. Separation using HIC ensures that ADC remains intact and does not get denatured as in RPLC. The loaded variants are separated in HIC and are used to define DAR.
HIC process involves the injection of sample into a buffer solution maintained at isoelectric point and then subsequently eluted with mobile phase from low to high concentration. Along with buffer and mobile phase, a separate organic modifier Usually isopropyl alcohol or acetonitrile is used in concentration of 5–15% to improves peak shape, resolution, and separation (Neupane and Bergquist 2017; Matsuda and Mendelsohn 2021).
HIC is a method of choice for determination of ADR and drug load distribution in ADCs manufactured by site-specific conjugation as the heterogenicity is low compared to the random conjugation. The limitation of HIC includes its inability to resolve positional isomers of ADCs. Further, owing to use of large concentration of salt, the HIC is not compatible with MS analysis (Wagh et al. 2018).
Reversed phase liquid chromatography (RPLC)
It is the most efficient technique which involves analysis of protein in ADC and helps to determine heterogeneity of conjugation site. Along with that, it performs peptide mapping and determines positional isomerism. It is the technique to determine free drug residues. It also performs the determination of DAR by complete separation of light and heavy chain fragments and shows percentage peak area for calculation of average DAR. Quantification of ADC can be done if it is hyphenated with mass spectrometer (Neupane and Bergquist 2017). The usual methodology for RPLC is pre-treatment with DL-dithiothreitol which break the covalent bond between the chains. Trifluoro acetic acid is usually used as a component of mobile phase to improve peak separation (Matsuda and Mendelsohn 2021). Chen et al. has reported MS compatible native reversed phase liquid chromatography as an added advantage to HIC for separation of a model ADC (Ab095-PZ) and brentuximab vedotin. The separation was achieved using 50mM ammonium acetate and water/isopropanol gradient for MS condition. Using the said method, positional isomers of the ADCs are well separated (Chen et al. 2019).
Size exclusion chromatography (SEC)
This technique is used to determine aggregation among ADCs. It occurs because of increased hydrophobicity due to attached payloads. The aggregates of ADC are usually found during synthesis and storage. Such aggregated product, upon administration, can provoke immune reaction and alter pharmacokinetic response. SEC uses a matrix along with elution medium to study the aggregation effect. An optimum pH is required to reduce the chance of tailing of the peak (Neupane and Bergquist 2017).
Size exclusion chromatography is a traditional technique which separates proteins based on differences in hydrodynamic volumes. It usually separates the proteins in 3 components. Recent advancement in the SEC are ultra-high performance size exclusion chromatography and SEC-MS (Bobály et al. 2018; Jones et al. 2020). Goyon et al. applied SEC for analysis of 30 different therapeutic proteins having molecular weights between 54 and 153 KDa. They also studied the feasibility of hyphenation of SEC with MS (Goyon et al. 2017).
Multidimensional chromatography
Multidimensional chromatography is also known as 2D chromatography that can be used for drug distribution evaluation, DAR, aggregation analysis, measurement of free small molecule drug content and stability studies. The limitations of existing chromatographic methods can be overcome using multidimensional chromatography. In this technique, usually, HIC is kept as the first dimension followed by RPLC which can be considered as second dimension. This approach was successfully applied for estimation of brentuximab vedotin (Bobály et al. 2018).
Mass spectrometry (MS)
MS is a quantification technique used for characterization of large mAbs. It has been adopted over UV-Visible spectroscopy due to the observation of photochemical degradation of loaded drug as in calichaemian linked ADC. MS needs ionization of antibody for spectral analysis. ADC has hydrophobic nature and its ionization can be possible when separated from the drug, however, resolution is not achieved against unconjugated antibody. Hence use of isotopic drug conjugated to mAb for their specific evaluation is required. Mass spectrometry is usually used in adjacent to liquid chromatography for providing proper DAR and payload distribution. In addition, MS can be used to identify the drug conjugation site through peptide mapping as well as to identify unconjugated antibody and payload metabolite in order to estimate its in vivo stability (Huang and Chen 2016).
Apart from the conventional information, LC-MS can provide the novel information about the adduct formation which affect DAR. In near future, LC-MS would be applied to measure ADR In-Vivo, charge heterogenicity and positional isomers (Zhu et al. 2020).
Bio analytical techniques
ELISA is one of the techniques to quantify the ADC using anti-drug antibody which gives result on the amount of drug conjugated to antibody. Such assay is usually performed to access pharmacokinetic and pharmacodynamics data for determining its efficacy and safety (Alley and Anderson 2013). Hybrid-Ligand Binding Immuno-Affinity capture followed by liquid chromatography coupled to mass spectrometry is considered as the gold standard owing to the strength of both the techniques in ADC bioanalysis. For quantification of ADCs, usually bottom-up strategy is used. Another method used for ADC bioanalysis is Nano-surface and Molecular Orientation Limited proteolysis. The advantage of this technique is that, it can quantify antibody drug in coexistence with antidrug antibodies. Further, different radiolabelling methods like non-invasive molecular imaging, Ex-Vivo cut and count technique, Dual radiolabelling can also be used for bioanalysis of ADCs (Cahuzac and Devel 2020).
Challenges and future opportunities
The development of ADCs has revolutionized the targeted cancer therapy (Aretin 2022) and has shown better results in improving the quality of life as well as overall lifespan and disease free survival of cancer patients. Looking at the current success rate of ADCs, many efforts are concentrated towards development of next generation ADCs (Mckertish and Kayser 2021; Menon et al. 2022). ADCs are like ray of hope for cancer patients and can surely improve the treatment outcome by reducing the side effects and prolonging their lifespan. Despite of all these, ADCs have their own limitations and challenges of structural design and clinical development (Dott et al. 2018; Lyon 2018; Dean et al. 2021).
Emergence of ADC resistance has started creating limitations and issues in the cancer treatment (Garcia-Alonso et al. 2018; Pander et al. 2022). Some of the cancer cells remove the portion of the cell surface antigen or cover the antigen site with some shields like mucin or hyaluronam (Lopus 2022) so that mAb of ADC can’t bind with it and tumor cells develop resistance. Many other major causes behind resistance against ADCs include downregulation of the antigen; alteration in the cancer cell membrane trafficking pathways; upregulation of drug-efflux pumps; mutations in the payload binding targets; dysregulation of apoptosis through overexpression of antiapoptotic molecules such as BCL-2; alteration of signaling pathways etc. (Loganzo et al. 2016). For example, resistance of transtuzumab emastine in HER2 positive cell line of both breast and gastric cancer has been reported and investigations showed downregulation of HER2 protein in these types of cells followed by upregulation of drug efflux pump which have limited their applications (Barok et al. 2014).
Resistance in ADC are usually observed with single warhead. To overcome this,
-
(a)
Dual drug delivery using ADC has become possible using hetero functional linker which provide site specificity and multidrug loading. Such linker involves N-aryl maleimide to provide flexibility to attach two different drugs (Kumar et al. 2018). By combining two distinct mechanisms through dual payload ADCs, it is possible to combat the resistance (Yamazaki et al. 2021).
-
(b)
Novel payloads are also being explored which will work by novel mechanism of action other than that of classical drugs. For example, oligonucliotides, short DNA or RNA fragments, which exerts cytotoxic effect by either inhibiting DNA transcription or RNA translation. Also, novel payloads that interact with immune system can also be explored (Beusker 2022). Other novel class of payload is bifunctional degraders. Pillow, T.H. et al. have discovered novel extremely potent (picomolar range) chimeric BET degrader, GNE-987 and designed the first degrader-antibody conjugate by attaching it to an anti-CLL1 antibody via a novel linker. The ADC exhibited sustained in vivo exposures on single I.V. dose and resulted in antigen-specific tumor regressions (Pillow et al. 2019).
