
Abstract
Histone deacetylase (HDAC) inhibitors are emerging as promising anticancer drugs. Because aberrant activity and expression of HDACs have been implicated in various cancer types, a wide range of HDAC inhibitors are being investigated as anticancer agents. Furthermore, due to the demonstrable anticancer activity in both in vitro and in vivo studies, numerous HDAC inhibitors have undergone a rapid phase of clinical development in various cancer types, either as a monotherapy or in combination with other anticancer agents. Although preclinical trials show that HDAC inhibitors have a variety of biological effects across multiple pathways, including regulation of gene expression, inducing apoptosis and cell cycle arrest, inhibiting angiogenesis, and regulation of DNA damage and repair, the mechanism by which the clinical activity is mediated remains unclear. Understanding the mechanisms of anticancer activity of HDAC inhibitors is essential not only for rational drug design for targeted therapies, but for the design of optimized clinical protocols. This paper describes the links between HDACs and cancer, and the underlying mechanisms of action of HDAC inhibitors against hematological malignancies and solid tumors. Further, this review presents the clinical outcomes of vorinostat, romidepsin, and belinostat, which are approved by the United States Food and Drug Administration for the treatment of lymphomas.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Histone deacetylase (HDAC) inhibitors are a class of compounds that regulate acetylation states of histone proteins and other non-histone proteins by inhibiting the activity of HDAC. Because of the demonstrable antitumor activity in both in vitro and in vivo studies, HDAC inhibitors have undergone a rapid phase of clinical development in a wide range of cancer types, either as a monotherapy or in combination with other anticancer agents (Younes et al. 2014; Seo 2012; El-Khoury et al. 2014; Ogura et al. 2014).
To date, three HDAC inhibitors are approved by the United States Food and Drug Administration (U.S. FDA) based on the good clinical activity and favorable toxicity profile: vorinostat, also called suberoylanilide hydroxamic acid (Zolinza®, Merck and Co., Inc.) for treating cutaneous T cell lymphoma (CTCL) in October 2006; romidepsin, also known as depsipeptide and FK228 (Istodax®, Gloucester Pharmaceuticals—a subsidiary of Celgene Corp) for the treatment of CTCL in November 2009 and for peripheral T-cell lymphoma (PTCL) in May 2011; belinostat, also called PXD101 (Beleodaq®, Spectrum Pharms, Inc.) for the treatment of patients with relapsed or refractory PTCL in July 2014 (Drugs@FDA).
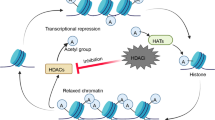
HDACs play a critical role towards the transcription regulation, removing the acetyl group from the ε-amino groups of the lysine residues on histones. While acetylation correlates with nucleosome remodeling and is generally associated with elevated gene transcription, deacetylation of histone tails induces transcriptional repression through chromatin condensation. This may be explained by the fact that acetylation neutralizes the positive charge of lysine residues and leads to relaxation of the chromatin structure, facilitating the accessibility of a variety of transcription factors to DNA (Norton et al. 1989; Grunstein 1997; Ropero and Esteller 2007). Besides regulating histone modification, HDACs also regulate the post-translational acetylation status of many non-histone proteins. HDAC-mediated deacetylation alters the transcriptional activity of nuclear transcription factors, E2F1, GATA1, GATA2, MyoD, nuclear factor κB (NF-κB), p53, runt-related transcription factor 3 (RUNX3), and Ying Yang 1 (YY1). In addition, proteins that regulate cellular cytoskeleton and protein stabilization are also being regulated by HDAC. HDAC6 deacetylates α-tubulin and aids in the remodeling of the synapse, which regulates the organization of adhesion and signaling molecules. Moreover, HDAC6 has been shown to regulate acetylation of Hsp90 that plays a major role in the proper wrapping and stabilization of several oncoproteins. It has also been shown that HDAC6 interacts with short heterodimer partner (SHP) and contributes to the intrinsic transrepressive activity of SHP (Table 1). These findings imply that antitumor activity of HDAC inhibitors includes effects on non-histone proteins as well as effects on chromatin modification. Through hyperacetylation of histone and non-histone targets, HDAC inhibitors can induce diverse biological effects, including regulation of gene expression, inducing apoptosis and cell cycle arrest, inhibiting angiogenesis, and regulation of DNA damage and repair pathway (Richon et al. 2000; Kim et al. 2014; Konstantinopoulos et al. 2014; Zhou et al. 2014). Therefore, HDAC inhibitors have great potential as anticancer drugs.
The present review describes the underlying mechanisms of anticancer activity of HDAC inhibitors, focusing on the agents that could have implications for the future use in cancer treatment.
The classification of HDACs
In humans, 18 HDAC enzymes have been identified and classified into four classes according to their homology to yeast HDACs (yHdas), their subcellular location, their tissue specificity and their enzymatic activity. Among them, the 11 enzymes, which require Zn2+ as a cofactor for their deacetylase activity, belong to classes I, II, and IV whereas the sirtuins (SIRT1-7) belong to class III HDACs and they are Zn2+-independent (de Ruijter et al. 2003; Gregoretti et al. 2004). The class III HDACs will not be discussed in this paper due to two reasons. Firstly, the role of some sirtuins in tumorigenesis is still controversial. For example, SIRT1 is expressed at a higher level in cancerous cells and promotes oncogenesis through deacetylation of lysine 382 in Burkitt lymphoma cells (Heltweg et al. 2006). However, in a colon cancer mouse model, increased SIRT1 expression suppressed cell proliferation and tumor formation (Firestein et al. 2008). The second reason is that a wide range of structures have been identified to be able to inhibit the activity of classes I, II, and IV HDACs, but not the NAD+-dependent class III enzymes (Marks et al. 2001; Johnstone 2002).
The class I HDACs are homologous to yeast (Saccharomyces cerevisiae) transcriptional regulator RPD3 and include HDAC1, -2, -3, and -8. They are usually located in the nucleus and ubiquitously expressed in various human tissues (Bertrand 2010). The class II HDACs have homology to yeast HDAC 1 (yHda1), and are further subdivided into two subclasses, IIa and IIb, based on sequence homology and domain organization. The class IIa HDACs, HDAC4, -5, -7, and -9, contain a highly conserved C-terminal deacetylase catalytic domain (~420 amino acids) homologous to yHda1 and share an N-terminal domain (~450–600 amino acids) with no similarity to HDACs in other classes. The N-terminal domains mediate interactions with myocyte enhancer factor 2 family of transcription factors, transcriptional corepressor C-terminal binding protein, and others. The class IIa HDACs are shuttled between the cytoplasm and the nucleus, and their expression is tissue-specific. The class IIb HDACs, HDAC6 and -10, are characterized by the presence of two catalytic HDAC domains arranged in tandem. The class IIb HDACs are mainly located in cytoplasm and are expressed in a restricted number of cell types (Bertos et al. 2001; Fischle et al. 2001; Verdin et al. 2003). HDAC11 is the sole member of class IV and resides in the nucleus; however, in activity assays, HDAC11 co-precipitates with the cytoplasmically localized HDAC6 (Bertos et al. 2001; Gao et al. 2002).
HDACs and cancer
HDACs regulate expression and activity of numerous proteins involved in both cancer initiation and cancer progression. Recruitment of HDACs to promoters causes a closed chromatin conformation that is inaccessible to transcription machinery or its mediators, resulting in transcriptional repression. Alterations in histone acetylation status have been involved in the development of cancer. In particular, a global loss of monoacetylation of lysine 16 and trimethylation of lysine 20 on histone H4 (H4K16ac and H4K20me3) has been found to be a common hallmark of human cancer (Fraga et al. 2005).
Aberrant expression and mutation of HDACs have been implicated in a variety of tumor types, making them attractive targets for anticancer drugs and therapies (Table 2). Overexpression of individual HDACs has been found to be significantly associated with poor disease-free, event-free, and overall survival (Weichert et al. 2008c; Oehme et al. 2009; Milde et al. 2010; Moreno et al. 2010; Minamiya et al. 2011; Quint et al. 2011). Furthermore, overexpressed HDACs have been correlated with aggressiveness, invasion and migration of cancer and have been found to be a poor prognosis indicator that is independent from other factors such as tumor type, age, sex, or comorbid condition (Song et al. 2005; Weichert et al. 2008a, b; Hayashi et al. 2010; Park et al. 2011; Wang et al. 2011; Müller et al. 2013; Li et al. 2014).
