
Abstract
The quintessential clinical diagnostic phenotype of human hypertrophic cardiomyopathy (HCM) is primary cardiac hypertrophy. Cardiac hypertrophy is also a major determinant of mortality and morbidity including the risk of sudden cardiac death (SCD) in patients with HCM. Reversal and attenuation of cardiac hypertrophy and its accompanying fibrosis is expected to improve morbidity as well as decrease the risk of SCD in patients with HCM.The conventionally used pharmacological agents in treatment of patients with HCM have not been shown to reverse or attenuate established cardiac hypertrophy and fibrosis. An effective treatment of HCM has to target the molecular mechanisms that are involved in the pathogenesis of the phenotype. Mechanistic studies suggest that cardiac hypertrophy in HCM is secondary to activation of various hypertrophic signaling molecules and, hence, is potentially reversible. The hypothesis is supported by the results of genetic and pharmacological interventions in animal models. The results have shown potential beneficial effects of angiotensin II receptor blocker losartan, mineralocorticoid receptor blocker spironolactone, 3-hydroxy-3-methyglutaryl-coenzyme A reductase inhibitors simvastatin and atorvastatin, and most recently, N-acetylcysteine (NAC) on reversal or prevention of hypertrophy and fibrosis in HCM. The most promising results have been obtained with NAC, which through multiple thiol-responsive mechanisms completely reversed established cardiac hypertrophy and fibrosis in three independent studies. Pilot studies with losartan and statins in humans have established the feasibility of such studies. The results in animal models have firmly established the reversibility of established cardiac hypertrophy and fibrosis in HCM and have set the stage for advancing the findings in the animal models to human patients with HCM through conducting large-scale efficacy studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite the modern recognition of hypertrophic cardiomyopathy (HCM) for over five decades and in spite of considerable advances in its phenotyping and molecular genetics, current pharmacological treatment of patients with HCM has remained largely unchanged and empiric. The primary emphasis in the management of patients with HCM has been on the risk of sudden cardiac death (SCD), as HCM is a common cause of SCD in the young [54]. Prudent use of automatic internal defibrillator/cardioverters (AICDs) has provided the physicians with an effective option for prevention of SCD in the high-risk patients [55, 56]. However, implantation of an AICD does not influence the underlying cardiac pathology, namely, myocyte hypertrophy, disarray, and interstitial fibrosis. Likewise, catheter-based septal ablation and surgical myectomy, while are highly effective in reducing the left ventricular outflow tract (LVOT) obstruction, alleviating symptoms (functional improvement), do not substantially alleviate the underlying arrhythmogenic substrate [53, 60, 78, 88, 95, 104, 115]. In contrast, the tissue scar at the site of percutaneous septal ablation has the potential to increase the risk of ventricular arrhythmias [7, 26, 59, 89, 95].
The effective treatment of HCM, whether pharmacological or otherwise, has to target the underlying mechanism(s) responsible for the clinical phenotype. The current phenotypic emphasis is on cardiac hypertrophy, as it, in the absence of a discernible cause, epitomizes the clinical diagnosis of HCM. The prominence of hypertrophy as the main target of therapy stems from the fact that adult cardiac myocytes are terminally differentiated cells and their primary response to pro-growth stimulants is increased size, i.e., hypertrophy [105]. Partial understanding of the molecular pathways involved in the pathogenesis of cardiac hypertrophy has further enhanced the interest in targeting hypertrophy for the effective treatment of HCM. The clinical significance of cardiac hypertrophy also legitimizes it as the primary therapeutic target. Underscoring the point is that pathological cardiac hypertrophy, regardless of the cause or the ethnic background, is an important determinant of mortality, morbidity, and the risk of SCD [4, 14, 20, 33, 34, 77, 96]. Likewise, cardiac hypertrophy is also an important determinant of diastolic dysfunction and failure as well as susceptibility to arrhythmias [18, 25, 58, 61, 68, 70, 71, 83, 120, 121].
Reversibility of Cardiac Phenotype in HCM
An important clinical question in HCM has been whether cardiac hypertrophy and fibrosis, once established, could be reversed or attenuated and whether evolving phenotype could be prevented. Conventionally used pharmacological agents have not proven to prevent or reverse established cardiac hypertrophy. The current evidence for potential regression of hypertrophy stems from observational studies following percutaneous septal ablation and surgical myectomy suggesting left ventricular remodeling and regression of cardiac hypertrophy upon relief of the LVOT obstruction [16, 30].
The most direct and robust evidence for the regression of established cardiac hypertrophy and fibrosis is derived from experimental studies in genetically modified animal models of HCM. We have advocated that cardiac hypertrophy in HCM is a secondary phenotype due to activation of pro-growth signaling molecules instigated by the functional defects imparted by the mutations and, hence, it is potentially reversible and preventable [46]. In proof-of-principle studies, we generated switch-on/switch-off bigenic mice, wherein expression of the transgene could be turned off and on, as desired [45]. We showed that turning off expression of the mutant transgene was associated with reversal and normalization of the induced phenotype [45]. Subsequently, we extended out studies by targeting signaling pathways involved in cardiac hypertrophic response [42, 51, 82, 91, 111]. Collectively, the findings, which are discussed in the present review, have established the potential to prevent the evolving and reverse the established cardiac hypertrophy and fibrosis in patients with HCM.
Ineffectiveness of Current Pharmacological Therapies in Reversing Cardiac Hypertrophy in HCM
Current pharmacological treatment of human HCM, while is effective for symptomatic improvement, has not been established to prevent, attenuate, or reverse cardiac hypertrophy in humans with HCM or even impact the prognosis [49, 50]. The most commonly used pharmacological agents are the β-blockers, which are the mainstay of therapy and the first choice in the absence of a contraindication. The proposed mechanisms of effects include improved ventricular relaxation and increased diastolic filling time and, hence, improved left ventricular end diastolic pressure as well as perfusion. In addition, treatment with β-blockers is expected to reduce ventricular and supra-ventricular arrhythmias. An observational retrospective study in a mixed pediatrics HCM population suggests potential beneficial effects of high-doses β-blockers on total survival [81]. Otherwise, the benefits of β-blockers on mortality and the risk of SCD in patients with HCM and their impact on prevention or reversal of cardiac hypertrophy in HCM remains to be established.
Verapamil and diltiazem exert their beneficial effects in part through their negative inotropic and chronotropic effects and in part through improving myocardial diastolic properties. These agents have been used for over two decades but not been proven to impart significant effects on cardiac hypertrophy or prognosis in patients with HCM. In a small study comprised of six mutation carriers without cardiac hypertrophy, treatment with diltiazem was found to improve myocardial tissue Doppler velocities [62]. Diltiazem also has been shown to improve cardiac systolic function and reduce interstitial fibrosis in the α-MyHC-Q403 mice, which exhibited reduced left ventricular systolic function [90]. However, the finding of improved systolic function with diltiazem in the mouse model may not have direct implications for human HCM, as global systolic function in HCM patients is enhanced or at least preserved.
