
Abstract
Lipid-soluble ginseng extracts (LSGE) is known to inhibit many types of cancer cells through arresting cell cycle and inducing apoptosis. Usually, normal cells are can also be damaged by anti-tumor reagents. The plasma membrane redox system (PMRS) is enhanced to compensate mitochondrial dysfunction and impaired energy metabolism. NADH-quinone oxidoreductase 1 (NQO1), a plasma membrane redox enzyme, is known to be induced by panaxytriol, one of components of lipid-soluble ginseng extracts (LSGE). The objective of this study was determine the mechanisms of NQO1 involved in neuroprotection in response to cytotoxicity induced by LSGE. Exposure of control SH-SY5Y cells to LSGE resulted in dramatic loss of cell viability in a dose-dependent manner. The loss of cell viability was significantly recovered in cells transfected with NQO1. LSGE-induced cell death occurred through apoptosis such as cell shrinkage, chromatin condensation and cleavage of poly (ADP-ribose) polymerase. These apoptotic features were significantly attenuated by overexpression of NQO1. Levels of oxidative/nitrative damage were highly elevated by LSGE in a dose-dependent manner. However, these elevated levels were greatly reduced by overexpression of NQO1. In addition, overexpression of NQO1 attenuated the decrease in mitochondrial complex I activity caused by LSGE. Taken together, these findings suggest that overexpressed NQO1 can protect cells against LSGE-induced cytotoxicity through lowering oxidative/nitrative damage and delaying apoptosis, supporting that stimulation of NQO1 activity could be a therapeutic targets in neurodegeration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ginseng (Panax ginseng C.A. Meyer) has long been used as an herbal medicine and dietary supplementary (Helms 2004). It has a wide range of protective effects against cancer, fatigue, inflammation in many types of cells including neurons (Park et al. 2004; Szeto et al. 2011). The most famous pharmacological function of ginseng is its anti-cancer effects (Park et al. 2009). Ginseng can be extracted by aqueous and lipophilic solutions. Water-soluble ginseng extracts (WSGE) contains ginsenosides Rh2 and Rg3, which show anti-cancer activities through inducing apoptotic cell death and decreasing telomerase activity in cancer cells (Oh et al. 1999; Yun 2003; Park et al. 2009). Similarly, lipid-soluble ginseng extracts (LSGE) possess panaxydol, panaxynol, and panaxytriol with the ability to induce expression of NQO1 in Hepa1c1c7 cells (Lee et al. 2009a). These compounds can inhibit proliferation of many types of human cancer cells and protect against sarcoma-180 and NCI-H460 human lung tumors in a dose-dependent manner (Matsunaga et al. 1990; Kim et al. 2002). LSGE also can slow the growth of NCI-H460 cells following xenograft in nude mice (Lee et al. 2010a, b). Recently, we have reported that LSGE arrest the cell cycle at the stage of G0/G1 phase, induces apoptotic death of NCI-H460 human lung cancer cells (Lee et al. 2010a, b), and inhibits invasion and metastasis of B16F10 melanoma cells (Yun et al. 2015)
Normal cells are also damaged by many anti-tumor reagents including LSGE due to non-specific cytotoxicity. Cytotoxic effects are usually accompanied by alteration in energy metabolism including mitochondrial dysfunction, decrease in antioxidant defense system (Lanius et al. 1993; Apostolski et al. 1998) and increase in oxidative damage to DNA, proteins and lipids (Jenner 2003; Moreira et al. 2005). The mitochondria produce major portion of ATP and free radicals (Mattson et al. 2008; Murphy 2009). Therefore, energy metabolism and oxidative stress are directly linked to the mitochondria. Mitochondrial dysfunction and altered energy metabolism are identified during the aging process and in many degenerative diseases during the aging process (Luft and Landau 1995; Kim et al. 2008). Enhanced glycolysis and plasma membrane redox system (PMRS) as alternative mechanisms can be induced when energy supply is limited (Hyun et al. 2006b).
The PMRS can modulate redox homeostasis via maintenance of high levels of NAD+/NADH ratio and reduced states of coenzyme Q (CoQ), a key electron shuttle in the plasma membrane (PM) (Alcain et al. 1991; del Castillo-Olivares et al. 2000; Hyun et al. 2006b). NADH-quinone oxidoreducatse 1 (NQO1), one of the key detoxifying enzymes located in the PM, is up-regulated as a compensatory mechanism in response to mitochondrial dysfunction (Beyer et al. 1996; Mataix et al. 1997; Navarro et al. 1998). Because NQO1 transfers 2 electron from NAD(P)H to oxidized CoQ, NQO1 does not produce semi-quinone radicals, which can generate production of superoxide (O •−2 ) (Merker et al. 2002).
