
Abstract
Histone deacetylase 6 (HDAC6) is a promising target for cancer treatment because it regulates cell mobility, protein trafficking, cell growth, apoptosis, and metastasis. However, the mechanism of HDAC6-induced anticancer drug resistance is unclear. In this study, we evaluated the anticancer effect of ACY-241, an HDAC6-selective inhibitor, on erlotinib-resistant pancreatic cancer cells that overexpress HDAC6. Our data revealed that ACY-241 hyperacetylated the HDAC6 substrate, α-tubulin, leading to a significant reduction in cell viability of erlotinib-resistant pancreatic cells, BxPC3-ER and HPAC-ER. Notably, a synergistic anticancer effect was observed in cells that received combined treatment with ACY-241 and erlotinib. Combined treatment effectively induced autophagy and inhibited autophagy through siLC3B, and siATG5 alleviated ACY-241-mediated cell death, as reflected by the recovery of PARP cleavage and apoptosis rates. In addition, combined ACY-241 and erlotinib treatment induced autophagy and subsequently, cell death by reducing AKT–mTOR activity and increasing phospho-AMPK signaling. Therefore, HDAC6 may be involved in the suppression of autophagy and acquisition of resistance to erlotinib in ER pancreatic cancer cells. ACY-241 to overcome erlotinib resistance could be an effective therapeutic strategy against pancreatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is the seventh leading cause of cancer-related deaths. The mortality rate of pancreatic cancer is as high as the incident rate, and a five-year survival rate of 10% (Sung et al. 2021; Siegel et al. 2021). The epidermal growth factor receptor (EGFR) is overexpressed in pancreatic cancer, with the receptor and ligands affect the aggressiveness and severity (Cohenuram and Saif 2007). Erlotinib, an EGFR-specific tyrosine kinase inhibitor (EGFR-TKI), in combination with gemcitabine, is the standard first-line treatment for pancreatic cancer. EGFR-TKIs, such as erlotinib and gefitinib, have dramatically improved survival rates in cancer patients. However, drug resistance occurs in most patients within 1–2 years. The T790M mutation in EGFR is the most frequent among several mechanisms conferring drug resistance (Kobayashi et al. 2005). The use of second and third generation TKIs is also not the solution against drug resistance because of problems associated with resistance (Nagano et al. 2018). Therefore, overcoming drug resistance remains a key challenge in pancreatic cancer therapy.
Post-translational modifications, such as acetylation and deacetylation, play roles in many cellular activities, including proliferation, differentiation, autophagy, and apoptosis (Chaudhary et al. 2019). In this context, it is reasonable to speculate that the overexpression of Histone deacetylases (HDAC) in cancer cells affects disease stage and prognosis (Witt et al. 2009). HDACs regulate gene expression by removing acetyl groups from histones and other proteins (Park and Han 2019). HDAC6, a class IIb HDAC, mainly remains in the cytoplasm and regulates the acetylation state of proteins, such as α-tubulin and histones (Zhang et al. 2015). Overexpression of HDAC6 is noticeable in cancer, and was reported to correlate with tumor progression and metastasis (Aldana-Masangkay and Sakamoto 2011). Consistently, the inhibition of HDAC6 can lead to therapeutic benefits, including inhibition of cancer cell proliferation, decrease in metastatic potential, and overcoming therapy resistance (Lee et al. 2008).
Pan-HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA, vorinostat), have been used for chemotherapy. However, their efficacy has been limited by severe adverse events when used in combination with other chemotherapeutic agents (Yoon and Eom 2016). More selective HDAC inhibitors have been developed to avoid the adverse effects related with the inhibition of class I HDACs (Bradner et al. 2010). ACY-241, a second generation HDAC6-selective inhibitor, exhibited clinical efficacy when used alone or in combination with other chemotherapies in a phase I trial (Pulya et al. 2021). ACY-241 has been reported to treat multiple myeloma and ovarian cancer by inducing cell-cycle arrest, promoting apoptosis, and inhibiting proliferation. The anticancer activity of ACY-241 looks promising, especially when used in combination with other chemotherapies (North et al. 2017; Bae et al. 2018; Ray et al. 2018; Yoo et al. 2021). Nonetheless, how ACY-241 overcomes drug resistance in pancreatic cancer is unclear.
Autophagy is an intracellular degradative process mediated by lysosomes in response to stressful conditions, such as organelle damage and starvation (Yun and Lee 2018). In mammalian cells, autophagy is largely divided into five stages: induction, vesicle nucleation, vesicle expansion, fusion, and degradation. Autophagy promotes cell survival by regulating metabolism and energy homeostasis. In cancer, autophagy affects the proliferation and death of cancer cells, with seemingly opposite roles—promotion and inhibition of cancer growth (Rosenfeldt and Ryan 2011; Tilija Pun et al. 2020). Thus, the regulation of autophagy has emerged as a new target for cancer therapy. The pharmacological inhibition of HDAC6 has been reported to regulate autophagy in various cancers, leading to cancer cell death or survival (Lee et al. 2010; Kaliszczak et al. 2018; Sharif et al. 2019). The regulation of autophagy can be exploited to increase the efficacy of chemotherapy or overcome resistance (Chang and Zou 2020). Studies on the function and role of HDAC6 in autophagy and apoptosis in EGFR-TKI-resistant cancer cells are limited.
In this study, we aimed to develop a therapy against erlotinib resistance. We observed that HDAC6 is overexpressed in erlotinib-resistant (ER) pancreatic cancer cells compared with their parental cells. We hypothesized that HDAC6 overexpression is responsible for erlotinib resistance in pancreatic cancer cells. To test our hypothesis, we investigated the role of HDAC6 by using an HDAC6 siRNA and HDAC6-specific inhibitor, ACY-241. We also evaluated whether combined treatment with ACY-241 and erlotinib could overcome erlotinib resistance in human ER pancreatic cancer cells.