Dihydropyridine calcium channel blockers, such as nifedipine are avoided in patients with HCM, because of the concern about the vasodilatory effects inducing hypotension, syncope, and possibly death. Overall, the clinical utility of Ca+2 channel blockers in treatment of patients with HCM is hindered by the risk of hypotension and syncope, at rest or particularly during exercise. This is not a trivial concern in patients with resting or provoked LVOT obstruction, who could comprise up to 70% of the patients with HCM [57, 79].
Disopyramide, a class I anti-arrhythmic drug, is used in conjunction with β-blockers to attenuate LVOT obstruction and improve symptoms [92]. The beneficial effects of disopyramide are largely due to its negative inotropic effects. Hence, it is most effective in patients with LVOT obstruction. Disopyramide does not reverse or attenuate cardiac hypertrophy in patients with HCM [92].
Mechanistic Approach to Effective Treatment of HCM
The seminal report of the R403Q mutation in MYH7 in a family with HCM by Dr. Seidman’s group in 1990 unraveled the genetic enigma of HCM [106]. The discovery led to subsequent identification of over a dozen causal genes for HCM [49, 66]. The known causal genes encode sarcomeric including the Z disk proteins. Accordingly, MYH7 and MYBPC3 have emerged as the two most common causal genes for HCM, together accounting for approximately 50% of the HCM cases. Mutations in TNNT2, TNNI3, TPM1, and ACTC1 collectively account for about 10% of the HCM cases and other causal genes, such as the Z disk proteins MYOZ2 and TCAP, encoding myozenin 2 and telethonin, respectively, are less common [31, 80]. Overall, the causal mutations in approximately two thirds of the patients with HCM have been identified.
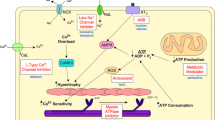
Mechanistic studies have elucidated a diverse array of functional defects imparted by the causal mutations in HCM. The diversity perhaps is best illustrated for the Ca2+ sensitivity of myofibrillar ATPase activity and force generation, which vary according to the causal genes and are context dependent [17, 19, 22, 64, 65, 69, 72, 73, 85, 86, 93, 98, 107, 108]. For example, mutations in the β-MyHC when engineered in the background of β-MyHC isoform, as in the human heart, decrease Ca+2 sensitivity of the myofilament force generation and ATPase activity [43, 69]. In contrast, the same mutation when engineered into α-MyHC isoform in the background of α-MyHC is associated with reduced Ca+2 sensitivity of the myofilament force generation [6]. It merits noting that in the human heart, the β-MyHC isoform encoded by the MYH7 comprises over 90% of the total myosin heavy chains. Likewise, in the rabbit heart, the β-MyHC isoform is the predominant isoform, comprising more than 85% of the total myosin heavy chain. This is in contrast to the mouse heart, which predominantly (>95%) contains the α-MyHC isoform, encoded by the MYH6. The α and β isoforms differ considerably in their ATPase activities and rate of actin displacement. The differences may influence delineating the responsible mechanisms for HCM caused by mutations in MYH7. In addition, TNNT2 mutations that cause HCM are associated with enhanced Ca2+ sensitivity of myofibrillar protein ATPase activity and force generation and so are mutations in TPM1 and TNNI3 that cause HCM [21, 23, 24, 41, 63, 84, 94, 98, 99, 110, 118]. Moreover, the pathogenesis of HCM caused by MYOZ2 mutations is likely to involve activation of the calcineurin protein phosphatase pathway [80].
The diversity of the mechanisms involved in the pathogenesis of HCM restricts the utility of targeting a specific pathway for the therapeutic benefits in HCM. The diversity could necessitate target-specific interventions, mandated by the functional phenotype of the causal mutations. An alternative approach is targeting mechanisms/pathways that are common to cardiac hypertrophic growth, such as oxidative stress pathways, which are common to various forms of cardiac hypertrophy including HCM [42, 51, 91, 103, 109]. We have shown that myocardial levels of lipid peroxides, markers of oxidative stress, as well as levels of oxidized mtDNA are increased in transgenic mouse and rabbit models of HCM [51, 91]. Thus, interventions aimed at mechanisms that regulate oxidative stress in the heart could emerge as desirable for reversal of cardiac hypertrophy and fibrosis in HCM as well as other hypertrophic conditions [42, 103]. The concept is further supported by the shared anti-oxidant properties of various pharmacological agents that have previously shown to effectively regress cardiac hypertrophy in animal models of HCM, namely losartan [37], spironolactone [111], statins [82, 91], and N-acetylcysteine (NAC) [42, 51]. Hence, therapies targeted at oxidative stress pathway have emerged as potentially effective agents in reversal of established cardiac hypertrophy and fibrosis in HCM.
Distinction between (sarcomeric) HCM and Phenocopy Conditions
Genetic discoveries have also elucidated the fallacies inherent to the clinical diagnosis of HCM, as many patients with the clinical diagnosis of HCM might have phenocopy conditions. The current clinical diagnosis of HCM, based on detection of cardiac hypertrophy, is neither sufficiently specific, because of the presence of phenocopy conditions [1, 9], nor sufficiently sensitive, because of low penetrance of certain causal mutations [74, 75]. Phenocopy conditions, such as storage diseases, are typically indistinguishable from true HCM [9]. Nevertheless, the distinction between HCM caused sarcomeric protein mutations, and phenocopy conditions are crucial for an effective treatment of HCM. The clinical significance of such distinction is highlighted by the differences in the treatments of the two conditions. For example, enzyme replacement therapy with human α-Gal A (agalsidase α) or recombinant human α-Gal A (agalsidase β) suppresses progression of Fabry disease [87, 114]. In contrast, conventional treatment for HCM in patients with phenocopy conditions is not expected to impart significant benefits [50]. The true incidence of phenocopy condition in patients with the clinical diagnosis of HCM is likely to be approximately 10% [47].
Specific Experimental Pharmacological Agents in HCM
Inhibitors of Rennin-Angiotensin-Aldosterone System (RAAS)
Given the well-established anti-hypertrophic and anti-fibrotic effects of the inhibitors of the rennin-angiotensin-aldosterone system (RAAS), we have tested the potential beneficial effects of treatment with losartan, an angiotensin II receptor blocker in the cTnT-Q92 mouse model [37]. Likewise, we have investigated the potential beneficial effects of spironolactone, a mineralocorticoid receptor inhibitor, in reversal of cardiac interstitial fibrosis in two independent (test and replication) randomized studies [111]. In these studies, treatment with losartan or spironolactone normalized myocardial collagen content, in part through their anti-oxidant effects and partly through inhibition of pro-fibrotic effects of transforming growth factor-β and protein kinase D (Table 1) [37, 111].