Among the PM redox enzymes, NQO1 is of particular interest because it is induced by activated Nrf2, a transcription factor involved in cytoprotection in response to oxidative and metabolic stresses (Jaiswal 2004; Johnson et al. 2008; Son et al. 2010). NQO1 expression can be induced when exposed to phytochemicals such as sulforaphane (Nioi et al. 2003). It has been reported that polyacetylents (e.g. panaxytriol) in ginseng extracts are the most potent NQO1 inducer (Lee et al. 2009b). In addition, lipophilic extracts of ginseng containing Z-ligusilide alkylate Keap1 can result in the high levels of free Nrf2 and ARE activation (Dietz et al. 2008).
Although protective effects of NQO1 were reported in many studies, it is yet to be investigated whether it can protect cells against LSGE. Previous reports have shown decreased levels of oxidative/nitrative damage by increased activity of NQO1 (Hyun et al. 2006a, 2007, 2010). These reports suggest a possible protective role of NQO1 against cytotoxicity induced by LSGE. In this study, we examine effects of NQO1 on the cellular responses in the presence of LSGE.
Materials and methods
Cell culture and transfection
SH-SY5Y human neuroblastoma cells were cultured in DMEM medium supplemented with 10 % fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 100 IU/ml penicillin (Invitrogen) and 100 μg/ml streptomycin (Invitrogen) in a humidified 5 % CO2/95 % air atmosphere in the presence or absence of LSGE. The cells were transfected with pBE8 vector containing the full-length NQO1 cDNA as described previously (Seow et al. 2004). Six different clones were selected using G-418 and their relative levels of NQO1 were established by immunoblot analysis (Hyun et al. 2012).
Preparation of lipid-soluble ginseng extracts
LSGE were prepared as described previously (Lee et al. 2010a, b). Red ginseng (first grade, 6 years old) was obtained from NH Hansamin (Jeungpyeong, South Korea). LSGE were prepared from the red ginseng powder (particle size <400 μm) using n-hexane as a solvent. Hexane was removed from LSGE using a rotary vacuum evaporator (Eyela, Tokyo, Japan) at 50 °C. LSGE were dissolved in dimethyl sulfoxide (DMSO) and further diluted with the culture media so that the final concentration of DMSO did not exceed 0.1 %.
Cell viability assay
Cell viability assay was performed by trypan blue exclusion method as described previously (Kelner et al. 1995; Lee et al. 2001). Briefly, cells were trypsinized, washed twice with PBS (Invitrogen). Trypan blue dye solution was added and the number of dye-excluding cells in triplicate dishes were counted using a haemocytometer.
Cell viability was also determined by evaluating mitochondrial activity using 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO, USA) (Kelner et al. 2000). Briefly, cell suspensions with equal numbers were transferred into a 96-well plate for 1 day. MTT solution (10 μl) was then added to each well. After incubation at 37 °C for 1 h, the absorbance of well was measured at 540 nm.
Assessment of NQO1 induction
Cells were cultured in the presence of indicated concentrations of LSGE for 24 h and lysed. In order to determine levels of NQO1 induction by LSGE, protein extracts were subjected to immunoblot analysis using anti-NQO1 antibody (1:2000, Abcam, Cambridge, MA, USA), as described previously (Hyun et al. 2010, 2012).
Examination of apoptotic features
Following exposure to the indicated concentration of LSGE for 24 h, cell membrane damage and chromatin condensation were assessed using propidium iodide (PI) and Hoechst 33258, as described previously (Negri et al. 1997; Chen et al. 2001). Cells were cultured in the presence of LSGE and harvested. The LSGE-treated cells were lysed. Cleaved form of poly (ADP-ribose) polymerase (PARP) was identified by immunoblot analysis using a PARP antibody (1:2000 dilution, Cell Signaling, Danvers, MA, USA).
Determination of levels of oxidative stress markers
Lipid peroxidation levels were assessed using 8-Isoprostane Assay Kit (OxisResearch, Portland, OR, USA), as described previously (Hyun et al. 2012). Briefly, following exposure to each mitochondrial toxins, cells were lysed and cell extracts (100 µl) were added to a 96-well plate and incubated with 100 µl horseradish peroxidase-conjugated antibody at room temperature for 1 h. After the incubation, 200 µl of substrate was added to the plate and it was incubated for 30 min. Absorbance was read at 450 nm after stopping the reaction by adding 50 µl of 3 M sulfuric acid. Carbonyl content was calculated as nmol/mg protein (Reznick and Packer 1994). Measurement of protein-bound nitrotyrosine content of isolated plasma membranes was performed using Nitrotyrosine Assay Kit (OxisResearch).