However, overexpression of HDACs is not always a poor prognostic marker. Interestingly, overexpressed HDAC1 and -6 have been associated with favorable outcome in Hodgkin’s lymphoma and in chronic lymphocytic leukemia, respectively (Marquard et al. 2008; Adams et al. 2010). What’s more, in non-small cell lung cancer, lower level of gene expression has been related with poorer prognosis (Osada et al. 2004). On top of that, overexpression of HDACs can serve as a molecular biomarker: up-regulation of HDAC2 involves in early events of colorectal carcinogenesis (Stypula-Cyrus et al. 2013) while high HDAC7 expression has been able to discriminate pancreatic adenocarcinomas from other pancreatic tumors (Ouaïssi et al. 2008). Overexpression of HDAC1 and -2 has been observed in many cancer types and in some cases it is significantly related to tumor cell growth with corresponding decrease in p21 expression (Halkidou et al. 2004; Xie et al. 2012; Jung et al. 2012). Indeed, the cyclin-dependent kinase (CDK) inhibitor p21 is a crucial target for HDAC inhibitors. In many cancer cell lines, HDAC inhibitors causes the transcriptional upregulation of this antiproliferative gene p21 and subsequently block the cyclin/CDK complexes, leading to cell G1 cycle arrest (Sandor et al. 2000). Moreover, in non-small cell lung cancer, HDAC2 inactivation resulted in regression of tumor cell growth via inductions of p53 and Bax expression and simultaneously suppressed Bcl-2 expression (Jung et al. 2012). Mutations of HDACs also contribute to tumorigenesis: the presence of the HDAC2 frame shift mutation renders cancer cells resistant to the anti-proliferative and proapoptotic effects of HDAC inhibitors in patients with hereditary nonpolyposis colorectal cancer (Ropero et al. 2006). Recently, overexpression of HDAC5 has been demonstrated in human hepatocellular carcinoma cell lines, which promotes tumor cell proliferation through up-regulation of Six1 (Feng et al. 2014).
In addition to altered expression of HDACs, the aberrant recruitment of HDACs to certain target genes through binding to oncogenic fusion proteins has been proposed as an important mechanism of tumorigenesis. For example, promyelocytic leukemia–retinoic acid receptor α (PML–RARα) and PML zinc finger (PLZF)–RARα, mutant forms of RARα produced by chromosomal translocations with either PML gene or PLZF gene, gain corepressor activity upon aberrant recruitment of HDAC complexes containing nuclear receptor corepressor (NCoR), and obtain subsequent leukemogenic potential upon aberrant recruitment of mSin3a and silencing mediator for retinoid and thyroid receptors (Hörlein et al. 1995; Lin et al. 1998; Kouzarides 1999). Similar phenomena have been described for acute myeloid leukemia 1-eight twenty-one (AML1–ETO), a fusion of the AML1 and ETO proteins (Gelmetti et al. 1998). Atsumi et al. (2006) have demonstrated that, in acute PML cells, HDAC3 is recruited to target promoters by PML–RARα, a component of the NCoR repressor complex, to repress transcription whereas Amann et al. (2001) have shown that the AML1–ETO fusion protein recruits HDAC1, -2, and -3 via ETO to repress transcription of leukemic cells.
Mechanisms of action of HDAC inhibitors
A large number of structurally diverse HDAC inhibitors have been identified from natural sources and developed synthetically, and many of them are undergoing clinical trials (Fig. 1). Numerous studies have shown that certain HDAC inhibitors selectively inhibit different HDACs (Table 3). Furthermore, the finding that normal cells are relatively resistant to HDAC inhibitor-induced cell death compared to tumor cells (Ungerstedt et al. 2005) is fundamental to the success in clinical application of HDAC inhibitors.

Four main chemical structures of HDAC inhibitors currently undergoing clinical trials
By inducing acetylation of histones and non-histone proteins, HDAC inhibitors are able to elicit a wide range of biological effects (Fig. 2).

The molecular targets of HDACs and the biological effects of HDAC inhibition. HDAC inhibitors induce acetylation of histones and non-histone proteins, leading to a wide range of biological effects, including regulation of gene expression, including apoptosis and cell cycle arrest, inhibiting angiogenesis, and regulation of DNA damage and repair pathway. AR, androgen receptor; ERα, estrogen receptor α; NF-kB, nuclear factor-kB; PLAG1, pleomorphic adenoma gene 1; PLAGL2, PLAG-like 2; RUNX3, runt-related transcription factor 3; SHP, short heterodimer partner; SMC3, structural maintenance of chromosomes 3; STAT3, signal transducer and activator of transcription 3; YY1, Ying Yang 1
Selective alterations of gene expression
There is accumulating evidence that anticancer activity of HDAC inhibitors is linked to their ability to regulate the expression of specific proliferative and/or apoptotic genes. After treatment with SAHA, in bladder carcinoma cells, a significant increase in p21 mRNA and concurrent accumulation of acetylated histones H3 and H4 were observed independently of p53. In addition, SAHA caused a marked decrease in HDAC1 in the protein complex bound to the p21 promoter in multiple myeloma (MM) cells. However, the expression of p27 gene that is actively expressed in MM cells was not altered (Richon et al. 2000; Gui et al. 2004). Further evidence of the effects of HDAC inhibitors on gene expression has been demonstrated. In breast cancer cells, FK228 caused downregulation of cyclin D1 and upregulation of CDK inhibitor p21, resulting in dephosphorylation of the tumor suppressor retinoblastoma (Rb) and growth arrest in the early G1 phase (Sandor et al. 2000). Moreover, by SAHA and FK228, multiple genes within the Myc, transforming growth factor-β1, cyclin/CDK, tumor-necrosis factor (TNF), Bcl-2, and caspase pathways were up- or down-regulated, inducing apoptosis and inhibiting cellular proliferation. Although, a substantial number of genes were regulated in common by both SAHA and FK228, some genes, including apoptosis-regulatory BNip family members BNip1 and BNip3L, and NF-κB-pathway genes IκB were differentially regulated (Peart et al. 2005).
The findings described above explain, at least in part, the selective effect of HDAC inhibitors in altering gene expression.
HDAC inhibitor-induced antitumor pathways
Activation of apoptotic pathways
The apoptotic process is accompanied by major changes in chromatin structure and gene expression. HDAC inhibitors induce apoptosis via both transcription-dependent and transcription-independent mechanisms in solid and hematological malignancies (Bolden et al. 2006). There are numerous studies reporting HDAC inhibitor-induced apoptosis via both the extrinsic and intrinsic pathways.
The extrinsic pathway of apoptosis is initiated upon binding of death ligands, the TNF superfamily receptors, including TNF-related apoptosis-inducing ligand (TRAIL), TNF-α, and Fas ligand (FasL), to their cognate death receptors, resulting in activation of caspase-8 and -10. The activated caspase-8 and -10 subsequently activate proteases caspase-3, -6, and -7, leading to apoptotic cell death (Ashkenazi 2002; Bolden et al. 2006).
By valproic acid, the TNF family ligands and receptors, including TRAIL, DR5, Fas and FasL, are transcriptionally activated, leading to initiation of the extrinsic apoptosis pathways in leukemic cells, but not in normal cells. That effect correlates with activation of caspase-8 and -3 (Insinga et al. 2005). Treatment of leukemia cells with FK228 also caused upregulation of TNF-α via hyperacetylation of histones H3 and H4 in its promoter region and induced activation of caspase-8 and -10, resulting in apoptotic cell death (Sutheesophon et al. 2005).
On the other hand, the intrinsic pathway, involving the mitochondria, is activated in response to lethal stimuli from inside the cell, such as DNA damage, oxidative stress, hypoxia, or chemotherapeutic drugs. Activation of mitochondrial apoptotic pathway causes release of pro-apoptotic proteins, including cytochrome c, from the inter-membrane space into the cytosol. Cytochrome c can then bind Apaf-1 forming the apoptosome and activating caspase-9. Once activated, caspase-9 cleaves and activates the same set of caspases that are activated through the extrinsic pathway by caspase-8 and -10 (Ashkenazi 2002; Burz et al. 2009). In response to suberoyl bis-hydroxamic acid, the expression of pro-apoptotic proteins including Bim, Bak, Bax, and caspase-3 increased while the expression of anti-apoptotic proteins including Bcl-2, Bcl-XL, Mcl-1, and X-linked inhibitor of apoptosis decreased in melanoma cells (Zhang et al. 2004).
Recently, we have demonstrated that MHY218, a hydroxamic acid derivative, induces apoptosis in colon cancer cells. By MHY218, Bax was markedly upregulated while Bcl-2 was downregulated, leading to a significant increase in Bax/Bcl-2 ratio in a concentration-dependent manner. In addition, the activity of caspase-3, -8 and -9 was significantly increased, suggesting MHY218 induces apoptosis via both the internal and external pathway in colon cancer cells (Kim et al. 2014).
Further evidence demonstrates that HDAC inhibitors alter the factors that mediate or regulate the intrinsic apoptosis pathway. SAHA caused apoptosis by promoting recruitment of E2F1 to the Bim promoter and inducing upregulation of Bim in colon cancer cells lacking p53 (Zhao et al. 2005). In addition, by SAHA, mitochondrial disruption was achieved by the cleavage of the BH3-only pro-apoptotic Bcl-2 family member Bid in leukemia cells, which was not blocked by caspase inhibitors or the overexpression of Bcl-2 (Ruefli et al. 2001). Moreover, SAHA treatment increased levels of Noxa and Puma in leukemia cells (Pérez-Perarnau et al. 2011).
In addition to SAHA, FK228 induced Bmf expression, concomitant with hyperacetylation of histones H3 and H4 at Bmf promoter region in squamous carcinoma cells (Zhang et al. 2006) while sodium butyrate increased the expression of the Bad protein in glioma cells with no changes in the levels of Bcl-2, Bcl-XL, Bax, and Fas (Sawa et al. 2001).
HDAC inhibitor-mediated apoptosis can be cell-type-dependent. Furthermore, it seems that different effects of HDAC inhibitors in the same cell type may be attributed to the structural features of diverse HDAC inhibitors.