-
(c)
Efforts are also going on to develop the ADCs that will work through novel promising targets/antigen sites of tumor. The in-silico approaches are being tried to identify the novel and promising antigens using RNA-sequencing and protein-expression data (Schettini et al. 2021). Some of the examples of such promising, novel antigenic sites identified by mAbs are; carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), HER3, Mesothelin, cMet, Folate receptor alpha (FRα), Tissue factor (TF) etc. (Criscitiello et al. 2021). Although Ganglioside GD2 is the established target in cancer immunotherapy, no any anti-GD2 ADC is in the market yet. Very recently in 2022, Kalinovsky D. V. et al. have published study on developing novel clinically relevant anti-GD2 ADCs and investigated its effect in-vitro on a wide panel of GD2-positive and GD2-negative tumor cell lines as well as in-vivo in GD2-positive solid syngeneic mouse models of B78-D14 melanoma and EL-4 lymphoma. The preliminary in-vivo study results indicated the potential for the human study. At the same time, it could not achieve complete tumor regression in treated mice because of its limited stability in plasma and immunogenicity. Overall, study suggested the future potential of anti-GD2 ADCs for the treatment of GD2-expressing tumors (Kalinovsky et al. 2022).
-
(d)
Several preclinical and early clinical studies suggested that, combination of ADCs with immunotherapy can also overcome the occurrence of resistance through additional interaction with immune cells and induction of tumor-specific adaptive immunity. Together they can increase the overall patient outcomes but the detailed studies on optimal dose of each, mechanism of synergistic action, risk benefit analysis etc. are yet to establish (Nicolò et al. 2022).
Another big challenge during the clinical development of ADCs is narrow therapeutic index due to many pharmacokinetic considerations. Many ADCs produce systemic toxicity due to uncoupling of linker or/and payload as observed in pyrrolobenzodiazepine (PBD) deconjugated with disulfide linker resulting in neurotoxicity (Nauseef et al. 2021). According to the reports, mild to moderately severe neutropenia, alopecia and GI side effects have been observed during the clinical trials of marketed ADCs like Trodelvy, Padcev etc. (Bardia et al. 2019; Powles et al. 2021). The innovations to design next generation ADCs are focussing on various aspects like improving individual ADC component, finding more stable linker technology in systemic circulation, finding new conjugation or targeting approaches, loading higher number of drugs, exploring novel ADC targets etc. Structural modification of linker is the most convenient approach as it governs the release of payload to the cancer cell. Stability, pharmacokinetics, DAR and toxicity of ADCs are directly or indirectly related to it. For an optimised ADC, physicochemical property of linker needs to be maintained. Hydrophilicity of linker is commonly managed through two methods; PEG or sulfonate moiety incorporated linker and having charged group within linker. Very recently in 2022, Zacharias, N et al. published a study of developing efficacious and homogeneous THIOMAB ADC by merging of the XTEN polypeptide scaffold with cysteine-engineered THIOMAB antibodies. The hydrophilic XTEN polypeptide was used as an alternative to PEG and resulted ADC exhibited DAR of up to 18 and increased half-life (Zacharias et al. 2022).
Efficient internalization of ADC into tumor cell and then into lysosome is also one of the major concerns of ADCs which decides the efficacy. Many protein and carbohydrate structures present on tumor cell surface may create the issues of tumor permeability. Another limitation as observed in HER2 targeted ADCs is the recycling of ADC, after internalization, back to the plasma membrane and so it cannot reach to lysosome. ADCs with bispecific antibody is the next generation technology to overcome these limitations where one binding arm domain provides tumor antigen specificity while other targets an existing fast internalising receptor to facilitate the lysosomal entry into tumor cell. Bispecific ADC can target two different tumor antigens or two different epitopes on the same antigen simultaneously and increases the efficacy as well as safety through efficient payload delivery. For example, bispecific ADCs directed against EGFR and cMET (rapidly internalises and efficiently reaches lysosomes where it is degraded) showed efficacious and more selective killing of cancer cells which overexpressed both EGFR and c-MET with reduced toxicity in only EGFR expressing normal cells (Lee et al. 2016; Maruani 2018). Another emerging technology to overcome the poor tissue penetration problem is the use of miniaturized antibodies or nanobodies (functional heavy-chain-only antibodies like caplacizumab, ozoralizumab, and vobarilizumab) to construct ADCs. In contrast to conventional mAbs having around 150 kDa molecular mass, nanobodies have nanoscale dimensions of around 3nM and molecular mass around 95 kDa (Jovčevska and Muyldermans 2020; Jin et al. 2022). Because of large difference in the molecular size and mass, nanobodies can easily diffuse across cell membrane and penetrate the tissue in much higher concentration and can be effectively used to target even brain tumors. Ian Nessler et al. has used series of prostate-specific membrane antigen–binding single domain ADC constructs and proved that constructs containing single-domain antibody and with lower in-vitro potency might resulted in higher in-vivo efficacy than other protein–drug conjugates. The only limitation of nanobody is rapid renal clearance which can be solved by fusion with albumin binding domain without compromising tissue penetration (Nessler et al. 2020).
Peptide‒drug conjugates (PDCs) are the emerging next generation targeted therapy with the core advantage of superior cellular permeability and selectivity as well as homogeneity over ADCs. In PDCs, peptides replace the antibodies and thereby overall molecular weight of tri-component system got reduced which enhances tumor cell permeability and penetration. Two PDCs, Lu-dotatate (lutathera) and melflufen, are currently approved by USFDA for the cancer treatment and many more are under investigation. Although, there is a long way to go to prove the clinical benefits of PDCs, it has given a new impulse to field of oncology (Hoppenz et al. 2020; Fu et al. 2022a).
Similar to PDCs, other novel and well established conjugation technologies are radionuclide drug conjugates (RDC) (Chatal et al. 2016) small molecule-drug conjugates (SMDC) (Patel et al. 2021) and immune-stimulating antibody conjugate (ISAC) (Ackerman et al. 2021; Mallet et al. 2021), whose related products are either in market or in the late stages of clinical development. Some other emerging technologies like antibody-oligonucleotide conjugates (AOC) (Dovgan et al. 2019; Dugal-Tessier et al. 2021), aptamer drug conjugates (ApDC) (Kim et al. 2021; Liu et al. 2022), virus-like drug conjugates (VDC) (Huisin’t Veld et al. 2022; Savinainen et al. 2022), antibody degraducer conjugates (ADeC) (BiopharmPEG; Debiopharm) etc. are either at conceptual or pre-clinical stage of development to evolve into more precisely targeted, safer and improved therapeutic approach for cancer treatment.
New delivery system needs to be searched to resolve problem of poor percolation of ADC into solid tumor. Antibody conjugated nanoparticles (ACNPs) are the emerging technology for targeted delivery of the encapsulated cargo payload precisely (Johnston and Scott 2018). Depending upon the solubility or lipophilicity of the cargo drug, nanoparticle carrier, ranges from liposomes to dendrimers, is chosen. In contrast to conventional ADCs, linker is required to conjugate antibody with polymer/lipid in ACNPs and release of drug is independent of this conjugation which avoids the decrease in payload potency till it reaches the target site. This technology also accommodates broader range of payloads and can offer high DAR around 100 which ensures internalization of much higher concentration of drug. Thus, even the payload with lower potency can also be used such as Camptothecin derivatives. Using ACNP, combination of payloads can also be explored. When targeting receptor agonism, ACNPs may also offer additional therapeutic value over ADCs. Currently, no any ACNPs based ADC is in market. It requires lot of careful control over nanoparticle size and shape as well as surface charge. Also, developing and manufacturing ACNPs is very challenging (Juan et al. 2020).
Due to the market availability of multiple ADCs targeting the same antigen and ADCs having same payload, the new emerging challenge is to choose the appropriate ADC or to optimize their sequencing during the treatment phase. Very less evidential data is available on this. However, as per some of the clinical studies data, sequential use of different ADCs targeting the same antigen (Modi et al. 2019) or sequential use of different ADCs with same payload (Tarantino et al. 2020) is effective. More research trials are needed in this area.
Out of three structural components of ADC, biological entity mAb and chemical small molecule entity payload are mainly responsible for the targeted drug delivery and action. Both types of entities have different physico-chemical properties and hence different analytical methods are required for qualitative and quantitative analysis of each. Apart from this, conjugated linker alters the chemical and/or physical properties of payload. Overall, the ADCs are predominantly heterogeneous analytes and present a big hurdle in their own development as it is very difficult to simultaneously analyse biological and chemical materials. Hence, hyphenated techniques and approaches are required to be used for the analysis of ADCs. ADC analyses are indeed very challenging and require lot of further advancements (Beck et al. 2019). These techniques are costlier than the conventional techniques (Källsten 2020). The cost of analysis of ADCs at the development/manufacturing stage, passes on to the patient, which ultimately increases the overall cost of treatment.