Isolation of the mitochondrial fractions and an assay of the complex I activity
Mitochondrial fractions were isolated from cells by centrifugation as described previously with minor modifications (Shim et al. 2011). Briefly, cells were washed with ice-cold PBS and homogenized in 10 mM Tris buffer (pH 7.6) with protease inhibitor cocktail (1.5 mM) (Sigma). The homogenates were centrifuged at 600g for 10 min at 4 °C. The supernatants were centrifuged again at 14,000g for 10 min at 4 °C. The resulting pellets were carefully removed and resuspended in the assay buffer (25 mM postassium phosphate, pH 7.4).
An activity of mitochondrial complex I was measured using decylubiquinone and dichloroindolphenol (DCIP), as described earlier (Janssen et al. 2007). Briefly, 10 µg of the isolated mitochondrial fractions were preincubated in the reaction buffer (70 µM decylubiquinone, 60 µM DCIP, 1 µM antimycin A, 0.35 % BSA, 25 mM potassium phosphate, pH 7.4) at 37 °C for 3 min. After adding substrates (5 mM NADH) to the mixture, absorbance was read at 600 nm for 5 min with 20 s intervals.
Statistical analysis
Statistical differences were determined by one-way analysis of variance (ANOVA). Multiple comparisons were performed with a post hoc Bonferroni t test. Statistical significance was considered when p value was less than 0.01.
Results
Effects of LSGE on the response of SH-SY5Y neuroblastoma cells
The proliferation rate of SH-SY5Y cells treated with LSGE was assessed by trypan blue exclusion and the MTT-reduction assay. LSGE significantly decreased the viability of these human neuroblastoma cells in a dose-dependent manner (Fig. 1a, b), consistent with previous results (Table 1).

Cytotoxic effects of LSGE on the viability of human neuoblstoma cells. Cells were treated with the indicated concentrations (micromolar) of LSGE for the indicated time periods (a) or for 24 h (b). Cell viability was measured by trypan blue exclusion (a) and MTT-reduction assay (b). Values are presented as mean ± SEM (n = 6). *p < 0.01 compared to the value for untransfected control cells under normal culture conditions
Levels of NQO1 in SH-SY5Y cell lines after exposure to LSGE were measured by immunoblot analysis. Levels of NQO1 expression in SH-SY5Y human neuroblastoma cell lines were increased following exposure to LSGE in a dose-dependent manner (Suppl. Fig. 1).
Overexpression of NQOQ in SH-SY5Y cells attenuate LSGE-induced cytotoxicity
It has been reported that NQO1 can be induced in response to stressed conditions as a survival mechanism to protect cells from a variety of insults (Hyun et al. 2006b). In order to investigate whether overexpressed NQO1 could protect cells against cytotoxic effects of LSGE, control cells and NQO1-transfected cells were treated with indicated concentration of LSGE. Our results revealed that overexpression of NQO1 made cells more resistant to cytotoxicity of LSGE based on trypan blue exclusion (Fig. 2a). The protection was clear during 3-days treatment with LSGE. MTT assay also showed that overexpressed NQO1 induced higher cell viability following exposure to LSGE (Fig. 2b). The higher cell viability of cells overexpressed NQO1 is also confirmed by cell morphology light microscope (Fig. 2c, d).

Cytotoxic effects of LSGE were attenuated by overexpressed NQO1. Cells (open circle control, filled circle NQO1) were exposed to the indicated concentrations (micromolar) of LSGE for 24 (A1, b), 48 (A2) or 72 h (A3). Cell viability was measured by trypan blue exclusion (a) and MTT-reduction assay (b). Images of cells under a light microscope in the absence or presence of LSGE are shown (c, d). Values are presented as mean ± SEM (n = 6). *p < 0.01 compared to the value for untransfected control cells under normal culture conditions. # p < 0.01 compared to the values of untransfected control cells under the same culture conditions. Scale bar 10 µm
Overexpression of NQO1 protects cells against apoptotic cell death caused by LSGE
In order to investigate whether overexpressed NQO1 might protect cells against apoptotic cell death caused by LSGE, apoptotic markers in control and NQO1-transfectants were assessed after exposure to different concentration of LSGE.
Cells membrane damage were assessed by PI-staining. Under normal culture conditions, approximately 5 % of control cells were identified as PI-positive cells. Similar levels of PI-positive cells were also found in NQO1 transfected cells. However, exposure of cells to LSGE significantly (p < 0.01) increased the frequency of PI-positive cells (Fig. 3a, b). Approximately 75 % of control cells were PI-positive after treatment with 0.05 % of LSGE (Fig. 3e). However, the percentage of PI-positive cells were decreased these levels were decreased (p < 0.01) to approximately 50 % following treatment with the same concentration of LSGE (0.05 %) after overexpression of NQO1 (Fig. 3e).