The studies in animal models were followed by pilot clinical studies in humans with HCM [2, 117]. The results in human studies, although preliminary, have been to a large extent unsatisfactory. Yamazaki et al. treated ten patients with HCM who did not have LVOT obstruction with 50 mg of losartan per day for 1 year and reported only a 7% change in LV mass, which was not significant [117]. Likewise, Araujo et al. treated 20 patients with non-obstructive HCM with 100 mg of losartan per day for 6 months and reported no significant changes in the echocardiographic indices of cardiac hypertrophy but modest improvement in selected indices of diastolic function [2]. The null findings of these two studies regarding regression of cardiac hypertrophy and given the potential risk of hypotension and syncope, particularly in those with resting or latent LVOT obstruction, have reduced the enthusiasm for the potential utility of the angiotensin II receptor blockers in treatment of human patients with HCM.
3-Hydroxy-3-methyglutaryl-coenzyme A (HMG-CoA) Reductase Inhibitors
HMG-Co A reductase inhibitors or statins are pleiotropic agents with various effects including anti-oxidant effects [8, 11, 36, 67, 76, 102]. We tested the potential effects of simvastatin in reversal of established cardiac hypertrophy and fibrosis in a randomized placebo-control study the β-MyHC-Q403 transgenic rabbits [82, 91]. We showed that treatment with simvastatin reduced left ventricular mass, wall thickness, myocyte size, and normalized interstitial fibrosis [82]. In addition, indices of cardiac diastolic function and filling pressure were improved [82].
In a subsequent study, we tested the effects of atorvastatin on prevention of cardiac hypertrophy and fibrosis in the β-MyHC-Q403 transgenic rabbits [91]. We showed administration of atorvastatin early and prior to the development of cardiac hypertrophy prevented cardiac hypertrophy [91]. However, interstitial fibrosis was not significantly affected, suggesting dissociation of mechanisms involved in the pathogenesis of hypertrophy and fibrosis in HCM. At mechanistic levels, treatment with statins reduced myocardial levels of lipid peroxides and oxidized mitochondrial DNA. In addition, treatment with statins restored expression levels of anti-oxidant genes and levels of activated membrane-bound Ras and pERK44/42. Studies in other animal models of cardiac hypertrophy and heart failure overall have corroborated the potential beneficial effects of statins in prevention and attenuation of hypertrophy and fibrosis [8, 28, 44, 102].
In contrast to animal studies, performed in a uniform population of HCM caused by a specific mutation, preliminary clinical studies in humans with HCM have not confirmed beneficial effects of statins in regression of established phenotype [3]. Bauersachs et al. reported no significant change in LV mass in 11 patients treated with 80 mg of atorvastatin for 9 months [3]. Likewise, we have performed a pilot feasibility study and have treated 21 HCM patients with established cardiac hypertrophy with escalating dose of atorvastatin, maximized at 80 mg per day for a total of 2 years. Fifteen and 11 patients completed 6 and 24 months of therapy. We detected no significant effects of treatment with atorvastatin on LV mass or ventricular wall thickness or other indices of cardiac hypertrophy and function.
N-acetylcysteine (NAC)
Oxidative stress has emerged as a common and important mediator of cardiac hypertrophic and fibrotic responses [103, 109]. Likewise, we have implicated increased oxidative stress and thiol-sensitive mechanisms in the pathogenesis of HCM and have shown that levels of lipid peroxides, oxidized mtDNA, and oxidized glutathione, markers of oxidative stress, are increased in the hearts of transgenic animal models of HCM [42, 51, 91]. It is also noteworthy that a common mechanism among losartan, aldosterone, simvastatin, and atorvastatin in inducing regression of cardiac hypertrophy and fibrosis is their actions against oxidative stress. In pursuit of testing the potential beneficial effects of NAC, we have performed placebo-control randomized studies in the β-MyHC-Q403 transgenic rabbits and cTnT-Q92 transgenic mice. NAC is a precursor to glutathione, which is the most abundant intracellular non-protein thiol pool against oxidative stress [119]. We have treated the genetically modified mice and rabbits with either a placebo or NAC for 3 to 14 months. We showed that treatment with NAC normalized myocardial and blood levels of oxidized glutathione (glutathione/glutathione disulfide ratio), which was increased in the transgenic rabbits treated with placebo. Treatment with NAC reversed cardiac and myocyte hypertrophy and interstitial fibrosis, prevented left ventricular systolic dysfunction, and improved arrhythmogenic propensity in our animal models (Fig. 1) [42, 51]. At mechanistic levels, we showed involvement of multiple mechanisms including effects on thiol-sensitive active protein kinase G, calcineurin target NFATc1 and phospho-p38, as well as glutathiolated sarcomeric protein cardiac α-actin. The finding of an anti-hypertrophic effect of NAC in our models is in accord with the results of a recent study in streptozotocin-induced diabetic cardiac hypertrophy [116]. Likewise, the observed anti-fibrotic effect of NAC is in accord with the findings in other models and organs [12, 27, 32, 39, 40]. The collective data indicating the anti-hypertrophic and anti-fibrotic effects of NAC in animal models of HCM and other forms of cardiac hypertrophy have set the stage for clinical trials to test potential salutary effects of this safe prodrug in reversal of cardiac hypertrophy and fibrosis in humans with HCM.

Effect of N-acetylcysteine (NAC) on left ventricular mass (LVM). LVM was increased in the β-myosin heavy chain-Q403 transgenic rabbits as compared to nontransgenic rabbits and continued to increase during the follow-up time (FU) in the placebo group. In contrast, LVM was reduced to normal in the NAC group at FU (N = 10 per group; dose, 500 mg/Kg/day; duration of treatment, 12 months)
Calcineurin Protein Phosphatase 2B Inhibitors
Inhibition of calcineurin-NFATc pathway has been established to reverse and prevent cardiac hypertrophy in various pathological states [10, 13, 38, 97, 100, 101]. However, Dr. Seidman’s group reported that treatment of the α-MyHC-Q403 mice with FK506 or cyclosporin A, inhibitors of calcineurin, worsened cardiac and myocyte hypertrophy, myocyte disarray, and interstitial fibrosis and increased mortality [15]. The observed deleterious effects of inhibition of calcineurin in the α-MyHC-Q403 mouse model of HCM have subdued the overall enthusiasm for the potential utility of calcineurin inhibitors in HCM. Nevertheless, given the diversity of the mechanisms involved in the pathogenesis of HCM and potential involvement of the calcineurin pathways in HCM due to MYOZ2 mutations, calcineurin inhibitors may impart beneficial effects only in a subset of HCM patients.