Materials and methods
Chemicals and reagents
Erlotinib was purchased from LC Laboratories (Woburn, MA, USA). ACY-241 was obtained from Cayman Chemical Company (Ann Arbor, MI, USA). Dimethyl sulfoxide (DMSO) and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Antibodies against HDAC1, HDAC2, HDAC3, HDAC6, PARP, Ac-α-tubulin, α-tubulin, Ac-histone-H3, histone-H3, caspase-3, LC3B, p-AKT, AKT, phospho-adenosine monophosphate kinase (p-AMPK), AMPK, and mammalian target of rapamycin (mTOR) and secondary antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against GAPDH and p-mTOR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell lines and cell culture
Human pancreatic adenocarcinoma BxPC3 and HPAC cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Atlas Biologicals, Fort Collins, CO, USA) and 1% penicillin–streptomycin (HyClone Laboratories, Logan, UT, USA) in a 37 °C humidified incubator containing 5% CO2. For maintenance, BxPC3-ER and HPAC-ER cell lines (Jang et al. 2017; Lee et al. 2017) were cultured in RPMI-1640 (10% FBS, 1% penicillin–streptomycin) with 1 µM erlotinib.
Cell viability assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded at 3 × 103 cells/well of a 96-well plate. After overnight incubation, cells were treated with drugs and incubated for 48 h. Then, the cells were incubated with 20 μL MTT for 2 h. Absorbance (570 nm) was detected using the FLUOstar Omega (BMG Labtech, Germany).
Colony formation assay
BxPC3-ER and HPAC-ER cells were plated at a density of 8 × 102 cells/well of a 6-well plate. After 14 d, cells were washed with Dulbecco’s phosphate-buffered saline (DPBS), fixed with methanol for 3 min, and stained with 5% crystal violet. Colony were counted using the ImageJ software.
Western blot analysis
Western blotting has been modified and performed as described previously (Zheng et al. 2021). Cells were seeded at a density of 5 × 105 cells in a 60-mm dish. After 24 h of incubation, cells were treated with drugs, such as erlotinib or ACY-241, for 24–72 h. The cells were then lysed with cold whole-cell lysis buffer or radio-immunoprecipitation assay lysis buffer (RIPA) supplemented with Halt™ Protease, phosphatase inhibitor cocktail, PMSF, and EDTA. Proteins were quantified using the BCA protein assay kit (Thermo Scientific, Rockford, IL, USA). Proteins were loaded onto sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to PVDF membranes (GE Healthcare, Chicago, IL, USA). Membranes were blocked with 2.5% skim milk in TBS-T, then incubated overnight at 4 °C with primary antibodies. Membranes were washed four times with TBS-T and incubated with species-specific horseradish peroxidase-conjugated secondary antibodies. Proteins were visualized using the SuperSignal® West Dura Extended Duration Substrate (Thermo Scientific, Waltham, MA) and developed with LAS-3000 (Fuji, Japan) according to the manufacturer's instructions.
RNA interference
Double-stranded siRNAs against human HDAC6 (NM_001321225.1), LC3B (NM_022818.4), and ATG5 (NM_001286107.1) were synthesized by Bioneer (Daejeon, Republic of Korea). Negative control siRNA was obtained from Bioneer (Cat. No.: SN-1003). Specific siRNA sequences used in this study are as follows: HDAC6 forward; 5′-CCGGUUUGCUGAAAAGGAA-3′ and reverse; 5′-UUCCUUUUCAGCAAACCGG-3′, LC3B forward; 5′-CAUAAAGACACCACUCAAA-3′ and reverse; 5′-UUUGAGUGGUGUCUUUAUG-3′, and ATG5 forward; 5′-CAGGAAAAAGAUUCCAUGU-3′ and reverse; 5′-ACAUGGAAUCUUUUUCCUG-3′. Cells were seeded at 3 × 103 cells/well of a 96-well plate or 35 × 104 cells in a 60-mm dish. After overnight incubation, the cells were transfected with siRNA or negative control siRNA using the Oligofectamine transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Transfection efficacy of siRNA was measured by evaluating cell viability or through western blotting. For HDAC6 overexpression, pBJ5-HDAC6 expression vector and jetPEI (Polyplus-transfection, Illkirch, France) transfection reagent were used.
Flow cytometry
The extent of apoptosis was detected using the FITC Annexin V apoptosis detection kit with 7-AAD (BioLegend, San Diego, CA). Cells were collected after drug treatment and resuspended in binding buffer. Annexin V and 7-AAD were added to the cell suspension and the cells were incubated for 15 min at RT in the dark. Autophagy was detected using acridine orange (Thermo Scientific, Rockford, IL, USA). The cells were stained with acridine orange for 20 min and washed two times with cold DPBS, according to the manufacturer’s protocol. The red fluorescence of acridine orange was quantified using the BD FACSVerse flow cytometer and BD FACSuite Software.
Immunofluorescence staining
Immunofluorescence stating has been modified and performed as described previously (Shou, J. et al. 2020). Cells were seeded at a density of 1.5 × 105 cells on the coverslips. Cells were washed once with PBS and then fixed methanol. The cells were then permeabilized using 0.1% Triton-X 100 and incubated at 4 °C for 18–20 h with LC3B antibody in a moist and humid chamber. After washing with PBS-T and incubated with an Alexa 488-conjugated secondary antibody for 1 h in the dark, nuclei were counterstained with 0.1 μg/mL DAPI. Images were then obtained by a Carl Zeiss LSM5 confocal laser microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed with Student's t-test or two-way ANOVA using GraphPad Prism 5. Data are presented as the mean ± standard deviation (SD).