LSGE-induced apoptotic features were delayed by overexpressed NQO1. Cells were incubated in the presence of the indicated concentrations (micromolar) of LSGE for 24 h and then stained by propidium iodide (a, b, e) or Hoechst 33258 (c, d, f) (open markers control, closed markers NQO1). Cells treated with LSGE were lysed and analyzed by immunoblot using a PARP antibody (white right pointing triangle intact PARP, black right pointing triangle cleaved PARP) (g). Values are presented as mean ± SEM (n = 6). *p < 0.01 compared to the value for untransfected control cells under normal culture conditions. # p < 0.01 compared to the values of untransfected control cells under the same culture conditions. Apoptotic cells were indicated by yellow arrows. Scale bar 10 µm
Chromatin condensation were also determined by staining with Hoechst 33258. Incubation of cells with LSGE significantly (p < 0.01) elevated the frequency of cells with chromatin condensation (Fig. 3c, d). Approximately 65 % of control cells were stained with Hoechst 33253 after treatment with 0.05 % LSGE (Fig. 3f). However, the percentage of cells with positive Hoechst 33258 staining after overexpression of NQO1 was decreased (p < 0.01) to less than 50 % at the same treatment (0.05 % LSGE) (Fig. 3f).
PARP cleavage was examined as one of apoptotic features by immunoblot analysis using Anti-PARP antibody. Treatment of control cells with LSGE (>0.02 %) induced a cleaved form of PARP (Fig. 3g). The generation of the cleaved PARP was delayed when NQO1 was overexpressed (Fig. 3g).
Increased levels of NQO1 decrease LSGE-induced oxidative damage
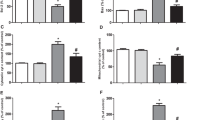
NQO1, an antioxidant enzyme, is expressed in response to oxidative stress (Hyun et al. 2006b). In order to investigate whether LSGE-induced oxidative damage could be attenuated by overexpressed NQO1, levels of oxidative/nitrative damage were measured. Lipid peroxidation was assessed as the formation of 8-isoprostane. Levels of 8-isoprostane were increased by treatment with LSGE in a dose-dependent manner (Fig. 4a). However, these levels were attenuated (p < 0.01) by overexpression of NQO1 (Fig. 4a).

Oxidative/nitrative damage and impaired mitochondrial complex I activity following addition of LSGE were attenuated by overexpressed NQO1. Cell extracts were used to assess levels of 8-isoprostane (a), protein carbonyls (b) and 3-nitrotyrosine (c) following exposure to the indicated concentrations (micromolar) of LSGE for 24 h (open markers control, closed markers NQO1). Values are presented as mean ± SEM (n = 6). *p < 0.01 compared to untransfected cells exposed to the same concentration of the toxin. Cells were cultured and then mitochondrial fractions were isolated by centrifugal fractionation. Mitochondrial complex activities were measured using NADH (c). Values are presented as mean ± SEM (n = 6). *p < 0.01 compared to the value for untransfected control cells under normal culture conditions. # p < 0.01 compared to the values of untransfected control cells under the same culture conditions
Protein oxidation was determined in the form of carbonylated proteins. Levels of protein carbonyls were significantly (p < 0.01) increased following exposure of control cells to LSGE (Fig. 4b). However, these levels of protein carbonyls were decreased (p < 0.01) in cells overexpressing NQO1 (Fig. 4b).
Protein nitration was examined in the form of protein-bound 3-NT, a biomarker of attack upon proteins by peroxinitrite and/or other reactive nitrogen species (Halliwell 1997; Greenacre and Ischiropoulos 2001). Addition of LSGE to the culture medium elevated (p < 0.01) the levels of 3-NT in control cells. However, overexpression of NQO1 significantly (p < 0.01) attenuated the levels of 3-NT (Fig. 4c).
Overexpression of NQO1 attenuate LSGE-induced mitochondrial dysfunction
Complexes I and III are a main site of reactive oxygen species (ROS) production during oxidative phosphorylation and mitochondrial functions are elevated by overexpressed NQO1 (Kim et al. 2013). In this study, the activity of mitochondrial complex I was determine using using decylubiquinone and DCIP. The activity of mitochondrial complex I was decreased by LSGE in a dose-dependent manner (Fig. 4d). However, the decrease in mitochondrial complex I activity following treatment with LSGE was attenuated by overexpression of NQO1 (Fig. 4d).