Challenges in Translating the Results of Experimental Therapies in Animal Models to Human Patients with HCM
Despite the remarkable results in the animal models of HCM documenting prevention, attenuation, and reversal of cardiac phenotype, the results of the preliminary studies with statins and losartan in humans have been less remarkable, if not completely null, as discussed earlier. We note that the human studies thus far have been conducted in a small number of patients and are considered feasibility studies that provide the preliminary data for designing the efficacy studies. Nevertheless, the discrepancy in the results of pharmacological interventions in humans and animal models of HCM is likely to reflect the genetic and phenotypic heterogeneity of HCM in humans as opposed to the genetically modified animals, wherein HCM is caused by a specific mutation in a more homogenous background. Accordingly, in a pharmacological intervention study, the animals share the same causal gene/mutation. In contrast, a group of human patients who are treated with the same pharmacological agent typically have a diverse array of causal genes and mutations. As noted earlier, the pathogenesis of HCM caused by different mutations could vary significantly. Therefore, diverse mechanisms are likely to be involved in a group of unrelated human HCM patients as opposed to more uniform mechanisms in the animal models. A pharmacological agent, such as statin or losartan, is unlikely to effectively block various mechanisms that may be involved in the pathogenesis of HCM in a group of unrelated human patients. The same agent, however, may effectively block the specific pathway involved in the pathogenesis of the phenotype in a specific animal model. For example, statin could inhibit geranyl-geranylation of Rho and Rac signaling, which may be involved in the pathogenesis of a subtype of HCM, represented by the β-MyHC-Q403 rabbits, but could not be activated in all cases of HCM caused by various mutations. It is also important to consider the potential confounding effects of the phenocopy conditions in humans that could render a specific pharmacological therapy ineffective or at least dilute its effect. In addition, the genetic background of animals is likely to be more homogenous, while human patients with HCM are expected to have heterogeneous genetic background. Moreover, the potential differences in the pharmacokinetic and pharmacodynamics of specific pharmacological agents in humans and animals merit considering when designing clinical trials in humans built on animal data.
Prospective
The current clinical diagnosis of HCM is based on detection of cardiac hypertrophy, typically on an echocardiogram [49, 52]. The clinical diagnosis, however, is neither sufficiently specific nor sufficiently sensitive [1, 9, 74, 75]. Phenocopy conditions, such as storage diseases, are typically indistinguishable from true (sarcomeric) HCM [9]. Likewise, individuals with the low penetrant mutations do not express cardiac hypertrophy despite carrying the causal mutations.
Given the limitation of the phenotype-based diagnosis, particularly for the early diagnosis, one would expect that DNA-based diagnosis would be used increasingly to complement the clinical diagnosis. The advent of deep sequencing technology, whether used in conjunction with target enrichment or whole genome sequencing, is expected to make DNA-based diagnosis practical, particularly in the early diagnosis and prior to and independent of the clinical phenotype. However, the DNA-based diagnosis is not without caveats, as no protein is perfect and each may carry a number of sequence variants that may not be responsible for the phenotype. The recent sequencing of individuals genomes clearly illustrates the presence of three to four million DNA sequence variants including several thousand insertions/deletions and copy number variants in each individual’s genome [5, 29, 35, 112, 113]. The challenge is to distinguish the DNA sequence variants that impart major phenotypic effects and/or are disease-causing variants and those that have modest if any biological effects [48].
In conjunction with the implementation of a DNA-based diagnosis, pharmacological treatment/prevention of HCM is also likely to evolve and become largely individualized. The notion is supported by the results of the mechanistic studies of causal mutations showing involvement of a diverse array of functional defects [17, 19, 22, 64, 65, 69, 72, 73, 85, 86, 93, 98, 107, 108]. The diversity of the responsible mechanisms perhaps is best illustrated for the Ca2+ sensitivity of myofibrillar ATPase activity, which varies according to the causal genes and is context dependent. The diversity of the responsible molecular pathways in HCM limits the potential utility of targeting a specific pathway. For example, Ca2+ channel blockers would be expected to be beneficial only in the subset of HCM that is caused by mutations that increase the Ca2+ sensitivity of myofibrillar force generation. In contrast, such agents could potentially be harmful in the subset that is caused by mutations that decrease the Ca2+ sensitivity of the myofibrils. Thus, one potential future approach would be to individualize the therapy based on the specific mechanisms involved in the pathogenesis of the phenotype.
The opposite end of the spectrum of approach to treatment of HCM is to broaden the application of a specific therapy to various subtypes of HCM by targeting mechanisms that are common to cardiac hypertrophic growth, the main clinical phenotype of HCM. An example of such common mechanism is increased oxidative stress, which is involved in the pathogenesis of cardiac hypertrophy in HCM as well as other pathological hypertrophy [42, 51, 91, 103, 109]. As discussed earlier, pharmacological interventions targeted to reduce oxidative stress have been used successfully to reverse cardiac hypertrophy and fibrosis in animal models of HCM as well as in other hypertrophic states [42, 103]. The findings in animal models of HCM beckon the need for clinical studies in humans to determine the potential beneficial effects of therapies targeted at oxidative stress pathway in prevention, attenuation, or reversal of cardiac phenotype in HCM.
Cardiac phenotype in human HCM evolves slowly and typically over a couple of decades and often even longer. The slow evolution of the phenotype may necessitate long-term pharmacological interventions in a relatively large number of patients in order to attain robust results with experimental therapies. Similarly, genetic-based diagnosis prior to expression of cardiac hypertrophy could afford the opportunity to intervene early to prevent the evolving phenotype. Whether prevention of the evolving phenotype or reversal of the established phenotype, the success will mandate recruitment of a large number of individuals to the study. Given that HCM is a relatively uncommon disease, collaborations among multiple centers would be essential for the design of adequately powered efficacy studies to test the potential salutary effects of experimental therapies in human patients with HCM.
References
Arad, M., Maron, B. J., Gorham, J. M., Johnson, W. H., Jr., Saul, J. P., Perez-Atayde, A. R., et al. (2005). Glycogen storage diseases presenting as hypertrophic cardiomyopathy. New England Journal of Medicine, 352, 362–372.
Araujo, A. Q., Arteaga, E., Ianni, B. M., Buck, P. C., Rabello, R., & Mady, C. (2005). Effect of Losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. American Journal of Cardiology, 96, 1563–1567.