Conclusions
ADCs are one of the most promising targeted cancer therapies which show tremendous treatment potential with minimal toxicity. USFDA has approved ADCs for varieties of liquid and solid tumors. Due to the last decade continuous efforts in the growing field of ADC discovery and development, we are witnessing remarkable improvement in their design and engineering through selection of better and novel payloads, promising linker chemistry and conjugation techniques. Currently, more than 100 different ADCs are at different stages of clinical trials for the treatment of either solid tumors or blood malignancies. Many ADCs are being tested either as monotherapy or in combination with differently targeted therapy like check point inhibitors or with immunotherapies. Despite its success, some of the major challenges like low payload potency, optimization of DAR, poor blood residency period, stable linker technology, emergence of resistance etc. are still raising major concerns which has opened up the door to explore various opportunities for the design of next generation ADCs. Further research needs to address the main concerns of stability of linker and conjugation site to reduce the chances of pre-breakdown of linker during systemic administration. Many emerging technologies like bispecific ADCs, nanobodies, ACNPs, PDCs, RDCs, SMDC, ISAC, AOC, ApDC, VDC etc. hold a profound potential to bring a paradigm shift in cancer treatment. Hopefully with the future advancement of research, ADCs may closely achieve Ehrlich’s vision of magic bullets in clinical practice.
Change history
26 June 2024
A Correction to this paper has been published: https://doi.org/10.1007/s12272-024-01502-4
References
Abdollahpour-Alitappeh M, Lotfinia M, Gharibi T, Mardaneh J, Farhadihosseinabadi B, Larki P, Faghfourian B, Sepehr KS, Abbaszadeh-Goudarzi K, Abbaszadeh-Goudarzi G, Johari B, Zali MR, Bagheri N (2019) Antibody-drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol 234:5628–5642. https://doi.org/10.1002/jcp.27419
Ackerman S, Pearson C, Gregorio J, Gonzalez J, Kenkel J, Hartmann F, Luo A, Ho P, LeBlanc H, Blum L, Kimmey S, Luo A, Nguyen M, Paik J, Sheu L, Ackerman B, Lee A, Li H, Melrose J, Laura R, Ramani V, Henning K, Jackson D, Safina B, Yonehiro G, Devens B, Carmi Y, Chapin S, Bendall S, Kowanetz M, Dornan D, Engleman E, Alonso M (2021) Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat Cancer 2:18–33. https://doi.org/10.1038/s43018-020-00136-x
Adair JR, Howard PW, Hartley JA, Williams DG, Chester KA (2012) Antibody–drug conjugates—a perfect synergy. Expert Opin Biol Ther 12:1191–1206. https://doi.org/10.1517/14712598.2012.693473
Adumeau P, Sharma SK, Brent C, Zeglis B (2016) Site-specifically labeled immunoconjugates for molecular imaging—part 1: cysteine residues and glycans. Mol Imaging Biol 18:1–17. https://doi.org/10.1007/s11307-015-0919-4
Agarwal P, Bertozzi CR (2015) Site-specific antibody–drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem 26:176–192. https://doi.org/10.1021/bc5004982
Albin N, Massaad L, Toussaint C, Mathieu M-C, Morizet J, Parise O, Gouyette A, Chabot GG (1993) Main drug-metabolizing enzyme systems in human breast tumors and peritumoral tissues. Cancer Res 53:3541–3546
Alley SC, Anderson KE (2013) Analytical and bioanalytical technologies for characterizing antibody-drug conjugates. Curr Opin Chem Biol 17:406–411
Aretin M-B (2022) Antibody–drug conjugates—the magic bullet? Memo-magazine. Eur Med Oncol 15:125–128. https://doi.org/10.1007/s12254-021-00780-8
Bardia A, Mayer I, Vahdat L, Tolaney S, Isakoff S, Diamond J, O’Shaughnessy J, Moroose R, Santin A, Abramson V, Shah N, Rugo H, Goldenberg D, Sweidan A, Iannone R, Washkowitz S, Sharkey R, Wegener W, Kalinsky K (2019) Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med 380:741–751. https://doi.org/10.1056/NEJMoa1814213
Bargh JD, Isidro-Llobet A, Parker JS, Spring DR (2019) Cleavable linkers in antibody–drug conjugates. Chem Soc Rev 48:4361–4374. https://doi.org/10.1039/C8CS00676H
Barok M, Joensuu H, Isola J (2014) Trastuzumab emtansine: mechanisms of action and drug resistance. Breast cancer Res 16:1–12. https://doi.org/10.1186/bcr3621
Beck A, D’atri V, Ehkirch A, Fekete S, Hernandez-Alba O, Gahoual R, Leize-Wagner E, Francois Y, Guillarme D, Cianférani S (2019) Cutting-edge multi-level analytical and structural characterization of antibody-drug conjugates: present and future. Expert Rev Proteomics 16:337–362. https://doi.org/10.1080/14789450.2019.1578215
Beerli RR, Grawunder U (2017) In: Grawunder U, Barth S (eds) Enzyme-based strategies to Generate Site-Specifically conjugated antibody drug conjugates BT - Next generation antibody drug conjugates (ADCs) and immunotoxins. Springer International Publishing, Cham, pp 85–106
Beusker PF. “Magic bullets” to “Trojan-Horses”: the evolution of next-generation antibody-drug conjugates. https://www.byonids.com/who-we-are/blogs/2022-03-28. Accessed 5 Aug 2022
BiopharmPEG 9 types of drug conjugates overview : ADC, RDC, ISAC, SMDC, AOC… https://www.biochempeg.com/article/266.html. Accessed 30 Aug 2022
Bobály B, Fleury-Souverain S, Beck A, Veuthey J-L, Guillarme D, Fekete S (2018) Current possibilities of liquid chromatography for the characterization of antibody-drug conjugates. J Pharm Biomed Anal 147:493–505. https://doi.org/10.1016/j.jpba.2017.06.022
Buecheler JW, Winzer M, Tonillo J, Weber C, Gieseler H (2018) Impact of payload hydrophobicity on the stability of antibody–drug conjugates. Mol Pharm 15:2656–2664. https://doi.org/10.1021/acs.molpharmaceut.8b00177
Cahuzac H, Devel L (2020) Analytical methods for the detection and quantification of ADCs in biological matrices. Pharmaceuticals 13:462. https://doi.org/10.3390/ph13120462
Carter PJ, Senter PD (2008) Antibody-drug conjugates for cancer therapy. Cancer J 14:154–169. https://doi.org/10.1097/ppo.0b013e318172d704
Chatal J-F, Kraeber-Bodéré F, Bodet-milin C, Rousseau C (2016) Therapeutic immunoconjugates. Which cytotoxic payload: chemotherapeutic drug (ADC) or Radionuclide (ARC) ? Curr Cancer Ther Rev 12:54–65. https://doi.org/10.2174/1573394712666160805121312
Chau CH, Steeg PS, Figg WD (2019) Antibody drug conjugates for cancer. Lancet 394:793–804. https://doi.org/10.1016/S0140-6736(19)31774-X
Chen T-H, Yang Y, Zhang Z, Fu C, Zhang Q, Williams JD, Wirth MJ (2019) Native reversed-phase liquid chromatography: a technique for LCMS of intact antibody–drug conjugates. Anal Chem 91:2805–2812. https://doi.org/10.1021/acs.analchem.8b04699
Criscitiello C, Morganti S, Curigliano G (2021) Antibody–drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol 14:20. https://doi.org/10.1186/s13045-021-01035-z
Dahl J, Marx K, Jabbour E (2016) Inotuzumab ozogamicin in the treatment of acute lymphoblastic leukemia. Expert Rev Hematol 9:329–334. https://doi.org/10.1586/17474086.2016.1143771
de Graaf M, Boven E, Scheeren HW, Haisma HJ, Pinedo HM (2002) Beta-glucuronidase-mediated drug release. Curr Pharm Des 8:1391–1403