Discussion
NQO1 is a detoxifying enzyme and its expression is increased in response to energetic and oxidative stress. It has been shown that red ginseng extracts have a variety of beneficial effects despite of some cytotoxicity at high doses (Park et al. 2004; Szeto et al. 2011). In addition, NQO1 expression can be induced by some ingredients (e.g. panaxytriol) (Lee et al. 2009a) in red ginseng extracts. In this study, we identified that NQO1 could protect cells against cytotoxicity caused by LSGE in terms of cell viability (Fig. 2), apoptotic features (Fig. 3) and oxidative/nitrative damage (Fig. 4). NQO1 also protected mitochondrial function in the presence of LSGE (Fig. 4). These protective effects of NQO1 suggest that NQO1 can delay apoptotic cell death through decreasing ROS production and subsequent oxidative/nitrative damage to cells under metabolic and oxidative stress. In fact, we found that human neuroblastoma cells transfected with NQO1 were more resistant to LSGE than control cells when treated with the same concentration of LSGE.
Panaxytriol is a very good inducer of Nrf2 with cytoprotective function including anti-inflammatory and immunomodulation activities (Ng et al. 2008; Chou et al. 2011). In this study, LSGE induced NQO1 expression in neuroblastoma cell lines (Fig. 1c). Other Nrf2 inducers such as sulforaphane, hydrocytyrosol and 3H-1,2-dithiole-3-thione can induce NQO1 expression and protect cells against oxidative stress (Jia et al. 2008, Zhu et al. 2008, 2009). Previously, we have reported that overexpressed NQO1 can elevate mitochondrial functions and attenuates ROS production (Kim et al. 2013), suggesting that overexpressed NQO1 in neuroblastoma cell lines can enhance efficient mitochondrial electron transport and decrease the production of O •−2 , thus promoting cell survival (Du et al. 2006; Gonzalez-Aragon et al. 2007). However, protective roles of NQO1 can be different in other cell types or depending on metabolic/redox states of the cells. NQO1 can accelerate apoptotic cell death in non-small-cell lung cancer cells (Bey et al. 2007). We have previously reported that exposure of human non-small cell lung (NCI-H460) cancer cells to LSGE caused apoptotic cell death showing significant anticancer effects (Kang et al. 2011).
LSGE caused drastic decrease in cell viability based on trypan blue exclusion (Fig. 2a) and MTT assay (Fig. 2b). These findings suggest that LSGE can affect neuroblastoma cell lines at very early stage because mitochondrial dysfunction is an early event of cell death based on declined MTT reduction. LSGE-induced apoptosis was confirmed by two different apoptotic features (cell shrinkage and chromatin condensation) (Fig. 3a, b) in this study. Overexpressed NQO1 was involved in survival after treatment with LSGE. Previously, we have found that LSGE treatment can cause caspase activation and then PARP cleavage (Kang et al. 2011). In this study, the formation of cleaved PARP was delayed by overexpression of NQO1 (Fig. 3c), indicating that decreased levels of oxidative/nitrative damage (Fig. 4a–c) was through enhanced mitochondrial function (Fig. 4d), consistent with our previous study (Kim et al. 2013).
ROS can be produced inevitably mainly in the mitochondrial complexes. ROS is especially involved in one-electron transfer via semi-quinone radicals and transition metals (Andreeva and Crompton 1994; James et al. 2004). Semi-quinones between complex I and III are the main source of mitochondrial O •−2 production during oxidative phosphorylation (Barja 1999; James et al. 2004). ROS levels are can be elevated in the presence of mitochondrial inhibitors (e.g. rotenone, antimycin A) or under pathogenic conditions (Kim et al. 2013).
NQO1 is a redox enzyme responsible for two-electron transfer without formation of semi-quinone radicals during electron transport in the mitochondria and converts oxidized CoQ to reduced CoQ (Hyun et al. 2006b). A reduced form of CoQ can neutralize semi-quinone radicals, resulting in decreased levels of oxidative/nitrative stress. In contrast, NRH-quinone oxidoreductase 2 (NQO2) can increase production of ROS (Wang et al. 2008), suggesting the potential differences between NQO1 and NQO2. A yeast form of NQO1, NQR1, play a key role in the transition from fermentation to respiration (Jiménez-Hidalgo et al. 2009). These are consistent with our findings that cells with overexpression of NQO1 had higher resistance to oxidative/nitrative stress (Fig. 4) and enhanced mitochondrial activity (Fig. 4) following exposure to LSGE.