Results
ACY-241, a selective HDAC6 inhibitor, reduces cell viability in ER pancreatic cancer cells
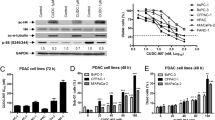
Previously, we established ER pancreatic cancer cells, BxPC3-ER and HPAC-ER (Jang et al. 2017; Lee et al. 2017). In this study, we compared HDAC6 expression between BxPC3-ER and HPAC-ER and their parental cells, BxPC3 and HPAC, respectively. HDAC overexpression was reported to be responsible for resistance to chemotherapy (Fantin and Richon 2007). Therefore, we determined the expression levels of HDAC1, 2, and 3 (HDAC family class I) and HDAC6 (HDAC family class II) through western blot analysis. Our data revealed that HDAC6 is significantly overexpressed in BxPC3-ER and HPAC-ER cells compared with parental cells (Fig. 1A). We next investigated whether increased HDAC6 in ER cells acts as a cause of resistance to erlotinib. The results show that inhibition of HDAC6 by siRNA in both types of ER cells significantly increases susceptibility to erlotinib, implying that HDAC6 can be one of the causes of acquired resistance to erlotinib (Fig. 1B). To assess the effect of HDAC6 inhibition, we treated ER cells with ACY-241 and determined the acetylation levels of histone H3, a substrate of HDAC family class I, and α-tubulin, an HDAC6-specific substrate. Treatment with low concentration of ACY-241 caused a dramatic increase in α-tubulin acetylation but not histone H3 acetylation. This result indicates that ACY-241 might be HDAC6-specific at concentrations lower than 5 μM (Fig. 1C). Our data also showed that dose-dependent treatment with ACY-241 caused a significant decrease in cell viability of both types of ER pancreatic cancer cells, while not causing a reduction in cell viability of their parental cells. These data suggest that decreased cell viability was achieved by selectively inhibiting HDAC6 with ACY-241 (Fig. 1D).

ACY-241 reduces cell viability in erlotinib-resistant (ER) pancreatic cancer cells. A Western blot analysis of histone deacetylases (HDACs) in erlotinib-sensitive (BxPC3 and HAPC) or resistant (BxPC3-ER and HPAC-ER) pancreatic cancer cells. B After knockdown of HDAC6, cells were treated with the indicated concentration of erlotinib for 48 h and cell viability was measured using the MTT assay. Statistical analysis was performed using two‐way ANOVA. Error bars represent mean ± SD (n = 6). ***p < 0.001. C Cells were treated with the indicated concentration of ACY-241 for 24 h, and levels of α-tubulin, acetyl-α-tubulin, H3, and acetyl-H3 expression were analyzed using western blotting. GAPDH was used as a loading control and SAHA was used as a pan-HDAC inhibitor control. D After treatment with the indicated concentration of ACY-241 for 48 h, cell viability was measured using the MTT assay. Statistical analysis was conducted using a t-test. Error bars represent mean ± SD (n = 6). *p < 0.05; ***p < 0.001
Synergistic anticancer effects of ACY-241 combined with erlotinib in ER pancreatic cancer cells
We next tested whether HDAC6 inhibition with ACY-241 could overcome erlotinib resistance. ER pancreatic cancer cells and their parental cells were treated with 5 μM erlotinib alone or in combination with 2.5–5 μM ACY-241 for 48 h. In parental cells, there was only an effect on elotinib, but no combined effect of the drugs was observed when ACY-241 and erlotinib were used together. But in ER pancreatic cancer cells, combined treatment with ACY-241 and erlotinib decreased cell viability more significantly than treatment with erlotinib alone (Fig. 2A). Consistent with cell viability data, the colony formation assay demonstrated that long-term ACY-241 treatment alone or in combination with erlotinib significantly decreased colony formation in both types of ER pancreatic cancer cells (Fig. 2B). Next, we determined the ratio of apoptotic cells after ACY-241 treatment alone or in combination with erlotinib using flow cytometry analysis. Our findings revealed that treatment with ACY-241 alone increased the number of Annexin V-positive cells, indicating apoptosis activation. Combined treatment with ACY-241 and erlotinib further increased the ratio of apoptotic cells by up to 50%, suggesting the synergism of this combination (Fig. 2C). The levels of apoptotic markers, such as cleaved caspase-3 and PARP (c-PARP), were determined by western blotting, and the result showed a clear increase of these proteins after combined ACY-241 and erlotinib treatment (Fig. 2D). Therefore, combined ACY-241 and erlotinib treatment synergistically reduces cell viability, colony formation, and induces apoptosis in ER pancreatic cell lines.