Dean AQ, Luo S, Twomey JD, Zhang B (2021) In: MAbs (ed) Targeting cancer with antibody-drug conjugates: promises and challenges. Taylor & Francis, p 1951427
Debiopharm Debiopharm and Ubix (2022) Therapeutics launch research to develop a new anti-cancer modality—antibody degraducer® conjugates. https://www.debiopharm.com/drug-development/press-releases/debiopharm-and-ubix-therapeutics-launch-research-to-develop-a-new-anti-cancer-modality-antibody-degraducer-conjugates/
Dennler P, Chiotellis, Aristeidis, Fischer E, Brégeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R (2014) Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjug Chem 25:569–578. https://doi.org/10.1021/bc400574z
Dennler P, Fischer E, Schibli R (2015) Antibody conjugates: from heterogeneous populations to defined reagents. Antibodies 4:197–224. https://doi.org/10.3390/antib4030197
Deonarain MP, Yahioglu G, Stamati I, Marklew J (2015) Emerging formats for next-generation antibody drug conjugates. Expert Opin Drug Discov 10:463–481. https://doi.org/10.1517/17460441.2015.1025049
DiJoseph JF, Popplewell A, Tickle S, Ladyman H, Lawson A, Kunz A, Khandke K, Armellino DC, Boghaert ER, Hamann PR, Zinkewich-Peotti K, Stephens S, Weir N, Damle NK (2005) Antibody-targeted chemotherapy of B-cell lymphoma using calicheamicin conjugated to murine or humanized antibody against CD22. Cancer Immunol Immunother 54:11–24. https://doi.org/10.1007/s00262-004-0572-2
Dornan D, Bennett F, Chen Y, Dennis M, Eaton D, Elkins K, French D, Go M, Jack A, Junutula J, Koeppen H, Lau J, McBride J, Rawstron A, Shi X, Yu N, Yu S, Yue P, Zheng B, Ebens A, Polson A (2009) Therapeutic potential of an anti-CD79b antibody–drug conjugate, anti–CD79b-vc-MMAE, for the treatment of non-hodgkin lymphoma. Blood 114:2721–2729. https://doi.org/10.1182/blood-2009-02-205500
Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD (2003) Erratum: development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 21:941. https://doi.org/10.1038/nbt0803-941a
Dorywalska M, Strop P, Melton-Witt J, Hasa-Moreno A, Farias S, Galindo Casas M, Delaria K, Lui V, Poulsen K, Sutton J, Bolton G, Zhou D, Moine L, Dushin R, Tran T, Liu S, Rickert M, Foletti D, Shelton D, Rajpal A (2015) Site-dependent degradation of a non-cleavable auristatin-based linker-payload in Rodent plasma and its effect on ADC efficacy. PLoS ONE 10:e0132282. https://doi.org/10.1371/journal.pone.0132282
Dott J, Abila B, Wuerthner JU (2018) Current trends in the clinical development of antibody-drug conjugates in oncology. Pharma Med 32:259–273. https://doi.org/10.1007/s40290-018-0238-6
Dovgan I, Koniev O, Kolodych S, Wagner A (2019) Antibody–Oligonucleotide Conjugates as Therapeutic, Imaging, and detection agents. Bioconjug Chem 30:2483–2501. https://doi.org/10.1021/acs.bioconjchem.9b00306
Drenkard D, Becke F, Langstein J, Spruss T, Kunz-Schughart L, Tan T, Lim Y, Schwarz H (2007) CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J 21:456–463. https://doi.org/10.1096/fj.05-4739com
Dugal-Tessier J, Barnscher S, Kanai A, Mendelsohn B (2017) Synthesis and evaluation of Dolastatin 10 Analogues containing heteroatoms on the amino acid side chains. J Nat Prod 80:2484–2491. https://doi.org/10.1021/acs.jnatprod.7b00359
Dugal-Tessier J, Thirumalairajan S, Jain N (2021) Antibody-oligonucleotide conjugates: a twist to antibody-drug conjugates. J Clin Med 10:838. https://doi.org/10.3390/jcm10040838
Falck G, Müller KM (2018) Enzyme-based labeling strategies for antibody–drug conjugates and antibody mimetics. Antibodies 7:4. https://doi.org/10.3390/antib7010004
Fang Q, Kanugula S, Pegg AE (2005) Function of domains of human O6-Alkylguanine-DNA alkyltransferase. Biochemistry 44:15396–15405. https://doi.org/10.1021/bi051460d
Fatima SW, Khare SK (2022) Benefits and challenges of antibody drug conjugates as novel form of chemotherapy. J Control Release 341:555–565. https://doi.org/10.1016/j.jconrel.2021.12.013
Feng Y, Zhu Z, Chen W, Prabakaran P, Lin K, Dimitrov DS (2014) Conjugates of small molecule drugs with antibodies and other proteins. Biomedicines 2:1–13
Fierer JO, Veggiani G, Howarth M (2014) SpyLigase peptide–peptide ligation polymerizes affibodies to enhance magnetic cancer cell capture. Proc Natl Acad Sci 111:E1176–E1181. https://doi.org/10.1073/pnas.1315776111
Firer MA, Gellerman G (2012) Targeted drug delivery for cancer therapy: the other side of antibodies. J Hematol Oncol 5:70. https://doi.org/10.1186/1756-8722-5-70
Fu C, Yu L, Miao Y, Liu X, Yu Z, Wei M (2022a) Peptide–drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm Sin B In Press. https://doi.org/10.1016/j.apsb.2022.07.020
Fu Z, Li S, Han S, Shi C, Zhang Y (2022b) Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther 7:1–25. https://doi.org/10.1038/s41392-022-00947-7
Fujii T, Matsuda Y, Seki T, Shikida N, Iwai Y, Ooba Y, Takahashi K, Isokawa M, Kawaguchi S, Hatada N (2023) AJICAP second generation: improved chemical site-specific conjugation technology for antibody-drug conjugate production
Garbaccio RM (2014) Chemistry of antibody–small molecule drug conjugates. In: Chemical linkers in antibody-drug conjugates (ADCs). Elsevier
Garcia-Alonso S, Ocana A, Pandiella A (2018) Resistance to antibody–drug conjugates. Cancer Res 78:2159–2165. https://doi.org/10.1158/0008-5472.CAN-17-3671
Gauzy-Lazo L, Sassoon I, Brun M-P (2020) Advances in antibody–drug conjugate design: current clinical landscape and future innovations. SLAS Discov Adv Sci Drug Discov 25:843–868. https://doi.org/10.1177/2472555220912955
Gaya AM, Rustin GJS (2005) Vascular disrupting agents: a new class of drug in cancer therapy. Clin Oncol 17:277–290. https://doi.org/10.1016/j.clon.2004.11.011
Gerber H-P, Koehn F, Abraham R (2013) The antibody-drug conjugate: an enabling modality for natural product-based cancer therapeutics. Nat Prod Rep 30:625–639. https://doi.org/10.1039/c3np20113a
Gerlach M, Stoschek T, Leonhardt H, Hackenberger CPR, Schumacher D, Helma J (2019) Tubulin tyrosine ligase-mediated modification of proteins. Methods Mol Biol 2012:327–355. https://doi.org/10.1007/978-1-4939-9546-2_17
Gianolio DA, Rouleau C, Bauta WE, Lovett D, Cantrell WR, Recio A, Wolstenholme-Hogg P, Busch M, Pan P, Stefano JE, Kramer HM, Goebel J, Krumbholz RD, Roth S, Schmid SM, Teicher BA (2012) Targeting HER2-positive cancer with dolastatin 15 derivatives conjugated to trastuzumab, novel antibody–drug conjugates. Cancer Chemother Pharmacol 70:439–449. https://doi.org/10.1007/s00280-012-1925-8
Goldenberg D, Sharkey R (2019) Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: a case study of anti-TROP-2 sacituzumab govitecan. MAbs 11:987–995. https://doi.org/10.1080/19420862.2019.1632115
Goldmacher VS, Kovtun YV (2011) Antibody–drug conjugates: using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther Deliv 2:397–416. https://doi.org/10.4155/tde.10.98
Goli N, Bolla PK, Talla V (2018) Antibody-drug conjugates (ADCs): potent biopharmaceuticals to target solid and hematological cancers- an overview. J Drug Deliv Sci Technol 48:106–117. https://doi.org/10.1016/j.jddst.2018.08.022