Induction of cell cycle arrest
HDAC inhibitors have been found to induce cell cycle arrest at G1 via upregulation of the CDK inhibitor p21 (Richon et al. 1996; Qiu et al. 2000). In addition, HDAC inhibitors reduce CDK activity through downregulation of cyclins, causing dephosphorylation of Rb and subsequently inhibiting E2F activities in the transcription of genes for G1 progression and G1/S transition (Rosato and Grant 2005; Zhao et al. 2005). In our study, MHY218, a hydroxamic acid derivative, induced G2/M phase arrest by p53-independent upregulation of p21 in colon cancer cells (Kim et al. 2014). SAHA caused predominantly G1 arrest at low concentration while at higher concentrations, both G1 and G2/M arrests were induced (Richon et al. 2000). The cytostatic activity of HDAC inhibitors at low doses is not restricted to tumour cells, since G1 arrest is also observed in normal cells. At higher doses, HDAC inhibitors are selectively cytotoxic, killing a wide range of cancer cells and transformed cells but not normal cells (Qiu et al. 2000; Burgess et al. 2004).
Although, HDAC inhibitor-induced arrest is largely associated with induction of p21, several reports have suggested the existence of a p21-independent pathway of growth arrest by HDAC inhibitors. Trichostatin A (TSA) activates the p15Ink4b gene, a member of INK4 family proteins, and induces cell growth inhibition of colon cancer cells lacking p21 (Hitomi et al. 2003). Recently it has been demonstrated that HDAC inhibitors can interfere with the interaction between HDACs and Aurora kinases involved in alteration of the G2–M cell cycle transition. In renal cancer cells, LBH589-mediated down-regulation of Aurora A and B induced G2/M cell cycle arrest through inhibition of HDAC3 and -6 (Cha et al. 2009). In the case of TSA, it increased levels of acetylated histones H3 and H4 in hepatoma cells, leading to G0/G1 phase arrest (Yamashita et al. 2003). What’s more, TSA increased levels of p21 gene and reduced levels of CyclinB1, Plk1, and Survivin, resulting in delay at the G2/M transition (Noh et al. 2009).
Collectively, HDAC inhibitors can affect cell cycle by interacting with cell cycle regulators, resulting in cell cycle arrest at certain phases.
Inhibition of angiogenesis
There is growing evidence that HDAC inhibitors can target tumor angiogenesis. The anti-angiogenic effect of HDAC inhibitors is associated with suppression of pro-angiogenic factors, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor, hypoxia-inducible factor-1α (HIF-1α), chemokine (C-X-C motif) receptor 4 (CXCR4), angiopoietin, tunica intima endothelial kinase 2, and endothelial nitric oxide synthase (eNOS).
HIF-1α plays a key role in the cellular adaptations to hypoxic microenvironment, which is critical for survival of tumor cells. Repression of HIF-1α activity by HDAC inhibitors has been reported by various mechanisms. The observation that TSA inhibits hypoxia-induced angiogenesis through upregulation of p53 and von Hippel–Lindau, and concurrent suppression of HIF-1α and VEGF suggests that HDAC modulation can be closely involved in hypoxia-induced angiogenesis (Kim et al. 2001). In addition, anti-angiogenic activity of FK228 has been demonstrated in Lewis lung carcinoma model. FK228 effectively inhibited the DNA binding activity of HIF-1α and the expression of VEGF mRNA under hypoxia (Lee et al. 2003). The reduced HIF-1α DNA-binding activity and decreased level of VEGF protein were also observed with butyrate. In colon cancer cells, HIF-1α nuclear sequestration was repressed through inhibition of nuclear translocation, which could be responsible for decreased VEGF expression and anti-angiogenic effects (Zgouras et al. 2003).
Furthermore, the activity of HIF-1α is regulated upon binding to the transcriptional co-activator cAMP-response element-binding protein-binding protein (CBP)/p300. HIF-1αCAD, the carboxyl-terminal transactivation domain of HIF-1α, provides the major transactivation activity. TSA induced hyperacetylation of p300 and repressed the HIF-1αp300 complex independently of direct acetylation of HIF-α (Fath et al. 2006).
Besides, HDAC inhibitors induce hyperacetylation of chaperone protein Hsp90 via inhibition of HDAC6, leading to increased affinity to HIF-1α. As a result, HIF-1α disrupts Hsp90 chaperone function and exposes HIF-1α to proteasomal degradation by Hsp70 (Kong et al. 2006).
Many studies have shown that the classes I and II HDACs are associated with HIF-1α activity, which are inhibited by different HDAC inhibitors: inhibition of HDAC1 and -3 by butyrate and valproic acid; inhibition of HDAC4 and -6 by LBH589 and valproic acid; inhibition of HDAC9 by SAHA (Qian et al. 2006; Kim et al. 2007; Hutt et al. 2014). HDAC inhibitors that inhibit HDAC7 may also contribute to anti-angiogenesis. Under hypoxic conditions, HDAC7 moves into the nucleus and increases transcriptional activity of HIF-1α through the formation of a complex with HIF-1α, HDAC7, and p300. HDAC inhibitors reduce the HIF-1α activity by inhibiting HDAC7 (Kato et al. 2004).
Taken together, HDAC inhibitors repress neovascularization by inhibiting positive factors of angiogenesis or altering angiogenesis signaling pathway.
Induction of DNA damage and inhibition of DNA repair
Changes in chromatin structure induced by HDAC inhibitors directly activate the DNA-damage pathway despite the fact that HDAC inhibitors alone do not induce double-strand breaks (DSBs; Bakkenist and Kastan 2003).
It has been well established that HDAC inhibitors can synergize with ionizing radiation (IR) and DNA-damaging agents to cause delay in tumor growth. The number of studies reporting the additive effect has grown exponentially. The combination of radiation and LBH589, SAHA, and butyrate increased the duration of γ-H2AX foci in irradiated cells (Geng et al. 2006; Munshi et al. 2005; Koprinarova et al. 2011). This finding suggests that HDAC inhibitors suppress DSB repair and/or render DNA more susceptible to IR-induced damage.
In addition to their action on chromatin structure during DSB repair, HDAC inhibitors have been shown to affect the expression of DNA repair proteins. Treatments with SAHA and MS-275 led to hyperacetylation of Ku70, a key non-homologous end joining component, concomitant with reduced DNA-binding affinity (Chen et al. 2007). HDAC inhibitors can also affect the expression of genes encoding homologous recombination (HR) components. SAHA induced the coordinated down-regulation of HR pathway genes, including RAD51 and BRCA1 in ovarian cancer cells (Konstantinopoulos et al. 2014).
A number of studies have reported that after treatment with HDAC inhibitors, reactive oxygen species (ROS) are accumulated, leading to DNA damage and oxidative stress. HDAC inhibition by SAHA and MS-275 caused accumulation of ROS and increased sensitivity to cell death induced by those agents in transformed but not normal cells (Ungerstedt et al. 2005).
HDAC inhibitors and clinical outcomes
HDAC inhibitors are undergoing extensive clinical evaluation as single agent or in combination with other agents. Among them, vorinostat, romidepsin, and belinostat have received approval from the U.S. FDA for the treatment of lymphoma.
Vorinostat
Vorinostat, also known as SAHA, has received approval from the U.S. FDA for the treatment of CTCL in patients with progressive, persistent, or recurrent disease on or following two systemic therapies. The major trial supporting approval was a phase II trial that recruited 74 patients with at least stage IB CTCL (61 had stage IIB or higher) who had failed two systemic therapies. The patients received 400 mg of oral vorinostat once daily as a single agent. The objective response rate (ORR) was 29.7 and 32 % of patients had pruritus relief. The common drug-related adverse events were diarrhea (49 %), fatigue (46 %), nausea (43 %), and anorexia (26 %) and most were grade 2 or lower. The 400 mg dose of vorinostat was generally well-tolerated (Olsen et al. 2007; Mann et al. 2007).
Vorinostat has also been investigated for other hematological malignancies and solid tumors. Recently, two phase II studies have reported favorable responses in relapsed/refractory indolent follicular lymphoma with acceptable safety profiles (Kirschbaum et al. 2011; Ogura et al. 2014). Despite the demonstrated effect in lymphomas, unfortunately, only a moderate effect was observed in solid tumors: squamous cell carcinoma of the head and neck (Blumenschein Jr et al. 2008), breast, colorectal, and non-small-cell lung cancer (Luu et al. 2008; Vansteenkiste et al. 2008; Traynor et al. 2009), ovarian and peritoneal (Modesitt et al. 2008), and prostate (Bradley et al. 2009). Although in the study by Bradley et al. (2009), 41 % of the patients discontinued therapy because of grade 2/3 toxicity including fatigue, nausea, and anorexia, oral vorinostat was generally well-tolerated at a dose of 400 mg daily.