Combination treatment with erlotinib and ACY-241 synergistically induces an anti-cancer effect in erlotinib-resistant (ER) pancreatic cancer cells. A After combined treatment with erlotinib and ACY-241 for 48 h, cell viability of parental and ER cells was measured using the MTT assay. Statistical analysis was performed using two‐way ANOVA. Error bars represent mean ± SD (n = 6). ***p < 0.001; ###p < 0.001. B Colony formation assay. Cells were treated with a combination of erlotinib and ACY-241 for 14 days and stained with crystal violet. Representative images from three independent sets of experiments and graph quantifying colony formation. Statistical analysis was performed using two-way ANOVA. Error bars show mean ± SD (n = 3). ##p < 0.01; ###p < 0.001. C Cells were treated with a combination of erlotinib and ACY-241 for 24 h, and the rate of apoptosis was measured by flow cytometric analysis with Annexin V staining (left). The quantification of apoptotic cells is shown as a graph (right). Statistical analysis was conducted using two-way ANOVA. Error bars show mean ± SD (n = 3). #p < 0.05; ##p < 0.01; ###p < 0.001. D Cells were treated with a combination of erlotinib and ACY-241 for 24 h, and cleavage of caspase‐3 and PARP was monitored through western blot analysis. GAPDH was used as a loading control
Combined ACY-241 and erlotinib treatment induces autophagy in ER pancreatic cancer cells
The inhibition of HDAC6 has been reported to induce various cellular responses, such as induction or inhibition of autophagy, depending on cancer cells and types of HDAC6 inhibitors (Lee et al. 2010; Kaliszczak et al. 2018; Sharif et al. 2019). Moreover, treating cells with EGFR-TKIs, such as erlotinib, can induce autophagy (Han et al. 2011; Fung et al. 2012). Based on these studies, we checked the expression of autophagy markers in both types of ER pancreatic cancer cells after treatment with erlotinib and ACY-241. ACY-241 treatment increased LC3BII levels, which increased significantly after combined ACY-241 and erlotinib treatment in both types of ER pancreatic cancer cells (Fig. 3A). Autophagy induction was also confirmed by immunofluorescence staining. Our data revealed that combined treatment with ACY-241 and erlotinib further increased LC3B expression in ER pancreatic cells (Fig. 3B). The induction of autophagy was reported to accompany an increase in acidic vesicular organelle (AVO) volume due to the increase in autolysosomes (Kanzawa et al. 2003). To determine whether ACY-241 induces autophagy in both types of ER pancreatic cancer cells, cells treated with a combination of ACY-241 and erlotinib were evaluated for the level of red fluorescence using flow cytometry. Our data revealed that dose-dependent treatment with ACY-241 increased the strength of red fluorescence. Moreover, combined ACY-241 and erlotinib treatment led to a large increase in red fluorescence (Fig. 3C). Thus, combined treatment with ACY-241 and erlotinib strongly increased the conversion of LC3BI into LC3BII, thereby inducing autophagy in both types of ER pancreatic cancer cells.

Combination treatment with erlotinib and ACY-241 induces autophagy in erlotinib-resistant (ER) pancreatic cancer cells. A After treatment with a combination of erlotinib and ACY-241 for 24 h, levels of LC3B were measured using western blot analysis. GAPDH was used as a loading control. Quantitation of protein intensity was performed using the ImageJ software. Statistical analysis was performed using two‐way ANOVA. Error bars represent mean ± SD (n = 3). **p < 0.01; ***p < 0.001; #p < 0.05; ##p < 0.01. B Immunofluorescence staining of LC3B. Cells were treated with a combination of erlotinib and ACY-241 for 24 h. LC3B-positive cells were detected via fluorescence microscopy, and intensity was quantified using the ImageJ software. Statistical analysis was conducted using Student's t-test. Error bars represent mean ± SD (n = 3). *p < 0.05. C Cells were treated with a combination of erlotinib and ACY-241 for 24 h. Acridine orange staining was performed to identify autophagic cells through FACS. Statistical analysis was performed using two‐way ANOVA. Error bars represent mean ± SD (n = 3). ##p < 0.01; ###p < 0.001
Inhibition of autophagy alleviated ACY-241-mediated apoptosis in ER pancreatic cancer cells
Autophagy and apoptosis play important roles in maintaining cellular homeostasis and are mutually regulated (Su et al. 2013). To investigate the role of autophagy on ACY-241-induced apoptosis, we silenced two autophagy related genes, LC3B and ATG5, in ER cells. The levels of cleaved PARP decreased significantly upon silencing LC3B and ATG5 (Fig. 4A, C). Consistently, flow cytometry analysis showed that knockdown of LC3B and ATG5 alleviated apoptotic cell death induced by combination treatment with ACY-241 and erlotinib (Fig. 4B, D). Thus, combination treatment with ACY-241 and erlotinib enhances apoptosis through the induction of autophagy in ER human pancreatic cancer cells.

Inhibition of autophagy alleviates apoptosis in erlotinib-resistant (ER) pancreatic cancer cells. A, C Cells were transfected with siLC3B or siATG5 and treated with a combination of erlotinib and ACY-241 for 24 h. Levels of LC3B, ATG5, and PARP expression were measured using western blot analysis. GAPDH was used as a loading control. B, D Flow cytometry analysis was performed to check the effect of siLC3B or siATG5 on apoptosis of ER cells. Statistical analysis was performed using two‐way ANOVA. Error bars represent mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001
HDAC6 might be involved in the suppression of autophagy and acquisition of resistance to erlotinib in ER pancreatic cancer cells
A study reported that defective autophagy is responsible for anticancer drug resistance and the activation of autophagy can reverse this resistance (Fung et al. 2012). In this study, we investigated how autophagy is regulated when erlotinib-sensitive and -resistant pancreatic cancer cells are treated with erlotinib. We evaluated LC3B conversion as a marker of autophagy. Erlotinib treatment led to an increase in the conversion of LC3BII in erlotinib-sensitive parental cells, whereas no significant LC3BII conversion was detected in BxPC3-ER and HPAC-ER cells. Concomitantly, erlotinib treatment led to reduced levels of p-AKT and p-mTOR and increased levels of p-AMPK in erlotinib-sensitive cells. This implies that erlotinib effectively induces autophagy by regulating upstream AKT–mTOR and AMPK signaling in erlotinib-sensitive cells. However, in ER pancreatic cancer cells, erlotinib did not induce autophagy and had no significant effect on these upstream signaling pathways (Fig. 5A). Based on this observation, we speculated that resistance to erlotinib can be caused by failure of autophagy induction. Therefore, we investigated the correlation between the failure to induce autophagy by erlotinib and HADC6 overexpression in ER cell lines. We used siRNA to knockdown HDAC6, the target of ACY-241, and then identified how it affected autophagy after erlotinib treatment in ER cells. Erlotinib treatment induced LC3B conversion in HDAC6 siRNA-treated ER cells through a decrease in p-AKT and p-mTOR and an increase in p-AMPK levels, while little change in LC3B conversion in control siRNA-treated ER cells (Fig. 5B). Conversely, HDAC6 overexpression in erlotinib sensitive-BxPC3 cells has been shown to reduce LC3B conversion increased by erlotinib treatment (Supplementary Figure). In addition, combined ACY-241 and erlotinib treatment strongly increased LC3BII conversion through the regulation of same upstream signaling pathways in both types of ER pancreatic cancer cells (Fig. 5C). Therefore, HDAC6 overexpression in ER pancreatic cancer cells might inhibit erlotinib-induced autophagy, thereby leading to erlotinib resistance. However, inhibition of HDAC6 by siRNA or ACY-241 led to erlotinib-induced autophagy and overcoming of erlotinib resistance in ER pancreatic cancer cells (Fig. 5D).