Goundry WRF, Parker JS (2022) Payloads for antibody–drug conjugates. Org Process Res Dev 26:2121–2123
Govindan SV, Sharkey RM, Goldenberg DM (2016) Prospects and progress of antibody-drug conjugates in solid tumor therapies. Expert Opin Biol Ther 16:883–893. https://doi.org/10.1517/14712598.2016.1173203
Govindan SV, Cardillo TM, Goldenberg DM (2019) Topoisomerase inhibitors as antibody–drug conjugate (ADC) payloads. In: Thurston DE, Jackson P (ed) Cytotoxic payloads for antibody–drug conjugates, p 166–186
Goyon A, D’Atri V, Colas O, Fekete S, Beck A, Guillarme D (2017) Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: feasibility assessment for future mass spectrometry hyphenation. J Chromatogr B 1065:35–43. https://doi.org/10.1016/j.jchromb.2017.09.027
Griebenow N, Dilmaç AM, Greven S, Bräse S (2016) Site-specific conjugation of peptides and proteins via rebridging of Disulfide Bonds using the thiol–yne coupling reaction. Bioconjug Chem 27:911–917. https://doi.org/10.1021/acs.bioconjchem.5b00682
Grünewald J, Brock A, Geierstanger BH (2019). In: Nuijens T, Schmidt M (eds) Site-specific antibody labeling using Phosphopantetheinyl transferase-catalyzed ligation BT—enzyme-mediated ligation methods. Springer, New York, pp 237–278
Hageman MJ, Morozowich W (2007). In: Stella VJ, Borchardt RT, Hageman MJ, Oliyai R, Maag H, Tilley JW (eds) Case study: irinotecan (CPT-11), a water-soluble prodrug of SN-38 BT—prodrugs: challenges and rewards part 1. Springer, New York, pp 1269–1279
Haque M, Forte N, Baker JR (2021) Site-selective lysine conjugation methods and applications towards antibody–drug conjugates. Chem Commun 57:10689–10702. https://doi.org/10.1039/D1CC03976H
Harris L, Tavares D, Rui L, Maloney E, Wilhelm A, Costoplus J, Archer K, Bogalhas M, Harvey L, Wu R, Chen X, Xu X, Connaughton S, Wang L, Whiteman K, Ab O, Hong E, Widdison W, Shizuka M, Miller M, Pinkas J, Keating T, Chari R, Fishkin N (2015) SeriMabs: N-terminal serine modification enables modular, site-specific payload incorporation into antibody-drug conjugates (ADCs). Cancer Res 75:647. https://doi.org/10.1158/1538-7445.AM2015-647
Hartley JA (2021) Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin Biol Ther 21:931–943. https://doi.org/10.1080/14712598.2020.1776255
Havaei S, Aucoin M, Jahanian-Najafabadi A (2021) Pseudomonas Exotoxin-Based immunotoxins: over three decades of efforts on Targeting Cancer cells with the Toxin. Front Oncol 11:781800–781817. https://doi.org/10.3389/fonc.2021.781800
Hoppenz P, Els-Heindl S, Beck-Sickinger AG (2020) Peptide-drug conjugates and their targets in Advanced Cancer Therapies. Front Chem 8:571. https://doi.org/10.3389/fchem.2020.00571
Huang RYC, Chen G (2016) Characterization of antibody-drug conjugates by mass spectrometry: advances and future trends. Drug Discov Today 21:850–855
Huisin’t Veld RV, Ma S, Kines R, Savinainen A, Rich CC, Ossendorp F, Jager M (2022) A novel virus-like drug conjugate (VDC) in combination with immune checkpoint inhibitors for the treatment of primary tumors and distant metastasis. J Clin Oncol 40:e14544–e14544. https://doi.org/10.1200/JCO.2022.40.16_suppl.e14544
Hussain AF, Grimm A, Sheng W, Zhang C, Al-Rawe M, Bräutigam K, Abu Mraheil M, Zeppernick F, Meinhold-Heerlein I (2021) Toward homogenous antibody drug conjugates using enzyme-based conjugation approaches. Pharmaceuticals 14:343. https://doi.org/10.3390/ph14040343
Jain N, Smith SW, Ghone S, Tomczuk B (2015) Current ADC linker chemistry. Pharm Res 32:3526–3540. https://doi.org/10.1007/s11095-015-1657-7
Jeffrey SC, De Brabander J, Miyamoto J, Senter PD (2010) Expanded utility of the β-Glucuronide linker: ADCs that deliver phenolic cytotoxic agents. ACS Med Chem Lett 1:277–280. https://doi.org/10.1021/ml100039h
Jin Y, Edalatian Zakeri S, Bahal R, Wiemer AJ (2022) New Technologies Bloom together for bettering Cancer Drug Conjugates. Pharmacol Rev 74:680–711. https://doi.org/10.1124/pharmrev.121.000499
Johnston MC, Scott CJ (2018) Antibody conjugated nanoparticles as a novel form of antibody drug conjugate chemotherapy. Drug Discov Today Technol 30:63–69. https://doi.org/10.1016/j.ddtec.2018.10.003
Jones J, Pack L, Hunter JH, Valliere-Douglass JF (2020). In: MAbs (ed) Native size-exclusion chromatography-mass spectrometry: suitability for antibody–drug conjugate drug-to-antibody ratio quantitation across a range of chemotypes and drug-loading levels. Taylor & Francis, p 1682895
Joubert N, Beck A, Dumontet C, Denevault-Sabourin C (2020) Antibody–drug conjugates: the last decade. Pharmaceuticals 13:245. https://doi.org/10.3390/ph13090245
Jovčevska I, Muyldermans S (2020) The therapeutic potential of nanobodies. BioDrugs 34:11–26. https://doi.org/10.1007/s40259-019-00392-z
Juan A, Cimas FJ, Bravo I, Pandiella A, Ocaña A, Alonso-Moreno C (2020) Antibody conjugation of nanoparticles as therapeutics for breast cancer treatment. Int J Mol Sci 21:6018. https://doi.org/10.3390/ijms21176018
Junutula J, Raab H, Clark S, Bhakta S, Leipold D, Weir S, Chen Y, Simpson M, Tsai S, Dennis M, Lu Y, Meng Y, Ng C, Yang J, Lee C, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer S, Lee W, Lowman H, Vandlen R, Sliwkowski M, Scheller R, Polakis P, Mallet W (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26:925–932. https://doi.org/10.1038/nbt.1480
Kalinovsky DV, Kibardin AV, Kholodenko IV, Svirshchevskaya EV, Doronin II, Konovalova MV, Grechikhina MV, Rozov FN, Larin SS, Deyev SM (2022) Therapeutic efficacy of antibody-drug conjugates targeting GD2-positive tumors. J Immunother cancer 10:e004646. https://doi.org/10.1136/jitc-2022-004646
Källsten M (2020) Development and evaluation of analytical techniques for antibodies and antibody-drug conjugates: From verification of conjugation to stability testing. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1901, Acta Universitatis Upsaliensis
Kang MS, Kong TWS, Khoo JYX, Loh T-P (2021) Recent developments in chemical conjugation strategies targeting native amino acids in proteins and their applications in antibody–drug conjugates. Chem Sci 12:13613–13647. https://doi.org/10.1039/D1SC02973H
Kern JC, Cancilla M, Dooney D, Kwasnjuk K, Zhang R, Beaumont M, Figueroa I, Hsieh S, Liang L, Tomazela D (2016) Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J Am Chem Soc 138:1430–1445. https://doi.org/10.1021/jacs.5b12547
Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J (2020) Antibody-drug conjugates: a comprehensive review. Mol Cancer Res 18:3–19. https://doi.org/10.1158/1541-7786.mcr-19-0582
Kim D-H, Seo J-M, Shin K-J, Yang S-G (2021) Design and clinical developments of aptamer-drug conjugates for targeted cancer therapy. Biomater Res 25:42. https://doi.org/10.1186/s40824-021-00244-4
Ko MJ, Song D, Kim J, Kim JY, Eom J, Sung B, Son Y-G, Kim YM, Lee SH, You W-K, Jung J (2021) N-terminal selective conjugation method widens the therapeutic window of antibody–drug conjugates by improving tolerability and stability. MAbs 13:1914885. https://doi.org/10.1080/19420862.2021.1914885