Taken together, our study suggests that NQO1 can be a target for therapeutic intervention and cancer treatment. This study found that overexpressed NQO1 could prevent cytotoxic effects of LSGE, suggesting that induction of NQO1 could be used as a therapeutic approach when LSGE containing a variety of bioactive compounds are used for treatment because high dose or long-term use of LSGE can be harmful. The extent of NQO1 induction by NQO1-modifying agents can be various depending on cell-types. Therefore, further work will be needed to develop cell-type specific NQO1 inducers for therapeutic intervention for cancers.
References
Alcain FJ, Buron MI, Villalba JM, Navas P (1991) Ascorbate is regenerated by HL-60 cells through the transplasmalemma redox system. Biochim Biophys Acta 1073:380–385
Andreeva L, Crompton M (1994) An ADP-sensitive cyclosporin-A-binding protein in rat liver mitochondria. Eur J Biochem 221:261–268
Apostolski S, Marinkovic Z, Nikolic A, Blagojevic D, Spasic MB, Michelson AM (1998) Glutathione peroxidase in amyotrophic lateral sclerosis: the effects of selenium supplementation. J Environ Pathol Toxicol Oncol 17:325–329
Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31:347–366
Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, Minna JD, Bornmann WG, Gao J, Boothman DA (2007) An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci USA 104:11832–11837
Beyer RE, Segura-Aguilar J, Di Bernardo S, Cavazzoni M, Fato R, Fiorentini D, Galli MC, Setti M, Landi L, Lenaz G (1996) The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc Natl Acad Sci USA 93:2528–2532
Chen HW, Jiang WS, Tzeng CR (2001) Nitric oxide as a regulator in preimplantation embryo development and apoptosis. Fertil Steril 75:1163–1171
Chou TC, Dong H, Zhang X, Lei X, Hartung J, Zhang Y, Lee JH, Wilson RM, Danishefsky SJ (2011) Multifaceted cytoprotection by synthetic polyacetylenes inspired by the ginseng-derived natural product, panaxytriol. Proc Natl Acad Sci USA 108:14336–14341
del Castillo-Olivares A, Nunez de Castro I, Medina MA (2000) Dual role of plasma membrane electron transport systems in defense. Crit Rev Biochem Mol Biol 35:197–220
Dietz BM, Liu D, Hagos GK, Yao P, Schinkovitz A, Pro SM, Deng S, Farnsworth NR, Pauli GF, van Breemen RB, Bolton JL (2008) Angelica sinensis and its alkylphthalides induce the detoxification enzyme NAD(P)H: quinone oxidoreductase 1 by alkylating Keap1. Chem Res Toxicol 21:1939–1948
Du J, Daniels DH, Asbury C, Venkataraman S, Liu J, Spitz DR, Oberley LW, Cullen JJ (2006) Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. J Biol Chem 281:37416–37426
Gonzalez-Aragon D, Ariza J, Villalba JM (2007) Dicoumarol impairs mitochondrial electron transport and pyrimidine biosynthesis in human myeloid leukemia HL-60 cells. Biochem Pharmacol 73:427–439
Greenacre SA, Ischiropoulos H (2001) Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res 34:541–581
Halliwell B (1997) What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett 411:157–160
Helms S (2004) Cancer prevention and therapeutics: Panax ginseng. Altern Med Rev 9:259–274
Hyun D-H, Emerson SS, Jo DG, Mattson MP, de Cabo R (2006a) Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci USA 103:19908–19912
Hyun D-H, Hernandez JO, Mattson MP, de Cabo R (2006b) The plasma membrane redox system in aging. Ageing Res Rev 5:209–220
Hyun D-H, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R (2007) Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem 100:1364–1374
Hyun D-H, Mughal MR, Yang H, Lee JH, Ko EJ, Hunt ND, de Cabo R, Mattson MP (2010) The plasma membrane redox system is impaired by amyloid beta-peptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp Neurol 225:423–429
Hyun D-H, Kim J, Moon C, Lim CJ, de Cabo R, Mattson MP (2012) The plasma membrane redox enzyme NQO1 sustains cellular energetics and protects human neuroblastoma cells against metabolic and proteotoxic stress. Age 34:359–370
Jaiswal AK (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36:1199–1207
James AM, Smith RA, Murphy MP (2004) Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch Biochem Biophys 423:47–56
Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–36 (discussion S36–28)