Bauersachs, J., Stork, S., Kung, M., Waller, C., Fidler, F., Hoyer, C., et al. (2007). HMG CoA reductase inhibition and left ventricular mass in hypertrophic cardiomyopathy: a randomized placebo-controlled pilot study. European Journal of Clinical Investigation, 37, 852–859.
Benjamin, E. J., & Levy, D. (1999). Why is left ventricular hypertrophy so predictive of morbidity and mortality? American Journal of the Medical Sciences, 317, 168–175.
Bentley, D. R., Balasubramanian, S., Swerdlow, H. P., Smith, G. P., Milton, J., Brown, C. G., et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature, 456, 53–59.
Blanchard, E., Seidman, C., Seidman, J. G., LeWinter, M., & Maughan, D. (1999). Altered crossbridge kinetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circulation Research, 84, 475–483.
Boltwood, C. M., Jr., Chien, W., & Ports, T. (2004). Ventricular tachycardia complicating alcohol septal ablation. New England Journal of Medicine, 351, 1914–1915.
Chen, M. S., Xu, F. P., Wang, Y. Z., Zhang, G. P., Yi, Q., Zhang, H. Q., et al. (2004). Statins initiated after hypertrophy inhibit oxidative stress and prevent heart failure in rats with aortic stenosis. Journal of Molecular and Cellular Cardiology, 37, 889–896.
Chimenti, C., Pieroni, M., Morgante, E., Antuzzi, D., Russo, A., Russo, M. A., et al. (2004). Prevalence of fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation, 110, 1047–1053.
De Windt, L. J., Lim, H. W., Haq, S., Force, T., & Molkentin, J. D. (2000). Calcineurin promotes protein kinase C and c-Jun NH2-terminal kinase activation in the heart. Cross-talk between cardiac hypertrophic signaling pathways. Journal of Biological Chemistry, 275, 13571–13579.
Delbosc, S., Cristol, J. P., Descomps, B., Mimran, A., & Jover, B. (2002). Simvastatin prevents angiotensin II-induced cardiac alteration and oxidative stress. Hypertension, 40, 142–147.
Demedts, M., Behr, J., Buhl, R., Costabel, U., Dekhuijzen, R., Jansen, H. M., et al. (2005). High-Dose acetylcysteine in idiopathic pulmonary fibrosis. New England Journal of Medicine, 353, 2229–2242.
Ding, B., Price, R. L., Borg, T. K., Weinberg, E. O., Halloran, P. F., & Lorell, B. H. (1999). Pressure overload induces severe hypertrophy in mice treated with cyclosporine, an inhibitor of calcineurin. Circulation Research, 84, 729–734.
Elliott, P. M., Gimeno, B., Jr., Mahon, N. G., Poloniecki, J. D., & McKenna, W. J. (2001). Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet, 357, 420–424.
Fatkin, D., McConnell, B. K., Mudd, J. O., Semsarian, C., Moskowitz, I. G., Schoen, F. J., et al. (2000). An abnormal Ca(2+) response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. Journal of Clinical Investigation, 106, 1351–1359.
Flores-Ramirez, R., Lakkis, N. M., Middleton, K. J., Killip, D., Spencer, W. H., 3rd, & Nagueh, S. F. (2001). Echocardiographic insights into the mechanisms of relief of left ventricular outflow tract obstruction after nonsurgical septal reduction therapy in patients with hypertrophic obstructive cardiomyopathy. Journal of the American College of Cardiology, 37, 208–214.
Fujita, H., Sugiura, S., Momomura, S., Omata, M., Sugi, H., & Sutoh, K. (1997). Characterization of mutant myosins of Dictyostelium discoideum equivalent to human familial hypertrophic cardiomyopathy mutants. Molecular force level of mutant myosins may have a prognostic implication. Journal of Clinical Investigation, 99, 1010–1015.
Gaasch, W. H., & Zile, M. R. (2004). Left ventricular diastolic dysfunction and diastolic heart failure. Annual Review of Medicine, 55(373–94), 373–394.
Georgakopoulos, D., Christe, M. E., Giewat, M., Seidman, C. M., Seidman, J. G., & Kass, D. A. (1999). The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation [see comments]. Natural Medicines, 5, 327–330.
Haider, A. W., Larson, M. G., Benjamin, E. J., & Levy, D. (1998). Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. Journal of the American College of Cardiology, 32, 1454–1459.
Haim, T. E., Dowell, C., Diamanti, T., Scheuer, J., & Tardiff, J. C. (2007). Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. Journal of Molecular and Cellular Cardiology, 42, 1098–1110.
Harada, K., & Potter, J. D. (2004). Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. Journal of Biological Chemistry, 279, 14488–14495.
Harada, K., Takahashi-Yanaga, F., Minakami, R., Morimoto, S., & Ohtsuki, I. (2000). Functional consequences of the deletion mutation deltaGlu160 in human cardiac troponin T. Journal of Biochemistry, 127, 263–268.
Hernandez, O. M., Szczesna-Cordary, D., Knollmann, B. C., Miller, T., Bell, M., Zhao, J., et al. (2005). F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. Journal of Biological Chemistry, 280, 37183–37194.
Ho, C. Y., Sweitzer, N. K., McDonough, B., Maron, B. J., Casey, S. A., Seidman, J. G., et al. (2002). Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation, 105, 2992–2997.
Hori, Y., Ueda, M., Nakayama, T., Saegusa, N., Uehara, M., Lee, K., et al. (2007). Occurrence of de novo sustained monomorphic ventricular tachycardia induced after percutaneous transluminal alcohol septal myocardial ablation for hypertrophic obstructive cardiomyopathy. International Journal of Cardiology, 119, 403–407.
Hoshikawa, Y., Ono, S., Suzuki, S., Tanita, T., Chida, M., Song, C., et al. (2001). Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. Journal of Applied Physiology, 90, 1299–1306.
Indolfi, C., Di Lorenzo, E., Perrino, C., Stingone, A. M., Curcio, A., Torella, D., et al. (2002). Hydroxymethylglutaryl coenzyme A reductase inhibitor simvastatin prevents cardiac hypertrophy induced by pressure overload and inhibits p21ras activation. Circulation, 106, 2118–2124.
Kim, J. I., Ju, Y. S., Park, H., Kim, S., Lee, S., Yi, J. H., et al. (2009). A highly annotated whole-genome sequence of a Korean individual. Nature, 460, 1011–1015.
Kitaoka, H., Kubo, T., Okawa, M., Hitomi, N., Furuno, T., & Doi, Y. L. (2006). Left ventricular remodeling of hypertrophic cardiomyopathy: longitudinal observation in rural community. Circulation, 70, 1543–1549.
Knoll, R., Hoshijima, M., Hoffman, H. M., Person, V., Lorenzen-Schmidt, I., Bang, M. L., et al. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell, 111, 943–955.