Kostova V, Désos P, Starck J-B, Kotschy A (2021) The chemistry behind ADCs. Pharmaceuticals 14:442. https://doi.org/10.3390/ph14050442
Kovtun YV, Goldmacher VS (2007) Cell killing by antibody–drug conjugates. Cancer Lett 255:232–240. https://doi.org/10.1016/j.canlet.2007.04.010
Krüger T, Dierks T, Sewald N (2019) Formylglycine-generating enzymes for site-specific bioconjugation. Biol Chem 400:289–297. https://doi.org/10.1515/hsz-2018-0358
Kulkarni PN, Blair AH, Ghose T, Mammen M (1985) Conjugation of methotrexate to IgG antibodies and their F(ab)2 fragments and the effect of conjugated methotrexate on tumor growth in vivo. Cancer Immunol Immunother 19:211–214. https://doi.org/10.1007/BF00199228
Kumar A, Kinneer K, Masterson L, Ezeadi E, Howard P, Wu H, Gao C, Dimasi N (2018) Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg Med Chem Lett 28:3617–3621. https://doi.org/10.1016/j.bmcl.2018.10.043
Lee A (2021) Loncastuximab tesirine: first approval. Drugs 81:1229–1233. https://doi.org/10.1007/s40265-021-01550-w
Lee JM, Lee SH, Hwang J-W, Oh SJ, Kim B, Jung S, Shim S, Lin PW, Lee SB, Cho M-Y, Koh YJ, Kim SY, Ahn S, Lee J, Kim K, Cheong KH, Choi J, Kim K-A (2016) Novel strategy for a bispecific antibody: induction of dual target internalization and degradation. Oncogene 35:4437–4446. https://doi.org/10.1038/onc.2015.514
Li X, Fang T, Boons G-J (2014) Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew Chem Int Ed Engl. https://doi.org/10.1002/anie.201402606
Liu P, Ga L, Aodeng G, Wang Y, Ai J (2022) Aptamer-drug conjugates: New probes for imaging and targeted therapy. Biosens Bioelectron X 10:100126. https://doi.org/10.1016/j.biosx.2022.100126
Liu-Shin L, Fung A, Malhotra A, Ratnaswamy G (2018). In: MAbs (ed) Influence of disulfide bond isoforms on drug conjugation sites in cysteine-linked IgG2 antibody-drug conjugates. Taylor & Francis, pp 583–595
Loganzo F, Sung M, Gerber H-P (2016) Mechanisms of resistance to antibody–drug conjugates. Mol Cancer Ther 15:2825–2834. https://doi.org/10.1158/1535-7163.MCT-16-0408
Lopus M (2022) Shooting Cancer with magic bullets. Promises and challenges of antibody-drug conjugates. Resonance 27:1127–1130. https://doi.org/10.1007/s12045-022-1409-z
Lu J, Jiang F, Lu A, Zhang G (2016) Linkers having a crucial role in antibody–drug conjugates. Int. J. Mol. Sci.17
Lyon R (2018) Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov Today Technol 30:105–109. https://doi.org/10.1016/j.ddtec.2018.10.001
Mack F, Ritchie M, Sapra P (2014) The next generation of antibody drug conjugates. Semin Oncol 41:637–652. https://doi.org/10.1053/j.seminoncol.2014.08.001
Maderna A, Doroski M, Subramanyam C, Porte A, Leverett C, Vetelino B, Cheng Z, Risley H, Parris K, Pandit J, Varghese A, Shanker S, Song C, Sukuru S, Farley K, Wagenaar M, Shapiro M, Musto S, Lam M, O’Donnell C (2014) Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J Med Chem. https://doi.org/10.1021/jm501649k
Majidi J, Barar J, Baradaran B, Abdolalizadeh J, Omidi Y (2009) Target therapy of cancer: implementation of monoclonal antibodies and nanobodies. Hum Antibodies 18:81–100. https://doi.org/10.3233/HAB-2009-0204
Mallet W, Gadkari R, Pearson C, Dulgeroff L, Luo A, Luo AA, Melrose J, Nolin J, Lee A, Zhou M, Anand P, Sarma G, Henning K, Blum L, Chapin S, Bogaert L, Ackerman S, Kudirka R, Shen Y, Dornan D (2021) 784 BDC-2034: discovery of a CEA-targeting immune-stimulating antibody conjugate (ISAC) for solid tumors. J Immunother Cancer 9:A819–A819. https://doi.org/10.1136/jitc-2021-SITC2021.784
Maruani A (2018) Bispecifics and antibody–drug conjugates: a positive synergy. Drug Discov Today Technol 30:55–61. https://doi.org/10.1016/j.ddtec.2018.09.003
Matsuda Y, Mendelsohn BA (2021) Recent advances in drug–antibody ratio determination of antibody–drug conjugates. Chem Pharm Bull 69:976–983. https://doi.org/10.1248/cpb.c21-00258
Mazor R, Onda M, Pastan I (2016) Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev 270:152–164. https://doi.org/10.1111/imr.12390
McCombs JR, Owen SC (2015) Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J 17:339–351. https://doi.org/10.1208/s12248-014-9710-8
Mcdonagh C, Turcott E, Westendorf L, Webster J, Alley S, Kim K, Andreyka J, Stone I, Hamblett K, Francisco J, Carter P (2006) Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel 19:299–307. https://doi.org/10.1093/protein/gzl013
Mckertish CM, Kayser V (2021) Advances and limitations of antibody drug conjugates for cancer. Biomedicines 9:872. https://doi.org/10.3390/biomedicines9080872
Mendelsohn BA, Barnscher SD, Snyder JT, An Z, Dodd JM, Dugal-Tessier J (2017) Investigation of hydrophilic auristatin derivatives for use in antibody drug conjugates. Bioconjug Chem 28:371–381. https://doi.org/10.1021/acs.bioconjchem.6b00530
Menon S, Parakh S, Scott A, Gan H (2022) Antibody-drug conjugates: beyond current approvals and potential future strategies. Explor Target anti-tumor Ther 3:252–277. https://doi.org/10.37349/etat.2022.00082
Meyer C, Liebscher S, Bordusa F (2016) Selective coupling of click anchors to proteins via trypsiligase. Bioconjug Chem 27:47–53. https://doi.org/10.1021/acs.bioconjchem.5b00618
Modi S, Saura C, Yamashita T, Park Y, Kim S, Tamura K, Andre F, Iwata H, Ito Y, Tsurutani J, Sohn J, Denduluri N, Perrin C, Aogi K, Tokunaga E, Im S, Lee K, Hurvitz S, Cortes J, Lee C, Chen S, Zhang L, Shahidi J, Yver A, Krop I (2019) Trastuzumab Deruxtecan in previously treated HER2-Positive breast Cancer. N Engl J Med 382:610–621. https://doi.org/10.1056/NEJMoa1914510
Müller P, Martin K, Theurich S, Schreiner J, Savic S, Terszowski G, Lardinois D, Heinzelmann-Schwarz VA, Schlaak M, Kvasnicka H-M, Spagnoli G, Dirnhofer S, Speiser DE, von Bergwelt-Baildon M, Zippelius A (2014) Microtubule-depolymerizing agents used in antibody–drug conjugates induce Antitumor Immunity by Stimulation of dendritic cells. Cancer Immunol Res 2:741–755. https://doi.org/10.1158/2326-6066.CIR-13-0198
Nadkarni DV (2020) Conjugations to endogenous cysteine residues: antibody-drug conjugates. Springer, pp 37–49
Nauseef JT, Bander NH, Tagawa ST (2021) Emerging prostate-specific membrane Antigen-based therapeutics: small molecules, antibodies, and Beyond. Eur Urol Focus 7:254–257. https://doi.org/10.1016/j.euf.2021.02.006
Nessler I, Khera E, Vance S, Kopp A, Qiu Q, Keating TA, Abu-Yousif AO, Sandal T, Legg J, Thompson L, Goodwin N, Thurber GM (2020) Increased tumor penetration of single-domain antibody–drug Conjugates improves in vivo efficacy in prostate Cancer models. Cancer Res 80:1268–1278. https://doi.org/10.1158/0008-5472.CAN-19-2295
Neupane R, Bergquist J (2017) Analytical techniques for the characterization of antibody drug conjugates: Challenges and prospects. Eur J Mass Spectrom 23:417–426. https://doi.org/10.1177/1469066717733919
Nicolò E, Giugliano F, Ascione L, Tarantino P, Corti C, Tolaney SM, Cristofanilli M, Curigliano G (2022) Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat Rev 106:102395. https://doi.org/10.1016/j.ctrv.2022.102395