Romidepsin
Romidepsin, also known as FK228 and depsipeptide, serves as a prodrug. Upon reduction of its disulfide bond, one of the sulfhydryl groups interacts with the zinc in the active site of the HDACs, preventing access of substrate. Romidepsin possesses stronger activity against HDAC1 and -2 than against HDAC4 and -6 (Furumai et al. 2002). In 2009, the U.S. FDA has granted approval to romidepsin for treating CTCL patients who have failed at least one prior systemic therapy based on the efficacy and safety evaluated in two phase II trials (U.S. FDA Drugs. Romidepsin; Piekarz et al. 2009; Whittaker et al. 2010). In those two studies, 71 and 96 patients with CTCL were included and treated with intravenous romidepsin at a dose of 14 mg/m2/day on days 1, 8, and 15 of a 28-day cycle. The ORRs were 35 and 34 % in each study and the complete response (CR) rate was 6 % in both studies. The most common drug related adverse events were nausea and fatigue being grade 2 or 3 in both studies. The serious adverse events reported in >2 % of the patients were leukopenia, lymphopenia, and granulocytopenia in the study by Piekarz et al. (2009), and nausea, asthenic conditions including fatigue, and anemia in the study by Whittaker et al. (2010).
Romidepsin was also approved by the U.S. FDA in 2011 for the treatment of relapsed or refractory PTCL. The approval was based on the results of a multinational phase II trial conducted in 130 patients who were refractory to at least one prior systemic therapy or for whom at least one prior systemic therapy failed. The patients received intravenous romidepsin at a dose of 14 mg/m2/day on days 1, 8, and 15 of a 28-day cycle. The ORR was 25 %, including 15 % with CR. The most common grade ≥3 drug-related adverse events were thrombocytopenia (23 %), neutropenia (18 %), and infections (6 %, Coiffier et al. 2012).
A series of phase II trials of romidepsin have also been conducted in patients with solid tumors, however, the single agent activity of romidepsin is disappointingly very low: renal cancer (Stadler et al. 2006), colorectal cancer (Whitehead et al. 2009), castration-resistant prostate cancer (Molife et al. 2010), small-cell lung cancer (Otterson et al. 2010), anaplastic glioma and glioblastoma multiforme (Iwamoto et al. 2011), and squamous cell carcinoma of the head and neck (Haigentz Jr et al. 2012). When romidepsin was administered at the dose of 14 mg/m2/day, the most common adverse events were GI disturbances, hematologic abnormalities, asthenic conditions, and infections.
Belinostat
Belinostat, also known as PXD101, is a hydroxamate pan-HDAC inhibitor that was approved by the U.S. FDA for the treatment of patients with relapsed or refractory PTCL. The approval was based on the result of the phase II trial conducted in 120 patients with PTCL that was refractory or had relapsed after prior treatment, including PTCL patients with baseline platelets <100,000/ml. Belinostat was administered by intravenous infusion at a dose of 1,000 mg/m2 once daily on days 1–5 of a 21-day cycle. The ORR was 25.8 % and median progression-free survival was 1.6 months. The most common grade ≥3 adverse events were thrombocytopenia (15 %), neutropenia (13 %), leukopenia (13 %), and anemia (12 %, ClinicalTrials.gov. BELIEF study; U.S. FDA Drugs. Belinostat).
Belinostat has also been investigated for other hematological malignancies and solid tumors. Recently, a phase II trial has reported favorable responses in recurrent/refractory PTCL and CTCL with acceptable safety profiles (Foss et al. 2014). Despite the demonstrated effect in lymphomas, unfortunately, only a moderate effect was observed in solid tumors: malignant pleural mesothelioma (Ramalingam et al. 2009), ovarian cancer (Mackay et al. 2010), and thymoma and thymic carcinoma (Giaccone et al. 2011).
The most common adverse events were anemia, nausea, QTc prolongation, and thrombocytopenia. Most of the toxicities were grades 2–3. In general, belinostat was well-tolerated at a dose of 1,000 mg/m2/day.
Safety issues with the use of HDAC inhibitors
Although toxicities are favorable and largely manageable, in some instances cardiotoxicities as well as hematologic and gastrointestinal adverse effects can be dose limiting. The most common dose-limiting toxicities related with vorinostat, romidepsin, and belinostat were constitutional and gastrointestinal effects, including anorexia, asthenia, diarrhea, fatigue, nausea, and vomiting, and hematologic effects, such as anemia, leucopenia, lymphopenia, neutropenia, and thrombocytopenia (Duvic et al. 2006; Garcia-Manero et al. 2008; Vansteenkiste et al. 2008; Steele et al. 2008; Lassen et al. 2010; Mwakwari et al. 2010).
As various HDAC inhibitors are being studied in clinical trials against cancers, accumulating evidence shows that use of HDAC inhibitors is associated with cardiotoxicity such as T-wave flattening, ST segment depression and QTc-prolongation (Piekarz et al. 2006). QT interval prolongation has been to date the most severe cardiac event in patients treated with HDAC inhibitors due to the risks for potentially life-threatening arrhythmia (Wolbrette 2004; Straus et al. 2006). HDAC inhibitors have been variably associated with QT prolongation. Romidepsin, previously called as FK228 and depsipeptide, has been associated with relatively frequent electrocardiogram (ECG) changes and QT prolongation, and rare sudden cardiac death (Bates et al. 2006; Shah et al. 2006; Mwakwari et al. 2010). The use of belinostat (PXD101) was also associated with QTc prolongation in patients treated with it (Lassen et al. 2010; Cashen et al. 2012). In case of vorinostat (SAHA), no QTc prolongation was reported in clinical trials (Kelly et al. 2005; Duvic et al. 2006; Bradley et al. 2009). However, recently, one case of torsade de pointes has been reported, drawing attention to the need for periodic monitoring of ECGs in patients using vorinostat (Lynch Jr et al. 2012).
Interference on human ether-a-go–go-related gene potassium ion (HERG K+) channels seems to be a common mechanism for these drugs (Strevel et al. 2007; Bagnes et al. 2010). The plausible hypothesis is that these HDAC inhibitors uniquely interact with HERG K+ channel, resulting in QTc prolongation. The activation of the HERG K+ channel leads to ventricular repolarization, thus blocking of this channel may result in QTc prolongation (Curran et al. 1995). In addition, mutations of HERG are presumed to be linked to QTc prolongation. In fact, increasing evidence suggests that QTc prolongation associated with HDAC inhibitors may be the result of altered gene expression and probably inhibition of specific HDAC isoforms (Montgomery et al. 2007). Therefore, changes in HERG expression or those of the coregulators of HERG activity may serve as another mechanism of QTc prolongation.
Collectively, although ECG abnormalities observed with the administration of HDAC inhibitors have not been associated with myocardial damage or altered left ventricular ejection fraction, the potential effect of heart rate-corrected QT interval prolongation remains under study.
Conclusion
HDAC inhibitors have shown good anticancer activity both in preclinical and clinical trials, with relatively little effect on normal tissues and favorable toxicity profile. Although preclinical trials show that HDAC inhibitors have a variety of biological effects across multiple pathways, the mechanism by which the clinical activity is mediated remains unclear. Accumulating evidence demonstrates the selective effect of HDAC inhibitors in altering gene expression and apoptosis, however, it has not yet been determined which inhibitor will be more therapeutically-effective against specific cancer types. What’s worse, comparing the anticancer activity and toxicity profile of pan-HDAC inhibitors, such as vorinostat and belinostat, and class I-selective HDAC inhibitors such as romidepsin, there are no significant differences between them in terms of inhibitory potency and adverse events. Nevertheless, certain biomarkers may indicate the potential for greater effect of HDAC inhibitors. Like other cancer therapies, a targeted therapy may be successful. The therapeutic response of HDAC inhibitors could be greater in those patients who strongly express HDACs in their cancer cells.
Positive therapeutic responses with some HDAC inhibitors have been shown consistently in hematologic malignancies, but the clinical outcomes in solid tumors are disappointing when used as single agents. Many clinical trials have examined combination therapies of HDAC inhibitors with chemotherapeutic and biotherapeutic agents in both solid and hematologic cancers. Even though none of these combinations is yet approved by the U.S. FDA to treat cancer, combination therapy with other medicines will yield improved clinical outcomes over those seen with single agents.
To conclude, understanding mechanisms of action of HDAC inhibitors is essential not only for intelligent drug design for targeted therapies in cancer, but for finding of predictive biomarkers in cancer initiation and progression, and further for the design of optimized clinical protocols.
References
Adams, H., F.R. Fritzsche, S. Dirnhofer, G. Kristiansen, and A. Tzankov. 2010. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opinion on Therapeutic Targets 14: 577–584.
Amann, J.M., J. Nip, D.K. Strom, B. Lutterbach, H. Harada, N. Lenny, J.R. Downing, S. Meyers, and S.W. Hiebert. 2001. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Molecular and Cellular Biology 21: 6470–6483.
Ashkenazi, A. 2002. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nature Reviews Cancer 2: 420–430.
Atsumi, A., A. Tomita, H. Kiyoi, and T. Naoe. 2006. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML–RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochemical and Biophysical Research Communications 345: 1471–1480.
Bagnes, C., P.N. Panchuk, and G. Recondo. 2010. Antineoplastic chemotherapy induced QTc prolongation. Current Drug Safety 5: 93–96.
Bakkenist, C.J., and M.B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506.
Bali, P., M. Pranpat, J. Bradner, M. Balasis, W. Fiskus, F. Guo, K. Rocha, S. Kumaraswamy, S. Boyapalle, P. Atadja, E. Seto, and K. Bhalla. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. Journal of Biological Chemistry 280: 26729–26734.