Kopp, J., Seyhan, H., Muller, B., Lanczak, J., Pausch, E., Gressner, A. M., et al. (2006). N-acetyl-L-cysteine abrogates fibrogenic properties of fibroblasts isolated from Dupuytren’s disease by blunting TGF-beta signalling. Journal of Cellular and Molecular Medicine, 10, 157–165.
Krumholz, H. M., Larson, M., & Levy, D. (1995). Prognosis of left ventricular geometric patterns in the Framingham Heart Study. Journal of the American College of Cardiology, 25, 879–884.
Levy, D., Garrison, R. J., Savage, D. D., Kannel, W. B., & Castelli, W. P. (1990). Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. New England Journal of Medicine, 322, 1561–1566.
Levy, S., Sutton, G., Ng, P. C., Feuk, L., Halpern, A. L., Walenz, B. P., et al. (2007). The diploid genome sequence of an individual human. PLoS Biology, 5, e254.
Liao, J. K., & Laufs, U. (2005). Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology, 45, 89–118.
Lim, D. S., Lutucuta, S., Bachireddy, P., Youker, K., Evans, A., Entman, M., et al. (2001). Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation, 103, 789–791.
Lim, H. W., De Windt, L. J., Mante, J., Kimball, T. R., Witt, S. A., Sussman, M. A., et al. (2000). Reversal of cardiac hypertrophy in transgenic disease models by calcineurin inhibition. Journal of Molecular and Cellular Cardiology, 32, 697–709.
Liu, D. D., Kao, S. J., & Chen, H. I. (2008). N-acetylcysteine attenuates acute lung injury induced by fat embolism. Critical Care Medicine, 36, 565–571.
Liu, R. M., Liu, Y., Forman, H. J., Olman, M., & Tarpey, M. M. (2004). Glutathione regulates transforming growth factor-{beta}-stimulated collagen production in fibroblasts. American Journal of Physiology. Lung Cellular and Molecular Physiology, 286, L121–L128.
Lombardi, R., Bell, A., Senthil, V., Sidhu, J., Noseda, M., Roberts, R., et al. (2008). Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies. Cardiovascular Research, 79, 109–117.
Lombardi, R., Rodriguez, G., Chen, S. N., Ripplinger, C. M., Li, W., Chen, J., et al. (2009). Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation, 119, 1398–1407.
Lowey, S., Lesko, L. M., Rovner, A. S., Hodges, A. R., White, S. L., Low, R. B., et al. (2008). Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. Journal of Biological Chemistry, 283, 20579–20589.
Luo, J. D., Zhang, W. W., Zhang, G. P., Guan, J. X., & Chen, X. (1999). Simvastatin inhibits cardiac hypertrophy and angiotensin-converting enzyme activity in rats with aortic stenosis. Clinical and Experimental Pharmacology and Physiology, 26, 903–908.
Lutucuta, S., Tsybouleva, N., Ishiyama, M., Defreitas, G., Wei, L., Carabello, B., et al. (2004). Induction and reversal of cardiac phenotype of human hypertrophic cardiomyopathy mutation cardiac troponin T-Q92 in switch on-switch off bigenic mice. Journal of the American College of Cardiology, 44, 2221–2230.
Marian, A. J. (2000). Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet, 355, 58–60.
Marian, A. J. (2007). Hypertrophic cardiomyopathy. In C. C. Liew & V. Dzau (Eds.), Cardiovascular genetics and genomics for the gardiologist (pp. 30–54). Hoboken, NJ:Wiley-Blackwell.
Marian, A. J. (2008). Clinical implications of the “personal” genome. Current Atherosclerosis Reports, 10, 361–363.
Marian, A. J. (2008). Genetic determinants of cardiac hypertrophy. Current Opinion in Cardiology, 23, 199–205.
Marian, A. J. (2008). Hypertrophic Cardiomyopathy. In R. E. Rackel & E. T. Bope (Eds.), Conn’s current therapy (pp. 333–335). Philadelphia: Saunders-Elsevier.
Marian, A. J., Senthil, V., Chen, S. N., & Lombardi, R. (2006). Antifibrotic effects of antioxidant N-acetylcysteine in a mouse model of human hypertrophic cardiomyopathy mutation. Journal of the American College of Cardiology, 47, 827–834.
Maron, B. J. (2002). Hypertrophic cardiomyopathy: a systematic review. Journal of the American Medical Association, 287, 1308–1320.
Maron, B. J., Dearani, J. A., Ommen, S. R., Maron, M. S., Schaff, H. V., Gersh, B. J., et al. (2004). The case for surgery in obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 44, 2044–2053.
Maron, B. J., Doerer, J. J., Haas, T. S., Tierney, D. M., & Mueller, F. O. (2009). Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation, 119, 1085–1092.
Maron, B. J., Shen, W. K., Link, M. S., Epstein, A. E., Almquist, A. K., Daubert, J. P., et al. (2000). Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. New England Journal of Medicine, 342, 365–373.
Maron, B. J., Spirito, P., Shen, W. K., Haas, T. S., Formisano, F., Link, M. S., et al. (2007). Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. Journal of the American Medical Association, 298, 405–412.
Maron, M. S., Olivotto, I., Zenovich, A. G., Link, M. S., Pandian, N. G., Kuvin, J. T., et al. (2006). Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation, 114, 2232–2239.
Matsumura, Y., Elliott, P. M., Virdee, M. S., Sorajja, P., Doi, Y., & McKenna, W. J. (2002). Left ventricular diastolic function assessed using Doppler tissue imaging in patients with hypertrophic cardiomyopathy: relation to symptoms and exercise capacity. Heart, 87, 247–251.
McGregor, J. B., Rahman, A., Rosanio, S., Ware, D., Birnbaum, Y., & Saeed, M. (2004). Monomorphic ventricular tachycardia: a late complication of percutaneous alcohol septal ablation for hypertrophic cardiomyopathy. American Journal of the Medical Sciences, 328, 185–188.
McLeod, C. J., Ommen, S. R., Ackerman, M. J., Weivoda, P. L., Shen, W. K., Dearani, J. A., et al. (2007). Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. European Heart Journal, 28, 2583–2588.
McMahon, C. J., Nagueh, S. F., Pignatelli, R. H., Denfield, S. W., Dreyer, W. J., Price, J. F., et al. (2004). Characterization of left ventricular diastolic function by tissue Doppler imaging and clinical status in children with hypertrophic cardiomyopathy. Circulation, 109, 1756–1762.
McTaggart, D. R. (2004). Diltiazem reverses tissue Doppler velocity abnormalities in pre-clinical hypertrophic cardiomyopathy. Heart, Lung & Circulation, 13, 39–40.