Pahl A, Lutz C, Hechler T (2018) Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov Today Technol. https://doi.org/10.1016/j.ddtec.2018.08.005
Pander G, Uhl P, Kühl N, Haberkorn U, Anderl J, Mier W (2022) Antibody–drug conjugates: what drives their progress? Drug Discov Today 27:103311. https://doi.org/10.1016/j.drudis.2022.06.011
Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR (2014) Site-specific antibody drug conjugates for cancer therapy. MAbs 6:34–45. https://doi.org/10.4161/mabs.27022
Patel TK, Adhikari N, Amin SA, Biswas S, Jha T, Ghosh B (2021) Small molecule drug conjugates (SMDCs): an emerging strategy for anticancer drug design and discovery. New J Chem 45:5291–5321. https://doi.org/10.1039/D0NJ04134C
PEG BioPharma (2020) The history of ADC drugs development. https://www.biochempeg.com/article/125.html. Accessed 11 Aug 2022
Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM (2014) Antibody–drug conjugates: current status and future directions. Drug Discov Today 19:869–881. https://doi.org/10.1016/j.drudis.2013.11.004
Pillow T, Adhikari P, Blake R, Chen J, Rosario G, Deshmukh G, Figueroa I, Gascoigne K, Kamath A, Kaufman S, Kleinheinz T, Kozak K, Latifi B, Leipold D, Li C, Li R, Mulvihill M, O’Donohue A, Rowntree R, Dragovich P (2019) Antibody conjugation of a chimeric BET degrader enables. Vivo Activity ChemMedChem. https://doi.org/10.1002/cmdc.201900497
Polson A, Calemine-Fenaux J, Chan P, Chang W, Christensen E, Clark S, de Sauvage F, Eaton D, Elkins K, Elliott J, Frantz G, Fuji R, Gray A, Harden K, Ingle G, Kljavin N, Koeppen H, Nelson C, Prabhu S, Raab H, Ross S, Stephan J, Scales S, Spencer S, Vandlen R, Wranik B, Yu S, Zheng B, Ebens A (2009) Antibody-drug conjugates for the treatment of non–Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res 69:2358–2364. https://doi.org/10.1158/0008-5472.CAN-08-2250
Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Durán I, Lee J-L, Matsubara N, Vulsteke C, Castellano D, Wu C, Campbell M, Matsangou M, Petrylak DP (2021) Enfortumab Vedotin in previously treated Advanced Urothelial Carcinoma. N Engl J Med 384:1125–1135. https://doi.org/10.1056/NEJMoa2035807
Qasba PK (2015) Glycans of antibodies as a specific site for drug conjugation using glycosyltransferases. Bioconjug Chem 26:2170–2175. https://doi.org/10.1021/acs.bioconjchem.5b00173
Ramesh M, Ahlawat P, Srinivas NR (2010) Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed Chromatogr 24:104–123. https://doi.org/10.1002/bmc.1345
Ricart A, Tolcher A (2007) Technology Insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Clin Pract Oncol 4:245–255. https://doi.org/10.1038/ncponc0774
Robak T, Robak E (2014) Current phase II antibody-drug conjugates for the treatment of lymphoid malignancies. Expert Opin Investig Drugs 23:911–924. https://doi.org/10.1517/13543784.2014.908184
Rosen CB, Francis MB (2017) Targeting the N terminus for site-selective protein modification. Nat Chem Biol 13:697–705. https://doi.org/10.1038/nchembio.2416
Sang H, Wan N, Lu G, Tian Y, Wang G, Ye H (2020). In: Tumey LN (ed) Conjugation site analysis of lysine-conjugated ADCs BT—antibody-drug conjugates: methods and protocols. Springer, New York, pp 235–250
Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK (2017) Advances in antibody–drug conjugates: a new era of targeted cancer therapy. Drug Discov Today 22:1547–1556
Savinainen A, Kines R, Rich CC (2022) A first in class Virus-Like Drug Conjugate (VDC) shows anti-tumor activity in cancers that commonly metastasize to the Choroid. Invest Ophthalmol Vis Sci 63:2616
Savoy EA, Olatunji FP, Yoon H, Mesbahi N, Knight JR, Berkman CE (2021) Acid-labile linkers. Chem Linkers Antibody-Drug Conjug 81:213. https://doi.org/10.1039/9781839165153
Schettini F, Barbao P, Brasó-Maristany F, Galván P, Martínez D, Paré L, De Placido S, Prat A, Guedan S (2021) Identification of cell surface targets for CAR-T cell therapies and antibody–drug conjugates in breast cancer. ESMO Open 6:100102. https://doi.org/10.1016/j.esmoop.2021.100102
Schumacher D, Hackenberger C, Leonhardt H, Helma-Smets J (2016) Current status: site-specific antibody drug conjugates. J Clin Immunol. https://doi.org/10.1007/s10875-016-0265-6
Shah B, Li M, Wypych J, Joubert MK, Zhang Z (2022) Observation of Heavy-Chain C-Terminal Amidation in Human endogenous IgG. J Pharm Sci 111:2445–2450. https://doi.org/10.1016/j.xphs.2022.06.012
Sheyi R, de la Torre BG, Albericio F (2022) Linkers: an assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 14:396. https://doi.org/10.3390/pharmaceutics14020396
Siegmund V, Piater B, Zakeri B, Eichhorn T, Fischer F, Deutsch C, Becker S, Toleikis L, Hock B, Betz UAK, Kolmar H (2016) Spontaneous isopeptide bond formation as a powerful tool for engineering site-specific antibody-drug conjugates. Sci Rep 6:39291. https://doi.org/10.1038/srep39291
Singh R, Erickson HK (2009). In: Dimitrov AS (ed) Antibody–cytotoxic agent conjugates: preparation and characterization BT—therapeutic antibodies: methods and protocols. Humana Press, Totowa, pp 445–467
Sochaj AM, Świderska KW, Otlewski J (2015) Current methods for the synthesis of homogeneous antibody–drug conjugates. Biotechnol Adv 33:775–784. https://doi.org/10.1016/j.biotechadv.2015.05.001
Sun MMC, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FGM, Ihle NC, Senter PD, Alley SC (2005) Reduction—alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 16:1282–1290. https://doi.org/10.1021/bc050201y
Sussman D, Westendorf L, Meyer D, Leiske C, Anderson M, Okeley N, Alley S, Lyon R, Sanderson R, Carter P, Benjamin D (2018) Engineered cysteine antibodies: an improved antibody-drug conjugate platform with a novel mechanism of drug-linker stability. Protein Eng Des Sel. https://doi.org/10.1093/protein/gzx067
Tan C (2015). In: Wang J, Shen W-C, Zaro JL (eds) Payloads of antibody-drug conjugates BT—antibody-drug conjugates: the 21st century magic bullets for Cancer. Springer International Publishing, Cham, pp 11–22
Tang H, Liu Y, Yu Z, Sun M, Lin L, Liu W, Han Q, Wei M, Jin Y (2019) The analysis of key factors related to adcs structural design. Front Pharmacol 10:373. https://doi.org/10.3389/fphar.2019.00373
Tarantino P, Hamilton E, Tolaney SM, Cortes J, Morganti S, Ferraro E, Marra A, Viale G, Trapani D, Cardoso F, Penault-Llorca F, Viale G, Andrè F, Curigliano G (2020) HER2-Low breast Cancer: pathological and clinical Landscape. J Clin Oncol 38:1951–1962. https://doi.org/10.1200/JCO.19.02488
Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, Cortes J, Soria J, Curigliano G (2022) Antibody–drug conjugates: Smart chemotherapy delivery across tumor histologies. CA Cancer J Clin 72:165–182. https://doi.org/10.3322/caac.21705
Tedeschini T, Campara B, Grigoletto A, Bellini M, Salvalaio M, Matsuno Y, Suzuki A, Yoshioka H, Pasut G (2021) Polyethylene glycol-based linkers as hydrophilicity reservoir for antibody-drug conjugates. J Control Release 337:431–447. https://doi.org/10.1016/j.jconrel.2021.07.041