Jia Z, Zhu H, Misra HP, Li Y (2008) Potent induction of total cellular GSH and NQO1 as well as mitochondrial GSH by 3H-1,2-dithiole-3-thione in SH-SY5Y neuroblastoma cells and primary human neurons: protection against neurocytotoxicity elicited by dopamine, 6-hydroxydopamine, 4-hydroxy-2-nonenal, or hydrogen peroxide. Brain Res 1197:159–169
Jiménez-Hidalgo M, Santos-Ocaña C, Padilla S, Villalba JM, López-Lluch G, Martín- Montalvo A, Minor RK, Sinclair DA, de Cabo R, Navas P (2009) NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast. Aging Cell 8:140–151
Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, Chen PC (2008) The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci 1147:61–69
Kang MR, Kim HM, Kang JS, Lee K, Lee SD, Hyun D-H, In MJ, Park SK, Kim DC (2011) Lipid-soluble ginseng extract induces apoptosis and G0/G1 cell cycle arrest in NCI-H460 human lung cancer cells. Plant Foods Hum Nutr 66:101–106
Kelner MJ, Bagnell R, Montoya M, Estes L, Uglik SF, Cerutti P (1995) Transfection with human copper-zinc superoxide dismutase induces bidirectional alterations in other antioxidant enzymes, proteins, growth factor response, and paraquat resistance. Free Radic Biol Med 18:497–506
Kelner GS, Lee M, Clark ME, Maciejewski D, McGrath D, Rabizadeh S, Lyons T, Bredesen D, Jenner P, Maki RA (2000) The copper transport protein Atox1 promotes neuronal survival. J Biol Chem 275:580–584
Kim JY, Lee KW, Kim SH, Wee JJ, Kim YS, Lee HJ (2002) Inhibitory effect of tumor cell proliferation and induction of G2/M cell cycle arrest by panaxytriol. Planta Med 68:119–122
Kim JA, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414
Kim J, Kim SK, Kim HK, Mattson MP, Hyun D-H (2013) Mitochondrial function in human neuroblastoma cells is up-regulated and protected by NQO1, a plasma membrane redox enzyme. PLoS One 8:e69030
Lanius RA, Krieger C, Wagey R, Shaw CA (1993) Increased [35S]glutathione binding sites in spinal cords from patients with sporadic amyotrophic lateral sclerosis. Neurosci Lett 163:89–92
Lee M, Hyun D-H, Jenner P, Halliwell B (2001) Effect of overexpression of wild-type and mutant Cu/Zn-superoxide dismutases on oxidative damage and antioxidant defences: relevance to Down’s syndrome and familial amyotrophic lateral sclerosis. J Neurochem 76:957–965
Lee LS, Stephenson KK, Fahey JW, Parsons TL, Lietman PS, Andrade AS, Lei X, Yun H, Soon GH, Shen P, Danishefsky S, Flexner C (2009a) Induction of chemoprotective phase 2 enzymes by ginseng and its components. Planta Med 75:1129–1133
Lee SB, Kim CY, Lee HJ, Yun JH, Nho CW (2009b) Induction of the phase II detoxification enzyme NQO1 in hepatocarcinoma cells by lignans from the fruit of Schisandra chinensis through nuclear accumulation of Nrf2. Planta Med 75:1314–1318
Lee SD, Yoo G, Chae HJ, In MJ, Oh NS, Hwang YK, Hwang WI, Kim DC (2009c) Lipid-soluble extracts as the main source of anticancer activity in ginseng and ginseng marc. J Am Oil Chem Soc 86:1065–1071
Lee ES, Kim HM, Lee SD, Lee KS, Park SK, Lee CW, Lee K, Lee KH, Lee J, Hwang WI, In MK, Kim DC (2010a) A lipid-soluble ginseng extract inhibits human large cell lung cancer (NCI-H460) cells xenograft in vivo and the proliferation of cancer cells in vitro. J Korean Soc Appl Biol Chem 53:375–378
Lee SD, Park SK, Lee ES, Kim HM, Lee CW, Lee K, Lee KH, Kang MR, Lee KS, Lee J, Hwang WI, Kim DC (2010b) A lipid-soluble red ginseng extract inhibits the growth of human lung tumor xenografts in nude mice. J Med Food 13:1–5
Luft R, Landau BR (1995) Mitochondrial medicine. J Intern Med 238:405–421
Mataix J, Manas M, Quiles J, Battino M, Cassinello M, Lopez-Frias M, Huertas JR (1997) Coenzyme Q content depends upon oxidative stress and dietary fat unsaturation. Mol Aspects Med 18(Suppl):S129–S135
Matsunaga H, Katano M, Yamamoto H, Fujito H, Mori M, Takata K (1990) Cytotoxic activity of polyacetylene compounds in Panax ginseng CA Meyer. Chem Pharm Bull (Tokyo) 38:3480–3482
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Merker MP, Bongard RD, Kettenhofen NJ, Okamoto Y, Dawson CA (2002) Intracellular redox status affects transplasma membrane electron transport in pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 282:L36–L43
Moreira PI, Honda K, Liu Q, Santos MS, Oliveira CR, Aliev G, Nunomura A, Zhu X, Smith MA, Perry G (2005) Oxidative stress: the old enemy in Alzheimer’s disease pathophysiology. Curr Alzheimer Res 2:403–408