Montgomery, D. E., Tardiff, J. C., & Chandra, M. (2001). Cardiac troponin T mutations: correlation between the type of mutation and the nature of myofilament dysfunction in transgenic mice. Journal of Physiology, 536, 583–592.
Morimoto, S., Lu, Q. W., Harada, K., Takahashi-Yanaga, F., Minakami, R., Ohta, M., et al. (2002). Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America, 99, 913–918.
Morimoto, S., Nakaura, H., Yanaga, F., & Ohtsuki, I. (1999). Functional consequences of a carboxyl terminal missense mutation Arg278Cys in human cardiac troponin T. Biochemical and Biophysical Research Communications, 261, 79–82.
Morita, H., Seidman, J., & Seidman, C. E. (2005). Genetic causes of human heart failure. Journal of Clinical Investigation, 115, 518–526.
Moriyama, T., Kawada, N., Nagatoya, K., Takeji, M., Horio, M., Ando, A., et al. (2001). Fluvastatin suppresses oxidative stress and fibrosis in the interstitium of mouse kidneys with unilateral ureteral obstruction. Kidney International, 59, 2095–2103.
Nagueh, S. F., Bachinski, L. L., Meyer, D., Hill, R., Zoghbi, W. A., Tam, J. W., et al. (2001). Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation, 104, 128–130.
Nagueh, S. F., Chen, S., Patel, R., Tsybouleva, N., Lutucuta, S., Kopelen, H. A., et al. (2004). Evolution of expression of cardiac phenotypes over a 4-year period in the beta-myosin heavy chain-Q403 transgenic rabbit model of human hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology, 36, 663–673.
Nagueh, S. F., Kopelen, H. A., Lim, D. S., Zoghbi, W. A., Quinones, M. A., Roberts, R., et al. (2000). Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation, 102, 1346–1350.
Nagueh, S. F., Lakkis, N. M., Middleton, K. J., Spencer, W. H., III, Zoghbi, W. A., & Quinones, M. A. (1999). Doppler estimation of left ventricular filling pressures in patients with hypertrophic cardiomyopathy. Circulation, 99, 254–261.
Nakaura, H., Morimoto, S., Yanaga, F., Nakata, M., Nishi, H., Imaizumi, T., et al. (1999). Functional changes in troponin T by a splice donor site mutation that causes hypertrophic cardiomyopathy. American Journal of Physiology, 277, C225–C232.
Nakaura, H., Yanaga, F., Ohtsuki, I., & Morimoto, S. (1999). Effects of missense mutations Phe110Ile and Glu244Asp in human cardiac troponin T on force generation in skinned cardiac muscle fibers. Journal of Biochemistry (Tokyo), 126, 457–460.
Niimura, H., Bachinski, L. L., Sangwatanaroj, S., Watkins, H., Chudley, A. E., McKenna, W., et al. (1998). Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. New England Journal of Medicine, 338, 1248–1257.
Niimura, H., Patton, K. K., McKenna, W. J., Soults, J., Maron, B. J., Seidman, J. G., et al. (2002). Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation, 105, 446–451.
Oi, S., Haneda, T., Osaki, J., Kashiwagi, Y., Nakamura, Y., Kawabe, J., et al. (1999). Lovastatin prevents angiotensin II-induced cardiac hypertrophy in cultured neonatal rat heart cells. European Journal of Pharmacology, 376, 139–148.
Olivotto, I., Gistri, R., Petrone, P., Pedemonte, E., Vargiu, D., & Cecchi, F. (2003). Maximum left ventricular thickness and risk of sudden death in patients with hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 41, 315–321.
Ommen, S. R., Maron, B. J., Olivotto, I., Maron, M. S., Cecchi, F., Betocchi, S., et al. (2005). Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 46, 470–476.
Ommen, S. R., Shah, P. M., & Tajik, A. J. (2008). Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy: past, present and future. Heart, 94, 1276–1281.
Osio, A., Tan, L., Chen, S. N., Lombardi, R., Nagueh, S. F., Shete, S., et al. (2007). Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circulation Research, 100, 766–768.
Ostman-Smith, I., Wettrell, G., & Riesenfeld, T. (1999). A cohort study of childhood hypertrophic cardiomyopathy: improved survival following high-dose beta-adrenoceptor antagonist treatment. Journal of the American College of Cardiology, 34, 1813–1822.
Patel, R., Nagueh, S. F., Tsybouleva, N., Abdellatif, M., Lutucuta, S., Kopelen, H. A., et al. (2001). Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation, 104, 317–324.
Ripplinger, C. M., Li, W., Hadley, J., Chen, J., Rothenberg, F., Lombardi, R., et al. (2007). Enhanced transmural fiber rotation and connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation Research, 101, 1049–1057.
Rust, E. M., Albayya, F. P., & Metzger, J. M. (1999). Identification of a contractile deficit in adult cardiac myocytes expressing hypertrophic cardiomyopathy-associated mutant troponin T proteins. Journal of Clinical Investigation, 103, 1459–1467.
Sarikas, A., Carrier, L., Schenke, C., Doll, D., Flavigny, J., Lindenberg, K. S., et al. (2005). Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovascular Research, 66, 33–44.
Sata, M., & Ikebe, M. (1996). Functional analysis of the mutations in the human cardiac beta-myosin that are responsible for familial hypertrophic cardiomyopathy. Implication for the clinical outcome. Journal of Clinical Investigation, 98, 2866–2873.
Schiffmann, R., Murray, G. J., Treco, D., Daniel, P., Sellos-Moura, M., Myers, M., et al. (2000). Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proceedings of the National Academy of Sciences of the United States of America, 97, 365–370.
Seggewiss, H. (2001). Current status of alcohol septal ablation for patients with hypertrophic cardiomyopathy. Current Cardiology Reports, 3, 160–166.
Seggewiss, H., Gleichmann, U., Faber, L., Fassbender, D., Schmidt, H. K., & Strick, S. (1998). Percutaneous transluminal septal myocardial ablation in hypertrophic obstructive cardiomyopathy: acute results and 3-month follow-up in 25 patients. Journal of the American College of Cardiology, 31, 252–258.
Semsarian, C., Ahmad, I., Giewat, M., Georgakopoulos, D., Schmitt, J. P., McConnell, B. K., et al. (2002). The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. Journal of Clinical Investigation, 109, 1013–1020.
Senthil, V., Chen, S. N., Tsybouleva, N., Halder, T., Nagueh, S. F., Willerson, J. T., et al. (2005). Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation Research, 97, 285–292.
Sherrid, M. V., Barac, I., McKenna, W. J., Elliott, P. M., Dickie, S., Chojnowska, L., et al. (2005). Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 45, 1251–1258.