The role of histone deacetylase (HDAC) 6 in suppressing autophagy and acquiring erlotinib resistance in erlotinib-resistant (ER) pancreatic cancer cells. A Erlotinib-sensitive (BxPC3 and HAPC) or resistant (BxPC3-ER and HPAC-ER) pancreatic cancer cells were treated with the indicated concentration of erlotinib for 24 h and level of protein expression was quantified using western blot analysis. GAPDH was used as a loading control. B Cells were transfected with siHDAC6 and treated with the indicated concentration of erlotinib for 72 h. Level of protein expression was measured using western blot analysis. C Cells were treated with a combination of erlotinib and ACY-241 for 24 h and level of protein expression was quantified using western blot analysis. GAPDH was used as a loading control. D Proposed mechanism of the anticancer effect of ACY-241 and erlotinib in erlotinib-sensitive and resistant pancreatic cancer cells
Discussion
Although various therapies have been developed to treat pancreatic cancer, drug resistance remains a challenge (Zhang et al. 2020). Combination therapy has emerged as a promising strategy to overcome drug resistance and the complexity of signaling pathways (Chen et al. 2014). In addition to EGFR-TKI, other drugs can be combined to inhibit other receptors, such as VEGFR, FGFR, and MET. Inhibitors of downstream signaling, such as PI3K–Akt–mTOR, Ras–Raf–MEK–ERK, and JAK–STAT, can be used in addition to EGFR-TKI treatment to overcome this resistance (Tong et al. 2017).
HDAC is a promising target for both single and combined treatments against cancer. HDAC inhibitors regulate the expression of various genes by modulating the acetylation states of histone protein (Eckschlager et al. 2017). Pan-HDAC inhibitors (panobinostat, belinostat, and vorinostat) and HDAC class I inhibitors (tucidinostat and mocetinostat) have been tested in clinical trials for treating various blood cancers (West and Johnstone 2014). HDAC inhibitors, romidepsin and MPT0E028, were reported to increase sensitivity to erlotinib in NSCLC (Zhang et al. 2009; Chen et al. 2013). A phase I trial is under way as vorinostat has been reported to induce apoptosis when combined with gefitinib (Nakagawa et al. 2013). Combined treatment with several pan-HDAC inhibitors and EGFR-TKIs can induce autophagic cell death in T790M mutant lung cancer (Lee et al. 2015). However, the clinical application of pan-HDAC inhibitors or HDAC class I inhibitors is limited, because they lack specificity and exhibit cytotoxicity when administered alone or in combination with other drugs (Deubzer et al. 2013; Koutsounas et al. 2013; Yoon and Eom 2016). Moreover, few studies have evaluated the use of HDAC inhibitors in treating pancreatic cancer. Therefore, we used ACY-241, a selective HDAC6 inhibitor, to minimize the side effects while increasing the effectiveness of a pan-HDAC inhibitor.
Autophagy is induced by AMPK, a key energy sensor that regulates metabolism to maintain energy homeostasis, and inhibited by mTOR, which integrates the upstream signaling of class I PI3K/AKT and AMPK (Kim et al. 2011). The absence of growth signals leads to the deactivation of AKT and accumulation of tuberous sclerosis protein 1/2 (TSC1/2), which inactivates mTOR by activating Ras homolog enriched in brain (Rheb) (Efeyan et al. 2013). Then, inactivated mTOR activates the Unc-51-like kinase (ULK) complex to induce autophagy (Li et al. 2020). When energy levels are low in cells, AMPK first activates TSC1/2, and then, inactivates mTOR and induces autophagy (Lacher et al. 2010). In general, autophagy induces drug resistance by protecting cancer cells from chemotherapy. The upregulation of autophagy can prevent DNA damage (White 2016) or increase drug resistance by increasing the expression of multidrug resistance (MDR) genes (Zhang et al. 2016). However, persistent and excessive autophagy can cause apoptotic cell death. Thus, the induction of autophagy by diverse therapeutic stresses has a dual role in promoting cell survival or death depending on cell type and environmental stress (Chang and Zou 2020). Baicalein, a flavonoid with anticancer properties, can induce autophagic cell death by activating AMPK/ULK1 and downregulating mTORC1 (Aryal et al. 2014). Pharmacological induction of autophagy in an EGFR-TKI-resistant cancer cell line was shown to increase the sensitivity of EGFR-TKI and induce autophagic cell death through the AKT–mTOR pathway (Fung et al. 2012).