Bantscheff, M., C. Hopf, M.M. Savitski, A. Dittmann, P. Grandi, A.M. Michon, J. Schlegl, Y. Abraham, I. Becher, G. Bergamini, M. Boesche, M. Delling, B. Dümpelfeld, D. Eberhard, C. Huthmacher, T. Mathieson, D. Poeckel, V. Reader, K. Strunk, G. Sweetman, U. Kruse, G. Neubauer, N.G. Ramsden, and G. Drewes. 2011. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nature Biotechnology 29: 255–265.
Bates, S.E., D.R. Rosing, T. Fojo, and R.L. Piekarz. 2006. Challenges of evaluating the cardiac effects of anticancer agents. Clinical Cancer Research 12: 3871–3874.
Bertos, N.R., A.H. Wang, and X.J. Yang. 2001. Class II histone deacetylases: Structure, function, and regulation. Biochemistry and Cell Biology 79: 243–252.
Bertrand, P. 2010. Inside HDAC with HDAC inhibitors. European Journal of Medicinal Chemistry 45: 2095–2116.
Blumenschein Jr., G.R., M.S. Kies, V.A. Papadimitrakopoulou, C. Lu, A.J. Kumar, J.L. Ricker, J.H. Chiao, C. Chen, and S.R. Frankel. 2008. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Investigational New Drugs 26: 81–87.
Bolden, J.E., M.J. Peart, and R.W. Johnstone. 2006. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery 5: 769–784.
Bradley, D., D. Rathkopf, R. Dunn, W.M. Stadler, G. Liu, D.C. Smith, R. Pili, J. Zwiebel, H. Scher, and M. Hussain. 2009. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 115: 5541–5549.
Bradner, J.E., N. West, M.L. Grachan, E.F. Greenberg, S.J. Haggarty, T. Warnow, and R. Mazitschek. 2010. Chemical phylogenetics of histone deacetylases. Nature Chemical Biology 6: 238–243.
Burgess, A., A. Ruefli, H. Beamish, R. Warrener, N. Saunders, R. Johnstone, and B. Gabrielli. 2004. Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene 23: 6693–6701.
Burz, C., I. Berindan-Neagoe, O. Balacescu, and A. Irimie. 2009. Apoptosis in cancer: Key molecular signaling pathways and therapy targets. Acta Oncologica 48: 811–821.
Cashen, A., M. Juckett, A. Jumonville, M. Litzow, P.J. Flynn, J. Eckardt, B. LaPlant, K. Laumann, C. Erlichman, and J. DiPersio. 2012. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Annals of Hematology 91: 33–38.
Cha, T.L., M.J. Chuang, S.T. Wu, G.H. Sun, S.Y. Chang, D.S. Yu, S.M. Huang, S.K. Huan, T.C. Cheng, T.T. Chen, P.L. Fan, and P.W. Hsiao. 2009. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2–M arrest and apoptosis of renal cancer cells. Clinical Cancer Research 15: 840–850.
Chen, L.f., W. Fischle, E. Verdin, and W.C. Greene. 2001. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 293: 1653–1657.
Chen, C.S., Y.C. Wang, H.C. Yang, P.H. Huang, S.K. Kulp, C.C. Yang, Y.S. Lu, S. Matsuyama, C.Y. Chen, and C.S. Chen. 2007. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Research 67: 5318–5327.
ClinicalTrials.gov. BELIEF study. http://clinicaltrials.gov/show/NCT00865969. Accessed 21 Nov 2014.
Coiffier, B., B. Pro, H.M. Prince, F. Foss, L. Sokol, M. Greenwood, D. Caballero, P. Borchmann, F. Morschhauser, M. Wilhelm, L. Pinter-Brown, S. Padmanabhan, A. Shustov, J. Nichols, S. Carroll, J. Balser, B. Balser, and S. Horwitz. 2012. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. Journal of Clinical Oncology 30: 631–636.
Curran, M.E., I. Splawski, K.W. Timothy, G.M. Vincent, E.D. Green, and M.T. Keating. 1995. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80: 795–803.
de Ruijter, A.J., A.H. van Gennip, H.N. Caron, S. Kemp, and A.B. van Kuilenburg. 2003. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochemical Journal 370: 737–749.
Decroos, C., C.M. Bowman, J.A. Moser, K.E. Christianson, M.A. Deardorff, and D.W. Christianson. 2014. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chemical Biology 9: 2157–2164.
Deubzer, H.E., M.C. Schier, I. Oehme, M. Lodrini, B. Haendler, A. Sommer, and O. Witt. 2013. HDAC11 is a novel drug target in carcinomas. International Journal of Cancer 132: 2200–2208.
Drugs@FDA. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. Accessed 21 Nov 2014.
Duvic, M., R. Talpur, X. Ni, C. Zhang, P. Hazarika, C. Kelly, J.H. Chiao, J.F. Reilly, J.L. Ricker, V.M. Richon, and S.R. Frankel. 2006. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109: 31–39.
El-Khoury, V., S. Pierson, E. Szwarcbart, N.H. Brons, O. Roland, S. Cherrier-De Wilde, L. Plawny, E. Van Dyck, and G. Berchem. 2014. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 28: 1636–1646.
Fath, D.M., X. Kong, D. Liang, Z. Lin, A. Chou, Y. Jiang, J. Fang, J. Caro, and N. Sang. 2006. Histone deacetylase inhibitors repress the transactivation potential of hypoxia-inducible factors independently of direct acetylation of HIF-alpha. Journal of Biological Chemistry 281: 13612–13619.
Feng, G.W., L.D. Dong, W.J. Shang, X.L. Pang, J.F. Li, L. Liu, and Y. Wang. 2014. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. European Review for Medical and Pharmacological Sciences 18: 811–816.
Firestein, R., G. Blander, S. Michan, P. Oberdoerffer, S. Ogino, J. Campbell, A. Bhimavarapu, S. Luikenhuis, R. de Cabo, C. Fuchs, W.C. Hahn, L.P. Guarente, and D.A. Sinclair. 2008. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE 3: e2020.
Fischle, W., V. Kiermer, F. Dequiedt, and E. Verdin. 2001. The emerging role of class II histone deacetylases. Biochemistry and Cell Biology 79: 337–348.
Foss, F., R. Advani, M. Duvic, K.B. Hymes, T. Intragumtornchai, A. Lekhakula, O. Shpilberg, A. Lerner, R.J. Belt, E.D. Jacobsen, G. Laurent, D. Ben-Yehuda, M. Beylot-Barry, U. Hillen, P. Knoblauch, G. Bhat, S. Chawla, L.F. Allen, and B. Pohlman. 2014. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma (Abstract). British Journal of Haematology. doi:10.1111/bjh.13222.
Fournel, M., C. Bonfils, Y. Hou, P.T. Yan, M.C. Trachy-Bourget, A. Kalita, J. Liu, A.H. Lu, N.Z. Zhou, M.F. Robert, J. Gillespie, J.J. Wang, H. Ste-Croix, J. Rahil, S. Lefebvre, O. Moradei, D. Delorme, A.R. Macleod, J.M. Besterman, and Z. Li. 2008. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Molecular Cancer Therapeutics 7: 759–768.
Fraga, M.F., E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero, K. Petrie, N.G. Iyer, A. Pérez-Rosado, E. Calvo, J.A. Lopez, A. Cano, M.J. Calasanz, D. Colomer, M.A. Piris, N. Ahn, A. Imhof, C. Caldas, T. Jenuwein, and M. Esteller. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics 37: 391–400.
Fritzsche, F.R., W. Weichert, A. Roske, V. Gekeler, T. Beckers, C. Stephan, K. Jung, K. Scholman, C. Denkert, M. Dietel, and G. Kristiansen. 2008. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 8: 381.
Furumai, R., A. Matsuyama, N. Kobashi, K.H. Lee, M. Nishiyama, H. Nakajima, A. Tanaka, Y. Komatsu, N. Nishino, M. Yoshida, and S. Horinouchi. 2002. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Research 62: 4916–4921.
Gao, L., M.A. Cueto, F. Asselbergs, and P. Atadja. 2002. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. Journal of Biological Chemistry 277: 25748–25755.
Garcia-Manero, G., H. Yang, C. Bueso-Ramos, A. Ferrajoli, J. Cortes, W.G. Wierda, S. Faderl, C. Koller, G. Morris, G. Rosner, A. Loboda, V.R. Fantin, S.S. Randolph, J.S. Hardwick, J.F. Reilly, C. Chen, J.L. Ricker, J.P. Secrist, V.M. Richon, S.R. Frankel, and H.M. Kantarjian. 2008. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 111: 1060–1066.
Gaughan, L., I.R. Logan, S. Cook, D.E. Neal, and C.N. Robson. 2002. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. Journal of Biological Chemistry 277: 25904–25913.
Gelmetti, V., J.S. Zhang, M. Fanelli, S. Minucci, P.G. Pelicci, and M.A. Lazar. 1998. Aberrant recruitment of the nuclear receptor corepressor–histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Molecular and Cellular Biology 18: 7185–7191.
Geng, L., K.C. Cuneo, A. Fu, T. Tu, P.W. Atadja, and D.E. Hallahan. 2006. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Research 66: 11298–11304.