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Navarro F, Navas P, Burgess JR, Bello RI, de Cabo R, Arroyo A, Villalba JM (1998) Vitamin E and selenium deficiency induces expression of the ubiquinone-dependent antioxidant system at the plasma membrane. FASEB J 12:1665–1673
Negri C, Donzelli M, Bernardi R, Rossi L, Bürkle A, Scovassi AI (1997) Multiparametric staining to identify apoptotic human cells. Exp Cell Res 234:174–177
Ng F, Yun H, Lei X, Danishefsky SJ, Fahey J, Stephenson K, Flexner C, Lee L (2008) (3R, 9R, 10R)-Panaxytriol: a molecular-based nutraceutical with possible application to cancer prevention and treatment. Tetrahedron Lett 49:7178–7179
Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD (2003) Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J 374:337–348
Oh M, Choi YH, Choi S, Chung H, Kim K, Kim SI, Kim DK, Kim ND (1999) Anti-proliferating effects of ginsenoside Rh2 on MCF-7 human breast cancer cells. Int J Oncol 14:869–875
Park EK, Choo MK, Han MJ, Kim DH (2004) Ginsenoside Rh1 possesses antiallergic and anti-inflammatory activities. Int Arch Allergy Immunol 133:113–120
Park SE, Park C, Kim SH, Hossain MA, Kim MY, Chung HY, Son WS, Kim GY, Choi YH, Kim ND (2009) Korean red ginseng extract induces apoptosis and decreases telomerase activity in human leukemia cells. J Ethnopharmacol 121:304–312
Reznick AZ, Packer L (1994) Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 233:357–363
Seow HA, Penketh PG, Belcourt MF, Tomasz M, Rockwell S, Sartorelli AC (2004) Nuclear overexpression of NAD(P)H:quinone oxidoreductase 1 in Chinese hamster ovary cells increases the cytotoxicity of mitomycin C under aerobic and hypoxic conditions. J Biol Chem 279:31606–31612
Shim JH, Yoon SH, Kim KH, Han JY, Ha JY, Hyun D-H, Paek SH, Kang UJ, Zhuang X, Son JH (2011) The antioxidant Trolox helps recovery from the familial Parkinson’s disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 11:707–715
Son TG, Camandola S, Arumugam TV, Cutler RG, Telljohann RS, Mughal MR, Moore TA, Luo W, Yu QS, Johnson DA, Johnson JA, Greig NH, Mattson MP (2010) Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J Neurochem 112:1316–1326
Szeto YT, Wong JW, Wong SC, Pak SC, Benzie IF (2011) DNA protective effect of ginseng and the antagonistic effect of Chinese turnip: a preliminary study. Plant Foods Hum Nutr 66:97–100
Wang W, Le WD, Pan T, Stringer JL, Jaiswal AK (2008) Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased susceptibility to Parkinson’s disease. J Gerontol A Biol Sci Med Sci 63:127–134
Yun TK (2003) Experimental and epidemiological evidence on non-organ specific cancer preventive effect of Korean ginseng and identification of active compounds. Mutat Res 523–524:63–74
Yun J, Kim BG, Kang JS, Park SK, Lee K, Hyun D-H, Kim HM, In MJ, Kim DC (2015) Lipid-soluble ginseng extract inhibits invasion and metastasis of B16F10 melanoma cells. J Med Food 18:102–108
Zhu H, Jia Z, Strobl JS, Ehrich M, Misra HP, Li Y (2008) Potent induction of total cellular and mitochondrial antioxidants and phase 2 enzymes by cruciferous sulforaphane in rat aortic smooth muscle cells: cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol 8:115–125
Zhu H, Jia Z, Zhou K, Misra HP, Santo A, Gabrielson KL, Li Y (2009) Cruciferous dithiolethione-mediated coordinated induction of total cellular and mitochondrial antioxidants and phase 2 enzymes in human primary cardiomyocytes: cytoprotection against oxidative/electrophilic stress and doxorubicin toxicity. Exp Biol Med (Maywood) 234:418–429
Acknowledgments
This study was supported by Grants (2010-0003064 and 2012R1A1A2039477) of the Basic Science Research Prcogram through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology, South Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors state have no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, HK., Son, T.G., Jo, DG. et al. Cytotoxicity of lipid-soluble ginseng extracts is attenuated by plasma membrane redox enzyme NQO1 through maintaining redox homeostasis and delaying apoptosis in human neuroblastoma cells. Arch. Pharm. Res. 39, 1339–1348 (2016). https://doi.org/10.1007/s12272-016-0817-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0817-6