HDAC6, a class IIb HDAC, is overexpressed in cancer and associated with increased tumorigenesis and cell survival (Aldana-Masangkay and Sakamoto 2011). Unlike that for other HDACs, knocking out HDAC6 in mice does not exhibit cytotoxicity, which is why active research is being conducted on HDAC6 as a target for cancer therapy (Zhang et al. 2008). Studies have shown that HDAC6 might regulate autophagy and cell death through various mechanisms. For example, C1A, an HDAC6 inhibitor, was reported to reduce cancer cell viability by blocking the fusion of autophagosomes and lysosomes in myc-positive neuroblastoma, KRAS-positive colorectal cancer, and multiple myeloma cells (Kaliszczak et al. 2018). In multiple myeloma, HDAC6 affects aggresome formation in cells; treatment with ACY-1215, an HDAC6 inhibitor, inhibits aggresome formation, which causes inhibition of autophagy and subsequently, cell death (Mishima et al. 2015). By contrast, treatment with another HDAC6 inhibitor, J22352, can induce autophagic cell death in glioblastoma (Liu et al. 2019). These are conflicting reports showing that HDAC6 inhibition by different inhibitors can induce apoptosis by inhibiting or promoting autophagy. Therefore, we tried to establish the relationship between autophagy and apoptosis in human pancreatic cancer cells, where HDAC6 is overexpressed. Although phase I/II clinical trials using ACY-1215, a commonly used HDAC6-selective inhibitor, have been conducted, the use of ACY-1215 in the clinic is a challenge owing to its low absorption (Hideshima et al. 2016). Therefore, we used ACY-241, a second-generation HDAC6-selective inhibitor that overcomes these shortcomings and increases selectivity for HDAC6 (Yoo et al. 2021). Our findings revealed that ACY-241 alone increased LC3B-II conversion; combined treatment with ACY-241 and erlotinib induced autophagy more strongly by regulating AKT–mTOR and AMPK signaling downstream.
Although extensive studies have been conducted on autophagy regulation through mTOR and AMPK signaling, studies on how HDAC6 controls autophagy to contribute to drug resistance have not been conducted. In this study, we observed increased expression of HDAC6 in two types of ER pancreatic cancer cells compared with their parental cells. We hypothesized that HDAC6 overexpression correlates with the acquisition of erlotinib resistance. Our results reveal several important points. First, HDAC6 is overexpressed in ER pancreatic cancer cells compared with parental cells. Second, erlotinib reduces cell viability by inducing autophagy in parental cells, but not in ER cell lines. Thus, the induction of autophagy by erlotinib can act as a prerequisite for cell death. Third, sensitivity to erlotinib in ER cells was synergistically enhanced when HDAC6 activity was inhibited using siRNA or a selective inhibitor. Fourth, combined ACY-241 and erlotinib treatment induced autophagic cell death by regulating AKT–mTOR and AMPK signaling. Therefore, whether autophagy is induced by erlotinib is one of the causes for the acquisition of erlotinib resistance and HDAC6 negatively regulates induction of autophagy in this process. In addition, autophagy can be induced by the selective inhibition of HDAC6, which can help overcome erlotinib resistance in ER pancreatic cancer cells. Consistent with this, the emerging data suggest the role of HDAC as a resistance regulator in EGFR-TKI-resistant cancer cells by showing that inhibition of HDAC sensitizes these resistant cells to EGFR-TKIs (Wang et al. 2016; Yu et al. 2017). However, there are no reports yet on how and why HDAC6 can be increased with the emergence of acquired resistance to erlotinib. We are still unaware of the answer to this question and will need to investigate how HDAC6 can be a new regulator of acquired drug resistance through follow-up studies.
Taken together, our findings suggest that ACY-241, a selective HDAC6 inhibitor, has anticancer effects in ER pancreatic cancer cell lines and combined ACY-241 and erlotinib treatment synergistically induces cell death in ER cancer cells. In addition, HDAC6 is implicated in the suppression of autophagy and acquisition of erlotinib resistance in ER pancreatic cancer cells. Therefore, combined ACY-241 and erlotinib treatment could be an effective therapeutic strategy against pancreatic cancer.