Giaccone, G., A. Rajan, A. Berman, R.J. Kelly, E. Szabo, A. Lopez-Chavez, J. Trepel, M.J. Lee, L. Cao, I. Espinoza-Delgado, J. Spittler, and P.J. Loehrer Sr. 2011. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. Journal of Clinical Oncology 29: 2052–2059.
Gobinet, J., S. Carascossa, V. Cavaillès, F. Vignon, J.C. Nicolas, and S. Jalaguier. 2005. SHP represses transcriptional activity via recruitment of histone deacetylases. Biochemistry 44: 6312–6320.
Gregoretti, I.V., Y.M. Lee, and H.V. Goodson. 2004. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. Journal of Molecular Biology 338: 17–31.
Grunstein, M. 1997. Histone acetylation in chromatin structure and transcription. Nature 389: 349–352.
Gui, C.Y., L. Ngo, W.S. Xu, V.M. Richon, and P.A. Marks. 2004. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proceedings of the National Academy of Sciences of USA 101: 1241–1246.
Haigentz Jr., M., M. Kim, C. Sarta, J. Lin, R.S. Keresztes, B. Culliney, A.G. Gaba, R.V. Smith, G.I. Shapiro, L.R. Chirieac, J.M. Mariadason, T.J. Belbin, J.M. Greally, J.J. Wright, and R.I. Haddad. 2012. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncology 48: 1281–1288.
Halkidou, K., L. Gaughan, S. Cook, H.Y. Leung, D.E. Neal, and C.N. Robson. 2004. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59: 177–189.
Hayashi, A., A. Horiuchi, N. Kikuchi, T. Hayashi, C. Fuseya, A. Suzuki, I. Konishi, and T. Shiozawa. 2010. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. International Journal of Cancer 127: 1332–1346.
Heltweg, B., T. Gatbonton, A.D. Schuler, J. Posakony, H. Li, S. Goehle, R. Kollipara, R.A. Depinho, Y. Gu, J.A. Simon, and A. Bedalov. 2006. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Research 66: 4368–4377.
Hitomi, T., Y. Matsuzaki, T. Yokota, Y. Takaoka, and T. Sakai. 2003. p15(INK4b) in HDAC inhibitor-induced growth arrest. FEBS Letters 554: 347–350.
Hörlein, A.J., A.M. Näär, T. Heinzel, J. Torchia, B. Gloss, R. Kurokawa, A. Ryan, Y. Kamei, M. Söderström, C.K. Glass, and M.G. Rosenfeld. 1995. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377: 397–404.
Hu, E., E. Dul, C.M. Sung, Z. Chen, R. Kirkpatrick, G.F. Zhang, K. Johanson, R. Liu, A. Lago, G. Hofmann, R. Macarron, M. de los Frailes, P. Perez, J. Krawiec, J. Winkler, and M. Jaye. 2003. Identification of novel isoform-selective inhibitors within class I histone deacetylases. Journal of Pharmacology and Experimental Therapeutics 307: 720–728.
Hutt, D.M., D.M. Roth, H. Vignaud, C. Cullin, and M. Bouchecareilh. 2014. The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS ONE 9: e106224.
Insinga, A., S. Monestiroli, S. Ronzoni, V. Gelmetti, F. Marchesi, A. Viale, L. Altucci, C. Nervi, S. Minucci, and P.G. Pelicci. 2005. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nature Medicine 11: 71–76.
Iwamoto, F.M., K.R. Lamborn, J.G. Kuhn, P.Y. Wen, W.K. Yung, M.R. Gilbert, S.M. Chang, F.S. Lieberman, M.D. Prados, and H.A. Fine. 2011. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neurooncology 13: 509–516.
Jin, Y.H., E.J. Jeon, Q.L. Li, Y.H. Lee, J.K. Choi, W.J. Kim, K.Y. Lee, and S.C. Bae. 2004. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. Journal of Biological Chemistry 279: 29409–29417.
Johnstone, R.W. 2002. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nature Reviews Drug Discovery 1: 287–299.
Jung, K.H., J.H. Noh, J.K. Kim, J.W. Eun, H.J. Bae, H.J. Xie, Y.G. Chang, M.G. Kim, H. Park, J.Y. Lee, and S.W. Nam. 2012. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. Journal of Cellular Biochemistry 113: 2167–2177.
Kato, H., S. Tamamizu-Kato, and F. Shibasaki. 2004. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. Journal of Biological Chemistry 279: 41966–41974.
Kawai, H., H. Li, S. Avraham, S. Jiang, and H.K. Avraham. 2003. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. International Journal of Cancer 107: 353–358.
Kelly, W.K., O.A. O’Connor, L.M. Krug, J.H. Chiao, M. Heaney, T. Curley, B. MacGregore-Cortelli, W. Tong, J.P. Secrist, L. Schwartz, S. Richardson, E. Chu, S. Olgac, P.A. Marks, H. Scher, and V.M. Richon. 2005. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. Journal of Clinical Oncology 23: 3923–3931.
Khan, N., M. Jeffers, S. Kumar, C. Hackett, F. Boldog, N. Khramtsov, X. Qian, E. Mills, S.C. Berghs, N. Carey, P.W. Finn, L.S. Collins, A. Tumber, J.W. Ritchie, P.B. Jensen, H.S. Lichenstein, and M. Sehested. 2008. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochemical Journal 409: 581–589.
Kim, M.S., H.J. Kwon, Y.M. Lee, J.H. Baek, J.E. Jang, S.W. Lee, E.J. Moon, H.S. Kim, S.K. Lee, H.Y. Chung, C.W. Kim, and K.W. Kim. 2001. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nature Medicine 7: 437–443.
Kim, S.H., J.W. Jeong, J.A. Park, J.W. Lee, J.H. Seo, B.K. Jung, M.K. Bae, and K.W. Kim. 2007. Regulation of the HIF-1alpha stability by histone deacetylases. Oncology Reports 17: 647–651.
Kim, M.K., Y.J. Kang, D.H. Kim, M.A. Hossain, J.Y. Jang, S.H. Lee, J.H. Yoon, P. Chun, H.R. Moon, H.S. Kim, H.Y. Chung, and N.D. Kim. 2014. A novel hydroxamic acid derivative, MHY218, induces apoptosis and cell cycle arrest through downregulation of NF-κB in HCT116 human colon cancer cells. International Journal of Oncology 44: 256–264.
Kirschbaum, M., P. Frankel, L. Popplewell, J. Zain, M. Delioukina, V. Pullarkat, D. Matsuoka, B. Pulone, A.J. Rotter, I. Espinoza-Delgado, A. Nademanee, S.J. Forman, D. Gandara, and E. Newman. 2011. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. Journal of Clinical Oncology 29: 1198–1203.
Kong, X., Z. Lin, D. Liang, D. Fath, N. Sang, and J. Caro. 2006. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Molecular and Cellular Biology 26: 2019–2028.
Konstantinopoulos, P.A., A.J. Wilson, J. Saskowski, E. Wass, and D. Khabele. 2014. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecologic Oncology 133: 599–606.
Koprinarova, M., P. Botev, and G. Russev. 2011. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amsterdam) 10: 970–977.
Kouzarides, T. 1999. Histone acetylases and deacetylases in cell proliferation. Current Opinion in Genetics and Development 9: 40–48.
Kovacs, J.J., P.J. Murphy, S. Gaillard, X. Zhao, J.T. Wu, C.V. Nicchitta, M. Yoshida, D.O. Toft, W.B. Pratt, and T.P. Yao. 2005. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Molecular Cell 18: 601–607.
Lassen, U., L.R. Molife, M. Sorensen, S.A. Engelholm, L. Vidal, R. Sinha, R.T. Penson, P. Buhl-Jensen, E. Crowley, J. Tjornelund, P. Knoblauch, and J.S. de Bono. 2010. A phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. British Journal of Cancer 103: 12–17.
Lee, Y.M., S.H. Kim, H.S. Kim, M. Jin Son, H. Nakajima, H. Jeong Kwon, and K.W. Kim. 2003. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1alpha activity. Biochemical and Biophysical Research Communications 300: 241–246.
Li, D., X. Sun, L. Zhang, B. Yan, S. Xie, R. Liu, M. Liu, and J. Zhou. 2014. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein and Cell 5: 214–223.
Lin, R.J., L. Nagy, S. Inoue, W.L. Shao, W.H. Miller, and R.M. Evans. 1998. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 391: 811–814.
Luo, J., F. Su, D. Chen, A. Shiloh, and W. Gu. 2000. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408: 377–381.
Luu, T.H., R.J. Morgan, L. Leong, D. Lim, M. McNamara, J. Portnow, P. Frankel, D.D. Smith, J.H. Doroshow, C. Wong, A. Aparicio, D.R. Gandara, and G. Somlo. 2008. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clinical Cancer Research 14: 7138–7142.
Lynch Jr., D.R., J.B. Washam, and L.K. Newby. 2012. QT interval prolongation and torsades de pointes in a patient undergoing treatment with vorinostat: A case report and review of the literature. Cardiology Journal 19: 434–438.
Mackay, H.J., H. Hirte, T. Colgan, A. Covens, K. MacAlpine, P. Grenci, L. Wang, J. Mason, P.A. Pham, M.S. Tsao, J. Pan, J. Zwiebel, and A.M. Oza. 2010. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. European Journal of Cancer 46: 1573–1579.