Sirenko, S. G., Potter, J. D., & Knollmann, B. C. (2006). Differential effect of troponin T mutations on the inotropic responsiveness of mouse hearts—role of myofilament Ca2+ sensitivity increase. Journal of Physiology (Online), 575, 201–213.
Solaro, R. J., Varghese, J., Marian, A. J., & Chandra, M. (2002). Molecular mechanisms of cardiac myofilament activation: modulation by pH and a troponin T mutant R92Q. Basic Research in Cardiology, 97(Suppl 1), I102–I110.
Sorajja, P., Valeti, U., Nishimura, R. A., Ommen, S. R., Rihal, C. S., Gersh, B. J., et al. (2008). Outcome of alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation, 118, 131–139.
Spirito, P., Bellone, P., Harris, K. M., Bernabo, P., Bruzzi, P., & Maron, B. J. (2000). Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. New England Journal of Medicine, 342, 1778–1785.
Sussman, M. A., Lim, H. W., Gude, N., Taigen, T., Olson, E. N., Robbins, J., et al. (1998). Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science, 281, 1690–1693.
Sweeney, H. L., Feng, H. S., Yang, Z., & Watkins, H. (1998). Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: insights into disease pathogenesis and troponin function. Proceedings of the National Academy of Sciences of the United States of America, 95, 14406–14410.
Szczesna, D., Zhang, R., Zhao, J., Jones, M., Guzman, G., & Potter, J. D. (2000). Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Journal of Biological Chemistry, 275, 624–630.
Taigen, T., De Windt, L. J., Lim, H. W., & Molkentin, J. D. (2000). Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proceedings of the National Academy of Sciences of the United States of America, 97, 1196–1201.
Takeda, Y., Yoneda, T., Demura, M., Usukura, M., & Mabuchi, H. (2002). Calcineurin inhibition attenuates mineralocorticoid-induced cardiac hypertrophy. Circulation, 105, 677–679.
Takemoto, M., Node, K., Nakagami, H., Liao, Y., Grimm, M., Takemoto, Y., et al. (2001). Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. Journal of Clinical Investigation, 108, 1429–1437.
Takimoto, E., & Kass, D. A. (2007). Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension, 49, 241–248.
Talreja, D. R., Nishimura, R. A., Edwards, W. D., Valeti, U. S., Ommen, S. R., Tajik, A. J., et al. (2004). Alcohol septal ablation versus surgical septal myectomy: comparison of effects on atrioventricular conduction tissue. Journal of the American College of Cardiology, 44, 2329–2332.
Tam, S. K., Gu, W., Mahdavi, V., & Nadal-Ginard, B. (1995). Cardiac myocyte terminal differentiation. Potential for cardiac regeneration. Annals of the New York Academy of Sciences, 752(72–9), 72–79.
Tanigawa, G., Jarcho, J. A., Kass, S., Solomon, S. D., Vosberg, H. P., Seidman, J. G., et al. (1990). A molecular basis for familial hypertrophic cardiomyopathy: an alpha/beta cardiac myosin heavy chain hybrid gene. Cell, 62, 991–998.
Tardiff, J. C., Factor, S. M., Tompkins, B. D., Hewett, T. E., Palmer, B. M., Moore, R. L., et al. (1998). A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. Journal of Clinical Investigation, 101, 2800–2811.
Tardiff, J. C., Hewett, T. E., Palmer, B. M., Olsson, C., Factor, S. M., Moore, R. L., et al. (1999). Cardiac troponin T mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. Journal of Clinical Investigation, 104, 469–481.
Tirouvanziam, R., Conrad, C. K., Bottiglieri, T., Herzenberg, L. A., Moss, R. B., & Herzenberg, L. A. (2006). High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proceedings of the National Academy of Sciences of the United States of America, 103, 4628–4633.
Tobacman, L. S., Lin, D., Butters, C., Landis, C., Back, N., Pavlov, D., et al. (1999). Functional consequences of troponin T mutations found in hypertrophic cardiomyopathy. Journal of Biological Chemistry, 274, 28363–28370.
Tsybouleva, N., Zhang, L., Chen, S., Patel, R., Lutucuta, S., Nemoto, S., et al. (2004). Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation, 109, 1284–1291.
Wang, J., Wang, W., Li, R., Li, Y., Tian, G., Goodman, L., et al. (2008). The diploid genome sequence of an Asian individual. Nature, 456, 60–65.
Wheeler, D. A., Srinivasan, M., Egholm, M., Shen, Y., Chen, L., McGuire, A., et al. (2008). The complete genome of an individual by massively parallel DNA sequencing. Nature, 452, 872–876.
Wilcox, W. R., Banikazemi, M., Guffon, N., Waldek, S., Lee, P., Linthorst, G. E., et al. (2004). Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. American Journal of Human Genetics, 75, 65–74.
Woo, A., Williams, W. G., Choi, R., Wigle, E. D., Rozenblyum, E., Fedwick, K., et al. (2005). Clinical and echocardiographic determinants of long-term survival after surgical myectomy in obstructive hypertrophic cardiomyopathy. Circulation, 111, 2033–2041.
Xia, Z., Kuo, K. H., Nagareddy, P. R., Wang, F., Guo, Z., Guo, T., et al. (2007). N-acetylcysteine attenuates PKCbeta2 overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats. Cardiovascular Research, 73, 770–782.
Yamazaki, T., Suzuki, J., Shimamoto, R., Tsuji, T., Ohmoto-Sekine, Y., Ohtomo, K., et al. (2007). A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an Angiotensin II receptor blocker. International Heart Journal, 48, 715–724.
Yanaga, F., Morimoto, S., & Ohtsuki, I. (1999). Ca2+ sensitization and potentiation of the maximum level of myofibrillar ATPase activity caused by mutations of troponin T found in familial hypertrophic cardiomyopathy. Journal of Biological Chemistry, 274, 8806–8812.
Zafarullah, M., Li, W. Q., Sylvester, J., & Ahmad, M. (2003). Molecular mechanisms of N-acetylcysteine actions. Cellular and Molecular Life Sciences, 60, 6–20.
Zile, M. R., Baicu, C. F., & Gaasch, W. H. (2004). Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle. New England Journal of Medicine, 350, 1953–1959.
Zile, M. R., & Brutsaert, D. L. (2002). New concepts in diastolic dysfunction and diastolic heart failure: Part I: diagnosis, prognosis, and measurements of diastolic function. Circulation, 105, 1387–1393.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Marian, A.J. Experimental Therapies in Hypertrophic Cardiomyopathy. J. of Cardiovasc. Trans. Res. 2, 483–492 (2009). https://doi.org/10.1007/s12265-009-9132-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-009-9132-7