References
Aldana-Masangkay GI, Sakamoto KM (2011) The role of HDAC6 in cancer. J Biomed Biotechnol 2011:875824. https://doi.org/10.1155/2011/875824
Aryal P, Kim K, Park PH, Ham S, Cho J, Song K (2014) Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J 281:4644–4658. https://doi.org/10.1111/febs.12969
Bae J, Hideshima T, Tai YT, Song Y, Richardson P, Raje N, Munshi NC, Anderson KC (2018) Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 32:1932–1947. https://doi.org/10.1038/s41375-018-0062-8
Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R (2010) Chemical phylogenetics of histone deacetylases. Nat Chem Biol 6:238–243. https://doi.org/10.1038/nchembio.313
Chang H, Zou Z (2020) Targeting autophagy to overcome drug resistance: further developments. J Hematol Oncol 13:159. https://doi.org/10.1186/s13045-020-01000-2
Chaudhary P, Ha E, Vo TTL, Seo JH (2019) Diverse roles of arrest defective 1 in cancer development. Arch Pharm Res 42:1040–1051. https://doi.org/10.1007/s12272-019-01195-0
Chen MC, Chen CH, Wang JC, Tsai AC, Liou JP, Pan SL, Teng CM (2013) The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis 4:e810. https://doi.org/10.1038/cddis.2013.330
Chen SM, Guo CL, Shi JJ, Xu YC, Chen Y, Shen YY, Su Y, Ding J, Meng LH (2014) HSP90 inhibitor AUY922 abrogates up-regulation of RTKs by mTOR inhibitor AZD8055 and potentiates its antiproliferative activity in human breast cancer. Int J Cancer 135:2462–2474. https://doi.org/10.1002/ijc.28880
Cohenuram M, Saif MW (2007) Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present and the future. JOP 8:4–15
Deubzer HE, Schier MC, Oehme I, Lodrini M, Haendler B, Sommer A, Witt O (2013) HDAC11 is a novel drug target in carcinomas. Int J Cancer 132:2200–2208. https://doi.org/10.1002/ijc.27876
Eckschlager T, Plch J, Stiborova M, Hrabeta J (2017) Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci 18:1414. https://doi.org/10.3390/ijms18071414
Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM (2013) Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493:679–683. https://doi.org/10.1038/nature11745
Fantin VR, Richon VM (2007) Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res 13:7237–7342. https://doi.org/10.1158/1078-0432.CCR-07-2114
Fung C, Chen X, Grandis JR, Duvvuri U (2012) EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol Ther 13:1417–1424. https://doi.org/10.4161/cbt.22002
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X, Wang X, Jin H (2011) EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 6:e18691. https://doi.org/10.1371/journal.pone.0018691
Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, West N, Colling ME, Estiu G, Mazitschek R, Perry JA, Ohguchi H, Cottini F, Mimura N, Görgün G, Tai YT, Richardson PG, Carrasco RD, Wiest O, Schreiber SL, Anderson KC, Bradner JE (2016) Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc Natl Acad Sci USA 113:13162–13167. https://doi.org/10.1073/pnas.1608067113
Jang WJ, Choi B, Song SH, Lee N, Kim DJ, Lee S, Jeong CH (2017) Multi-omics analysis reveals that ornithine decarboxylase contributes to erlotinib resistance in pancreatic cancer cells. Oncotarget 8:92727–92742. https://doi.org/10.18632/oncotarget.21572
Kaliszczak M, van Hechanova E, Li Y, Alsadah H, Parzych K, Auner HW, Aboagye EO (2018) The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br J Cancer 119:1278–1287. https://doi.org/10.1038/s41416-018-0232-5
Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I (2003) Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 63:2103–2108
Kim J, Kundu M, Viollet B, Guan K (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. https://doi.org/10.1038/ncb2152
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 352:786–792. https://doi.org/10.1056/NEJMoa044238
Koutsounas I, Giaginis C, Theocharis S (2013) Histone deacetylase inhibitors and pancreatic cancer: are there any promising clinical trials? World J Gastroenterol 19:1173–1181. https://doi.org/10.3748/wjg.v19.i8.1173
Lacher MD, Pincheira R, Zhu Z, Camoretti-Mercado B, Matli M, Warren RS, Castro AF (2010) Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: implications for cell proliferation and tumorigenesis. Oncogene 29:6543–6556. https://doi.org/10.1038/onc.2010.393
Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP (2008) The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res 68:7561–7569. https://doi.org/10.1158/0008-5472.CAN-08-0188
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29:969–980. https://doi.org/10.1038/emboj.2009.405
Lee TG, Jeong EH, Kim SY, Kim HR, Kim CH (2015) The combination of irreversible EGFR TKIs and SAHA induces apoptosis and autophagy-mediated cell death to overcome acquired resistance in EGFR T790M-mutated lung cancer. Int J Cancer 136:2717–2729. https://doi.org/10.1002/ijc.29320
Lee S, Jang WJ, Choi B, Joo SH, Jeong CH (2017) Comparative metabolomic analysis of HPAC cells following the acquisition of erlotinib resistance. Oncol Lett 13:3437–3444. https://doi.org/10.3892/ol.2017.5940
Li X, He S, Ma B (2020) Autophagy and autophagy-related proteins in cancer. Mol Cancer 19:12. https://doi.org/10.1186/s12943-020-1138-4
Liu JR, Yu CW, Hung PY, Hsin LW, Chern JW (2019) High-selective HDAC6 inhibitor promotes HDAC6 degradation following autophagy modulation and enhanced antitumor immunity in glioblastoma. Biochem Pharmacol 163:458–471. https://doi.org/10.1016/j.bcp.2019.03.023
Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, O’Donnell E, Selig MK, Quayle SN, Arastu-Kapur S, Kirk C, Boise LH, Jones SS, Raje N (2015) Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br J Haematol 169:423–434. https://doi.org/10.1111/bjh.13315
Nagano T, Tachihara M, Nishimura Y (2018) Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells 7:212. https://doi.org/10.3390/cells7110212
Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, Nanjo S, Ishikawa D, Sato M, Hasegawa Y, Sekido Y, Yano S (2013) EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res 73:2428–2434. https://doi.org/10.1158/0008-5472.CAN-12-3479
North BJ, Almeciga-Pinto I, Tamang D, Yang M, Jones SS, Quayle SN (2017) Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 12:e0173507. https://doi.org/10.1371/journal.pone.0173507
Park JW, Han JW (2019) Targeting epigenetics for cancer therapy. Arch Pharm Res 42:159–170. https://doi.org/10.1007/s12272-019-01126-z
Pulya S, Amin SA, Adhikari N, Biswas S, Jha T, Ghosh B (2021) HDAC6 as privileged target in drug discovery: a perspective. Pharmacol Res 163:105274. https://doi.org/10.1016/j.phrs.2020.105274
Ray A, Das DS, Song Y, Hideshima T, Tai YT, Chauhan D, Anderson KC (2018) Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 32:843–846. https://doi.org/10.1038/leu.2017.322
Rosenfeldt MT, Ryan KM (2011) The multiple roles of autophagy in cancer. Carcinogenesis 32:955–963. https://doi.org/10.1093/carcin/bgr031
Sharif T, Martell E, Dai C, Ghassemi-Rad MS, Hanes MR, Murphy PJ, Margam NN, Parmar HB, Giacomantonio CA, Duncan R, Lee PWK, Gujar S (2019) HDAC6 differentially regulates autophagy in stem-like versus differentiated cancer cells. Autophagy 15:686–706. https://doi.org/10.1080/15548627.2018.1548547
Shou J, Wang M, Cheng X, Wang X, Zhang L, Liu Y, Fei C, Wang C, Gu F, Xue F, Li J, Zhang K (2020) Tizoxanide induces autophagy by inhibiting PI3K/Akt/mTOR pathway in RAW264.7 macrophage cells. Arch Pharm Res 43:257–270. https://doi.org/10.1007/s12272-019-01202-4
Siegel RL, Miller KD, Fuchs HE, Jemal A (2021) Cancer statistics. CA Cancer J Clin 71:7–33. https://doi.org/10.3322/caac.21654
Su M, Mei Y, Sinha S (2013) Role of the crosstalk between autophagy and apoptosis in cancer. J Oncol 2013:102735. https://doi.org/10.1155/2013/102735
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. https://doi.org/10.3322/caac.21660
Tilija Pun N, Jang WJ, Jeong CH (2020) Role of autophagy in regulation of cancer cell death/apoptosis during anti-cancer therapy: focus on autophagy flux blockade. Arch Pharm Res 43:475–488. https://doi.org/10.1007/s12272-020-01239-w
Tong CWS, Wu WKK, Loong HHF, Cho WCS, To KKW (2017) Drug combination approach to overcome resistance to EGFR tyrosine kinase inhibitors in lung cancer. Cancer Lett 405:100–110. https://doi.org/10.1016/j.canlet.2017.07.023
Wang Z, Tang F, Hu P, Wang Y, Gong J, Sun S, Xie C (2016) HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncol Rep 36:589–597. https://doi.org/10.3892/or.2016.4811
West AC, Johnstone RW (2014) New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 124:30–39. https://doi.org/10.1172/JCI69738
White E (2016) Autophagy and p53. Cold Spring Harb Perspect Med 6:a026120. https://doi.org/10.1101/cshperspect
Witt O, Deubzer HE, Lodrini M, Milde T, Oehme I (2009) Targeting histone deacetylases in neuroblastoma. Curr Pharm Des 15:436–447. https://doi.org/10.2174/138161209787315774
Yoo J, Jeon YH, Lee DH, Kim GW, Lee SW, Kim SY, Park J, Kwon SH (2021) HDAC6-selective inhibitors enhance anticancer effects of paclitaxel in ovarian cancer cells. Oncol Lett 21:201. https://doi.org/10.3892/ol.2021.12462
Yoon S, Eom GH (2016) HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med J 52:1–11. https://doi.org/10.4068/cmj.2016.52.1.1
Yu W, Lu W, Chen G, Cheng F, Su H, Chen Y, Liu M, Pang X (2017) Inhibition of histone deacetylases sensitizes EGF receptor-TK inhibitor-resistant non-small-cell lung cancer cells to erlotinib in vitro and in vivo. Br J Pharmacol 174:3608–3622. https://doi.org/10.1111/bph.13961
Yun CW, Lee SH (2018) The roles of autophagy in cancer. Int J Mol Sci 19:3466. https://doi.org/10.3390/ijms19113466
Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P (2008) Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol 28:1688–1701. https://doi.org/10.1128/MCB.01154-06
Zhang W, Peyton M, Xie Y, Soh J, Minna JD, Gazdar AF, Frenkel EP (2009) Histone deacetylase inhibitor romidepsin enhances anti-tumor effect of erlotinib in non-small cell lung cancer (NSCLC) cell lines. J Thorac Oncol 4:161–166. https://doi.org/10.1097/JTO.0b013e318194fae7
Zhang L, Liu S, Liu N, Zhang Y, Liu M, Li D, Seto E, Yao TP, Shui W, Zhou J (2015) Proteomic identification and functional characterization of MYH9, Hsc70, and DNAJA1 as novel substrates of HDAC6 deacetylase activity. Protein Cell 6:42–54. https://doi.org/10.1007/s13238-014-0102-8
Zhang LH, Yang AJ, Wang M, Liu W, Wang CY, Xie XF, Chen X, Dong JF, Li M (2016) Enhanced autophagy reveals vulnerability of P-gp mediated epirubicin resistance in triple negative breast cancer cells. Apoptosis 21:473–488. https://doi.org/10.1007/s10495-016-1214-9
Zhang X, Zegar T, Weiser T, Hamdan FH, Berger BT, Lucas R, Balourdas DI, Ladigan S, Cheung PF, Liffers ST, Trajkovic-Arsic M, Scheffler B, Joerger AC, Hahn SA, Johnsen SA, Knapp S, Siveke JT (2020) Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int J Cancer 147:2847–2861. https://doi.org/10.1002/ijc.33137
Zheng X, Liu H, Ma M, Ji J, Zhu F, Sun L (2021) Anti-thrombotic activity of phenolic acids obtained from Salvia miltiorrhiza f. alba in TNF-alpha-stimulated endothelial cells via the NF-kappaB/JNK/p38 MAPK signaling pathway. Arch Pharm Res 44:427–438. https://doi.org/10.1007/s12272-021-01325-7
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant Nos., NRF-2016R1A6A1A03011325, NRF-2018R1D1A3B07048623).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Park, SJ., Joo, S.H., Lee, N. et al. ACY-241, an HDAC6 inhibitor, overcomes erlotinib resistance in human pancreatic cancer cells by inducing autophagy. Arch. Pharm. Res. 44, 1062–1075 (2021). https://doi.org/10.1007/s12272-021-01359-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01359-x