Mal, A., and M.L. Harter. 2003. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proceedings of the National Academy of Sciences of USA 100: 1735–1739.
Mal, A., M. Sturniolo, R.L. Schiltz, M.K. Ghosh, and M.L. Harter. 2001. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: Inhibition of the myogenic program. EMBO Journal 20: 1735–1739.
Mann, B.S., J.R. Johnson, M.H. Cohen, R. Justice, and R. Pazdur. 2007. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12: 1247–1252.
Marks, P., R.A. Rifkind, V.M. Richon, R. Breslow, T. Miller, and W.K. Kelly. 2001. Histone deacetylases and cancer: Causes and therapies. Nature Reviews Cancer 1: 194–202.
Marquard, L., L.M. Gjerdrum, I.J. Christensen, P.B. Jensen, M. Sehested, and E. Ralfkiaer. 2008. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 53: 267–277.
Martínez-Balbás, M.A., U.M. Bauer, S.J. Nielsen, A. Brehm, and T. Kouzarides. 2000. Regulation of E2F1 activity by acetylation. EMBO Journal 19: 662–671.
Milde, T., I. Oehme, A. Korshunov, A. Kopp-Schneider, M. Remke, P. Northcott, H.E. Deubzer, M. Lodrini, M.D. Taylor, A. von Deimling, S. Pfister, and O. Witt. 2010. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clinical Cancer Research 16: 3240–3252.
Minamiya, Y., T. Ono, H. Saito, N. Takahashi, M. Ito, M. Mitsui, S. Motoyama, and J. Ogawa. 2011. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 74: 300–304.
Modesitt, S.C., M. Sill, J.S. Hoffman, D.P. Bender, and Gynecologic Oncology Group. 2008. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecologic Oncology 109: 182–186.
Molife, L.R., G. Attard, P.C. Fong, V. Karavasilis, A.H. Reid, S. Patterson, C.E. Riggs Jr., C. Higano, W.M. Stadler, W. McCulloch, D. Dearnaley, C. Parker, and J.S. de Bono. 2010. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Annals of Oncology 21: 109–113.
Montgomery, R.L., C.A. Davis, M.J. Potthoff, M. Haberland, J. Fielitz, X. Qi, J.A. Hill, J.A. Richardson, and E.N. Olson. 2007. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes and Development 21: 1790–1802.
Moreno, D.A., C.A. Scrideli, M.A. Cortez, R. de Paula Queiroz, E.T. Valera, V. da Silva Silveira, J.A. Yunes, S.R. Brandalise, and L.G. Tone. 2010. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. British Journal of Haematology 150: 665–673.
Müller, B.M., L. Jana, A. Kasajima, A. Lehmann, J. Prinzler, J. Budczies, K.J. Winzer, M. Dietel, W. Weichert, and C. Denkert. 2013. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—Overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 13: 215.
Munshi, A., J.F. Kurland, T. Nishikawa, T. Tanaka, M.L. Hobbs, S.L. Tucker, S. Ismail, C. Stevens, and R.E. Meyn. 2005. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clinical Cancer Research 11: 4912–4922.
Mwakwari, S.C., V. Patil, W. Guerrant, and A.K. Oyelere. 2010. Macrocyclic histone deacetylase inhibitors. Current Topics in Medicinal Chemistry 10: 1423–1440.
Noh, E.J., D.S. Lim, G. Jeong, and J.S. Lee. 2009. An HDAC inhibitor, trichostatin A, induces a delay at G2/M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner. Biochemical and Biophysical Research Communications 378: 326–331.
Norton, V.G., B.S. Imai, P. Yau, and E.M. Bradbury. 1989. Histone acetylation reduces nucleosome core particle linking number change. Cell 57: 449–457.
Oehme, I., H.E. Deubzer, D. Wegener, D. Pickert, J.P. Linke, B. Hero, A. Kopp-Schneider, F. Westermann, S.M. Ulrich, A. von Deimling, M. Fischer, and O. Witt. 2009. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clinical Cancer Research 15: 91–99.
Ogura, M., K. Ando, T. Suzuki, K. Ishizawa, S.Y. Oh, K. Itoh, K. Yamamoto, W.Y. Au, H.F. Tien, Y. Matsuno, T. Terauchi, K. Yamamoto, M. Mori, Y. Tanaka, T. Shimamoto, K. Tobinai, and W.S. Kim. 2014. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. British Journal of Haematology 165: 768–776.
Olsen, E.A., Y.H. Kim, T.M. Kuzel, T.R. Pacheco, F.M. Foss, S. Parker, S.R. Frankel, C. Chen, J.L. Ricker, J.M. Arduino, and M. Duvic. 2007. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology 25: 3109–3115.
Osada, H., Y. Tatematsu, H. Saito, Y. Yatabe, T. Mitsudomi, and T. Takahashi. 2004. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. International Journal of Cancer 112: 26–32.
Otterson, G.A., L. Hodgson, H. Pang, E.E. Vokes, and Cancer and Leukemia Group B. 2010. Phase II study of the histone deacetylase inhibitor Romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304). Journal of Thoracic Oncology 5: 1644–1648.
Ouaïssi, M., I. Sielezneff, R. Silvestre, B. Sastre, J.P. Bernard, J.S. Lafontaine, M.J. Payan, L. Dahan, N. Pirrò, J.F. Seitz, E. Mas, D. Lombardo, and A. Ouaissi. 2008. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Annals of Surgical Oncology 15: 2318–2328.
Ozawa, Y., M. Towatari, S. Tsuzuki, F. Hayakawa, T. Maeda, Y. Miyata, M. Tanimoto, and H. Saito. 2001. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 98: 2116–2123.
Park, S.Y., J.A. Jun, K.J. Jeong, H.J. Heo, J.S. Sohn, H.Y. Lee, C.G. Park, and J. Kang. 2011. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncology Reports 25: 1677–1681.
Peart, M.J., G.K. Smyth, R.K. van Laar, D.D. Bowtell, V.M. Richon, P.A. Marks, A.J. Holloway, and R.W. Johnstone. 2005. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of USA 102: 3697–3702.
Pérez-Perarnau, A., L. Coll-Mulet, C. Rubio-Patiño, D. Iglesias-Serret, A.M. Cosialls, D.M. González-Gironès, M. de Frias, A.F. de Sevilla, E. de la Banda, G. Pons, and J. Gil. 2011. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics 6: 1228–1235.
Piekarz, R.L., A.R. Frye, J.J. Wright, S.M. Steinberg, D.J. Liewehr, D.R. Rosing, V. Sachdev, T. Fojo, and S.E. Bates. 2006. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clinical Cancer Research 12: 3762–3773.
Piekarz, R.L., R. Frye, M. Turner, J.J. Wright, S.L. Allen, M.H. Kirschbaum, J. Zain, H.M. Prince, J.P. Leonard, L.J. Geskin, C. Reeder, D. Joske, W.D. Figg, E.R. Gardner, S.M. Steinberg, E.S. Jaffe, M. Stetler-Stevenson, S. Lade, A.T. Fojo, and S.E. Bates. 2009. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. Journal of Clinical Oncology 27: 5410–5417.
Qian, D.Z., S.K. Kachhap, S.J. Collis, H.M. Verheul, M.A. Carducci, P. Atadja, and R. Pili. 2006. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Research 66: 8814–8821.
Qiu, L., A. Burgess, D.P. Fairlie, H. Leonard, P.G. Parsons, and B.G. Gabrielli. 2000. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Molecular Biology of the Cell 11: 2069–2083.
Quint, K., A. Agaimy, P. Di Fazio, R. Montalbano, C. Steindorf, R. Jung, C. Hellerbrand, A. Hartmann, H. Sitter, D. Neureiter, and M. Ocker. 2011. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Archives 459: 129–139.
Ramalingam, S.S., C.P. Belani, C. Ruel, P. Frankel, B. Gitlitz, M. Koczywas, I. Espinoza-Delgado, and D. Gandara. 2009. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. Journal of Thoracic Oncology 4: 97–101.
Richon, V.M., Y. Webb, R. Merger, T. Sheppard, B. Jursic, L. Ngo, F. Civoli, R. Breslow, R.A. Rifkind, and P.A. Marks. 1996. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proceedings of the National Academy of Sciences of USA 93: 5705–5708.
Richon, V.M., T.W. Sandhoff, R.A. Rifkind, and P.A. Marks. 2000. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proceedings of the National Academy of Sciences of USA 97: 10014–10019.
Ropero, S., and M. Esteller. 2007. The role of histone deacetylases (HDACs) in human cancer. Molecular Oncology 1: 19–25.
Ropero, S., M.F. Fraga, E. Ballestar, R. Hamelin, H. Yamamoto, M. Boix-Chornet, R. Caballero, M. Alaminos, F. Setien, M.F. Paz, M. Herranz, J. Palacios, D. Arango, T.F. Orntoft, L.A. Aaltonen, S. Schwartz Jr., and M. Esteller. 2006. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nature Genetics 38: 566–569.
Rosato, R.R., and S. Grant. 2005. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert Opinion on Therapeutic Targets 9: 809–924.