Tiberghien AC, Levy J-N, Masterson LA, Patel NV, Adams LR, Corbett S, Williams DG, Hartley JA, Howard PW (2016) Design and synthesis of Tesirine, a clinical antibody–drug conjugate pyrrolobenzodiazepine dimer payload. ACS Med Chem Lett 7:983–987. https://doi.org/10.1021/acsmedchemlett.6b00062
Tsuchikama K, An Z (2018) Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 9:33–46. https://doi.org/10.1007/s13238-016-0323-0
U.S.F.D.A (2022) FDA D.I.S.C.O. Burst edition: FDA approval of Elahere (mirvetuximab soravtansine-gynx) for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-elahere-mirvetuximab-soravtansine-gynx-fra-positive-platinum. Accessed 31 Jan 2023
van Berkel SS, van Delft FL (2018) Enzymatic strategies for (near) clinical development of antibody-drug conjugates. Drug Discov Today Technol 30:3–10. https://doi.org/10.1016/j.ddtec.2018.09.005
Vankemmelbeke M, Durrant L (2016) Third-generation antibody drug conjugates for cancer therapy–a balancing act. Ther Deliv 7:141–144. https://doi.org/10.4155/tde-2016-0002
Wagh A, Song H, Zeng M, Tao L, Das TK (2018) In: MAbs (ed) Challenges and new frontiers in analytical characterization of antibody-drug conjugates. Taylor & Francis, pp 222–243
Wakanka A, Chen Y, Gokarn Y, Jacobson FS (2011) In: MAbs (ed) Analytical methods for physicochemical characterization of antibody drug conjugates. Taylor & Francis, pp 161–172
Waldum HL, Sandvik AK, Brenna E, Fossmark R, Qvigstad G, Soga J (2008) Classification of tumours. J Exp Clin Cancer Res 27:70. https://doi.org/10.1186/1756-9966-27-70
Walsh SJ, Bargh JD, Dannheim FM, Hanby AR, Seki H, Counsell AJ, Ou X, Fowler E, Ashman N, Takada Y, Isidro-Llobet A, Parker JS, Carroll JS, Spring DR (2021) Site-selective modification strategies in antibody–drug conjugates. Chem Soc Rev 50:1305–1353. https://doi.org/10.1039/D0CS00310G
Widdison WC, Chari RVJ (2013) In: Phillips GL (ed) Factors involved in the design of cytotoxic payloads for antibody–drug Conjugates BT - antibody-drug conjugates and immunotoxins: from pre-clinical development to therapeutic applications. Springer, New York, pp 93–115
Woitok M, Klose D, Niesen J, Richter W, Abbas M, Stein C, Fendel R, Bialon M, Püttmann C, Fischer R (2016) The efficient elimination of solid tumor cells by EGFR-specific and HER2-specific scFv-SNAP fusion proteins conjugated to benzylguanine-modified auristatin F. Cancer Lett 381:323–330. https://doi.org/10.1016/j.canlet.2016.08.003
Wolf P, Elsässer-Beile U (2009) Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol 299:161–176. https://doi.org/10.1016/j.ijmm.2008.08.003
Wu T, Zhu J (2021) Recent development and optimization of pseudomonas aeruginosa exotoxin immunotoxins in cancer therapeutic applications. Int Immunopharmacol 96:107759. https://doi.org/10.1016/j.intimp.2021.107759
Yamada K, Shikida N, Shimbo K, Ito Y, Khedri Z, Matsuda Y, Mendelsohn BA (2019) AJICAP: affinity peptide mediated regiodivergent functionalization of native antibodies. Angew Chemie 131:5648–5653
Yamazaki CM, Yamaguchi A, Anami Y, Xiong W, Otani Y, Lee J, Ueno NT, Zhang N, An Z, Tsuchikama K (2021) Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat Commun 12:1–13. https://doi.org/10.1038/s41467-021-23793-7
Yin J, Liu F, Li X, Walsh CT (2004) Labeling proteins with small molecules by site-specific posttranslational modification. J Am Chem Soc 126:7754–7755. https://doi.org/10.1021/ja047749k
You J, Zhang J, Wang J, Jin M (2021) Cysteine-based coupling: challenges and solutions. Bioconjug Chem 32:1525–1534. https://doi.org/10.1021/acs.bioconjchem.1c00213
Yuan D, Zhang Y, Lim KH, Leung SKP, Yang X, Liang Y, Lau WCY, Chow KT, Xia J (2022) Site-selective lysine acetylation of human immunoglobulin G for immunoliposomes and bispecific antibody complexes. J Am Chem Soc 144:18494–18503
Zacharias N, Podust VN, Kajihara KK, Leipold D, Del Rosario G, Thayer D, Dong E, Paluch M, Fischer D, Zheng K (2022) A homogeneous high-DAR antibody–drug conjugate platform combining THIOMAB antibodies and XTEN polypeptides. Chem Sci 13:3147–3160. https://doi.org/10.1039/D1SC05243H
Zhang Y, Park K-Y, Suazo KF, Distefano MD (2018) Recent progress in enzymatic protein labelling techniques and their applications. Chem Soc Rev 47:9106–9136. https://doi.org/10.1039/C8CS00537K
Zhang J, Jain A, Milhas S, Williamson DJ, Mysliwy J, Lodge A, Thirlway J, Al Nakeeb M, Miller A, Rabbitts TH (2021) An antibody-drug conjugate with intracellular drug release properties showing specific cytotoxicity against CD7-positive cells. Leuk Res 108:106626. https://doi.org/10.1016/j.leukres.2021.106626
Zhao P, Zhang Y, Li W, Jeanty C, Xiang G, Dong Y (2020) Recent advances of antibody drug conjugates for clinical applications. Acta Pharm Sin B 10:1589–1600. https://doi.org/10.1016/j.apsb.2020.04.012
Zhou L, Xu N, Sun Y, Liu X, Margaret (2014a) Targeted biopharmaceuticals for cancer treatment. Cancer Lett 352:145–151. https://doi.org/10.1016/j.canlet.2014.06.020
Zhou Q, Stefano JE, Manning C, Kyazike J, Chen B, Gianolio DA, Park A, Busch M, Bird J, Zheng X, Simonds-Mannes H, Kim J, Gregory RC, Miller RJ, Brondyk WH, Dhal PK, Pan CQ (2014b) Site-specific antibody–drug conjugation through Glycoengineering. Bioconjug Chem 25:510–520. https://doi.org/10.1021/bc400505q
Zhu X, Huo S, Xue C, An B, Qu J (2020) Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. J Pharm Anal 10:209–220. https://doi.org/10.1016/j.jpha.2020.05.008
Zou S (2023) Directions for next generation antibody-drug conjugates. https://www.eposters.net/poster/directions-for-next-generation-antibody-drug-conjugates#. Accessed 31 Jan 2023
Zuberbühler K, Casi G, Bernardes GJL, Neri D (2012) Fucose-specific conjugation of hydrazide derivatives to a vascular-targeting monoclonal antibody in IgG format. Chem Commun 48:7100–7102. https://doi.org/10.1039/C2CC32412A
Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L (2016) Current challenges in cancer treatment. Clin Ther 38:1551–1566. https://doi.org/10.1016/j.clinthera.2016.03.026
Acknowledgements
Authors, Ritwik Maiti, Bhumika Patel and Nrupesh Patel are thankful to Nirma University, Ahmedabad, Gujarat, India for providing resourceful support to carry out literature review for the present work. The efforts of author, Dr. NIrav Dhanesha for this publication were supported by grants from the NHLBI/NIH (R01HL15854601), and by the Career Development Award (20CDA35260123) from American Heart Association.
Funding
This work is supported by the National Institute of Health, NHLBI/NIH (R01HL15854601), and American Heart Association, Career Development Award (20CDA35260123) to Nirav Dhanesha
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Declared None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised to correct the grant number for Author Nirav Dhanesha
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Maiti, R., Patel, B., Patel, N. et al. Antibody drug conjugates as targeted cancer therapy: past development, present challenges and future opportunities. Arch. Pharm. Res. 46, 361–388 (2023). https://doi.org/10.1007/s12272-023-01447-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-023-01447-0