Ruefli, A.A., M.J. Ausserlechner, D. Bernhard, V.R. Sutton, K.M. Tainton, R. Kofler, M.J. Smyth, and R.W. Johnstone. 2001. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proceedings of the National Academy of Sciences of USA 98: 10833–10838.
Sandor, V., A. Senderowicz, S. Mertins, D. Sackett, E. Sausville, M.V. Blagosklonny, and S.E. Bates. 2000. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. British Journal of Cancer 83: 817–825.
Sawa, H., H. Murakami, Y. Ohshima, T. Sugino, T. Nakajyo, T. Kisanuki, Y. Tamura, A. Satone, W. Ide, I. Hashimoto, and H. Kamada. 2001. Histone deacetylase inhibitors such as sodium butyrate and trichostatin A induce apoptosis through an increase of the Bcl-2-related protein Bad. Brain Tumor Pathology 18: 109–114.
Seo, S.Y. 2012. Multi-targeted hybrids based on HDAC inhibitors for anti-cancer drug discovery. Archives of Pharmacal Research 35: 197–200.
Serrador, J.M., J.R. Cabrero, D. Sancho, M. Mittelbrunn, A. Urzainqui, and F. Sánchez-Madrid. 2004. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity 20: 417–428.
Shah, M.H., P. Binkley, K. Chan, J. Xiao, D. Arbogast, M. Collamore, Y. Farra, D. Young, and M. Grever. 2006. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clinical Cancer Research 12: 3997–4003.
Song, J., J.H. Noh, J.H. Lee, J.W. Eun, Y.M. Ahn, S.Y. Kim, S.H. Lee, W.S. Park, N.J. Yoo, J.Y. Lee, and S.W. Nam. 2005. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113: 264–268.
Stadler, W.M., K. Margolin, S. Ferber, W. McCulloch, and J.A. Thompson. 2006. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clinical Genitourinary Cancer 5: 57–60.
Steele, N.L., J.A. Plumb, L. Vidal, J. Tjørnelund, P. Knoblauch, A. Rasmussen, C.E. Ooi, P. Buhl-Jensen, R. Brown, T.R. Evans, and J.S. DeBono. 2008. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clinical Cancer Research 14: 804–810.
Straus, S.M., J.A. Kors, M.L. De Bruin, C.S. van der Hooft, A. Hofman, J. Heeringa, J.W. Deckers, J.H. Kingma, M.C. Sturkenboom, B.H. Stricker, and J.C. Witteman. 2006. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. Journal of the American College of Cardiology 47: 362–367.
Strevel, E.L., D.J. Ing, and L.L. Siu. 2007. Molecularly targeted oncology therapeutics and prolongation of the QT interval. Journal of Clinical Oncology 25: 3362–3371.
Stypula-Cyrus, Y., D. Damania, D.P. Kunte, M.D. Cruz, H. Subramanian, H.K. Roy, and V. Backman. 2013. HDAC up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS ONE 8: e64600.
Sutheesophon, K., N. Nishimura, Y. Kobayashi, Y. Furukawa, M. Kawano, K. Itoh, Y. Kano, H. Ishii, and Y. Furukawa. 2005. Involvement of the tumor necrosis factor (TNF)/TNF receptor system in leukemic cell apoptosis induced by histone deacetylase inhibitor depsipeptide (FK228). Journal of Cellular Physiology 203: 387–397.
Traynor, A.M., S. Dubey, J.C. Eickhoff, J.M. Kolesar, K. Schell, M.S. Huie, D.L. Groteluschen, S.M. Marcotte, C.M. Hallahan, H.R. Weeks, G. Wilding, I. Espinoza-Delgado, and J.H. Schiller. 2009. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: A Wisconsin Oncology Network phase II study. Journal of Thoracic Oncology 4: 522–526.
Ungerstedt, J.S., Y. Sowa, W.S. Xu, Y. Shao, M. Dokmanovic, G. Perez, L. Ngo, A. Holmgren, X. Jiang, and P.A. Marks. 2005. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of USA 102: 673–678.
U.S. Food and Drug Administration. Drugs. Belinostat. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm403960.htm. Accessed 21 Nov 2014.
U.S. Food and Drug Administration. Drugs. Romidepsin. http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm189466.htm. Accessed 21 Nov 2014.
Vansteenkiste, J., E. Van Cutsem, H. Dumez, C. Chen, J.L. Ricker, S.S. Randolph, and P. Schöffski. 2008. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investigational New Drugs 26: 483–488.
Verdin, E., F. Dequiedt, and H.G. Kasler. 2003. Class II histone deacetylases: Versatile regulators. Trends in Genetics 19: 286–293.
Wang, J.C., M.I. Kafeel, B. Avezbakiyev, C. Chen, Y. Sun, C. Rathnasabapathy, M. Kalavar, Z. He, J. Burton, and S. Lichter. 2011. Histone deacetylase in chronic lymphocytic leukemia. Oncology 81: 325–329.
Watamoto, K., M. Towatari, Y. Ozawa, Y. Miyata, M. Okamoto, A. Abe, T. Naoe, and H. Saito. 2003. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene 22: 9176–9184.
Weichert, W., A. Röske, V. Gekeler, T. Beckers, M.P. Ebert, M. Pross, M. Dietel, C. Denkert, and C. Röcken. 2008a. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncology 9: 139–148.
Weichert, W., A. Roske, S. Niesporek, A. Noske, A.C. Buckendahl, M. Dietel, V. Gekeler, M. Boehm, T. Beckers, and C. Denkert. 2008b. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clinical Cancer Research 14: 1669–1677.
Weichert, W., A. Röske, V. Gekeler, T. Beckers, C. Stephan, K. Jung, F.R. Fritzsche, S. Niesporek, C. Denkert, M. Dietel, and G. Kristiansen. 2008c. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. British Journal of Cancer 98: 604–610.
Whitehead, R.P., C. Rankin, P.M. Hoff, P.J. Gold, K.G. Billingsley, R.A. Chapman, L. Wong, J.H. Ward, J.L. Abbruzzese, and C.D. Blanke. 2009. Phase II trial of romidepsin (NSC-630176) in previously treated colorectal cancer patients with advanced disease: A Southwest Oncology Group study (S0336). Investigational New Drugs 27: 469–475.
Whittaker, S.J., M.F. Demierre, E.J. Kim, A.H. Rook, A. Lerner, M. Duvic, J. Scarisbrick, S. Reddy, T. Robak, J.C. Becker, A. Samtsov, W. McCulloch, and Y.H. Kim. 2010. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. Journal of Clinical Oncology 28: 4485–4491.
Wolbrette, D. 2004. Drugs that cause torsades de pointes and increase the risk of sudden cardiac death. Current Cardiology Reports 6: 379–384.
Xie, H.J., J.H. Noh, J.K. Kim, K.H. Jung, J.W. Eun, H.J. Bae, M.G. Kim, Y.G. Chang, J.Y. Lee, H. Park, and S.W. Nam. 2012. HDAC1 inactivation induces mitotic defect and caspase independent autophagic cell death in liver cancer. PLoS ONE 7: e34265.
Yang, W.M., Y.L. Yao, J.M. Sun, J.R. Davie, and E. Seto. 1997. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. Journal of Biological Chemistry 272: 28001–28007.
Yamashita, Y., M. Shimada, N. Harimoto, T. Rikimaru, K. Shirabe, S. Tanaka, and K. Sugimachi. 2003. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. International Journal of Cancer 103: 572–576.
Younes, A., Y. Oki, R.G. Bociek, J. Kuruvilla, M. Fanale, S. Neelapu, A. Copeland, H. Zhou, S. Jiang, J. Chen, and S.B. Su. 2014. Suberoylanilide hydroxamic acid suppresses inflammation-induced neovascularization. Canadian Journal of Physiology and Pharmacology 92: 879–885.
Yuan, Z.L., Y.J. Guan, D. Chatterjee, and Y.E. Chin. 2005. STAT3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307: 269–273.
Zgouras, D., A. Wächtershäuser, D. Frings, and J. Stein. 2003. Butyrate impairs intestinal tumor cell-induced angiogenesis by inhibiting HIF-1alpha nuclear translocation. Biochemical and Biophysical Research Communications 300(4): 832–838.
Zhang, X.D., S.K. Gillespie, J.M. Borrow, and P. Hersey. 2004. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Molecular Cancer Therapeutics 3: 425–435.
Zhang, Y., M. Adachi, R. Kawamura, and K. Imai. 2006. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death and Differentiation 13: 129–140.
Zhao, Y., J. Tan, L. Zhuang, X. Jiang, E.T. Liu, and Q. Yu. 2005. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proceedings of the National Academy of Sciences of USA 102: 16090–16095.
Zheng, G., and Y.C. Yang. 2005. Sumoylation and acetylation play opposite roles in the transactivation of PLAG1 and PLAGL2. Journal of Biological Chemistry 280: 40773–40781.
Zhou, H., S. Jiang, J. Chen, and S.B. Su. 2014. Suberoylanilide hydroxamic acid suppresses inflammation-induced neovascularization. Canadian Journal of Physiology and Pharmacology 92: 879–885.
Acknowledgments
This work was supported by the 2013 Inje University research grant.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chun, P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch. Pharm. Res. 38, 933–949 (2015). https://doi.org/10.1007/s12272-015-0571-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0571-1