
Abstract
Hypertrophic cardiomyopathy is the most common inherited heart disease. Although it was first described over 50 years ago, there has been little in the way of novel disease-specific therapeutic development for these patients. Current treatment practice largely aims at symptomatic control using old drugs made for other diseases and does little to modify the disease course. Septal reduction by surgical myectomy or percutaneous alcohol septal ablation are well-established treatments for pharmacologic-refractory left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients. In recent years, there has been a relative surge in the development of innovative therapeutics, which aim to target the complex molecular pathophysiology and resulting hemodynamics that underlie hypertrophic cardiomyopathy. Herein, we review the new and emerging therapeutics for hypertrophic cardiomyopathy, which include pharmacologic attenuation of sarcomeric calcium sensitivity, allosteric inhibition of cardiac myosin, myocardial metabolic modulation, and renin-angiotensin-aldosterone system inhibition, as well as structural intervention by percutaneous mitral valve plication and endocardial radiofrequency ablation of septal hypertrophy. In conclusion, while further development of these therapeutic strategies is ongoing, they each mark a significant and promising advancement in treatment for hypertrophic cardiomyopathy patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common genetic heart disease with a prevalence in the general adult population of approximately 1 in 500 [1]. It is a common cause of sudden cardiac death in the young and a prevalent cause of morbidity and mortality in all ages [2, 3]. HCM is characterized by complex pathophysiology, which manifests in a heterogeneous clinical presentation. Typical features of HCM include asymmetric septal hypertrophy, diastolic dysfunction, a left ventricular outflow tract (LVOT) gradient, microvascular ischemia, altered cardiac myocyte energetics, and arrhythmogenicity [4, 5].
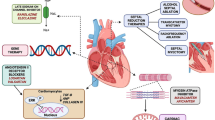
Since HCM was first described over 50 years ago, there has been little in the way of novel therapeutic development and the disease has largely been treated using old drugs developed for the treatment of other diseases [6, 7]. Currently used drugs in HCM are all negative inotropic agents focused on improving symptoms. In pharmacologic refractory obstructive HCM, septal reduction by surgical myectomy and percutaneous transcoronary alcohol septal ablation are well-established and effective therapies [8,9,10]. Recently, however, there has been a surge in the development of novel, disease-specific treatment in HCM. Here, we review pharmacologic treatment strategies which include attenuation of sarcomeric calcium sensitivity, allosteric inhibition of cardiac myosin, myocardial metabolic modulation, and renin-angiotensin-aldosterone system inhibition as well as structural interventions including percutaneous mitral valve plication and endocardial radiofrequency ablation of septal hypertrophy. Targets of each pharmacologic treatment strategy are summarized in Fig. 1 and Table 1. Structural interventions are summarized in Table 1.

Targets of pharmacologic therapies for hypertrophic cardiomyopathy. Examples discussed in the text are written beneath each target. AMPK adenosine monophosphate-activated protein kinase, ARB angiotensin receptor blocker, AT1 angiotensin receptor 1, ATP adenosine triphosphate, ADP adenosine diphosphate, CaMKII calcium/calmodulin-dependent protein kinase II, I CaL late calcium channel, I NaL late sodium channel, NAC N-acetyl cysteine, NCX sodium-calcium exchanger, P i inorganic phosphate, ROS reactive oxygen species
Pharmacologic therapies
Attenuation of sarcomeric calcium sensitivity
Dysregulation of calcium signaling in the cardiac myocyte plays a critical role in the HCM disease state. It contributes to both diastolic dysfunction and arrhythmogenicity, which oftentimes precede the development of hypertrophy and symptoms in HCM patients [31,31,32,33,34,36]. Mutations in sarcomeric proteins including cardiac myosin binding protein C (cMyBP-C) and cardiac troponin I and C (cTnI and cTnC) have been shown to cause enhanced calcium affinity in cardiac myofilaments in human and animal models of HCM [37,37,39]. It is hypothesized that increased myofilament Ca affinity leads to “calcium trapping” in the mutant sarcomere causing significant alterations in intracellular calcium handling with activation of the calcium-calmodulin kinase II (CaMKII) pathway [40, 41].
The delayed sodium channel is a downstream target of CaMKII, which is oftentimes upregulated in HCM and leads to further increases in intracellular calcium by the Na-Ca exchange protein setting up a vicious cycle as shown in Fig. 1 [13]. These electrophysiologic alterations cause an increased action potential duration as well as increased incidence of early and delayed after depolarizations (EADs and DADs) that underlie enhanced arrhythmogenicity and furthermore result in higher diastolic calcium concentrations leading to impaired myocardial relaxation [31,31,32,34].
In a mouse model of HCM (Arg403Gln missense mutation in the α cardiac myosin heavy chain), early administration of the L-type calcium channel inhibitor, diltiazem resulted in long-term (39-week) attenuation of hypertrophic pathology and improved cardiac function in pre-hypertrophic mice [11]. It is thought that by blocking the L-type calcium channel before development of the HCM phenotype, diltiazem prevents calcium trapping within the mutant sarcomere, thereby disrupting progression of pathologic hypertrophy. These findings were applied clinically in a double-blinded pilot study of diltiazem versus placebo as a disease modifying agent in pre-hypertrophic sarcomeric HCM mutation carriers over a 3-year follow-up. Compared to controls, diltiazem-treated MYBPC3 mutation carriers (n = 12) but not MYH7 mutation carriers (n = 21) exhibited significantly less increase in echocardiographic left ventricular (LV) wall thickness and cardiac magnetic resonance (CMR)-measured LV mass index as well as improved LV filling pressures as represented by a decrease in E/E′ [12]. By identifying at risk mutation carriers, use of diltiazem as a disease-modifying agent may potentially attenuate dysregulated calcium cycling and subsequent HCM phenotypic expression. Future large-scale trials are required however to explore this strategy further.
Ranolazine is a late Na channel inhibitor currently used as an antianginal drug. When administered to cardiac myocytes isolated from HCM patients in vitro, ranolazine was shown topartially reverse dysregulated calcium signaling, arrhythmogenicity, and diastolic dysfunction [13]. Furthermore, ranolazine improves markers of diastolic dysfunction in a hypertensive mouse (deoxycorticosterone acetate (DOCA)-salt) model [42]. Similar promising findings were seen in a transgenic mouse model of HCM (Mybpc3-targeted knock-in), though some of the beneficial effects seen in these studies are thought to be due to ranolazine’s beta blocker properties and to a lesser degree by blockage of the late sodium current [42, 43]. In a small, a single-center, open-label pilot study, ranolazine resulted inimproved symptoms and quality of life at 2-month follow-up in 11 symptomatic patients with HCM despite maximum tolerated medical therapy [14]. RESTYLE-HCM, a multicenter placebo-controlled trial, looks to study the efficacy of ranolazine on improving exercise tolerance in symptomatic HCM patients by measurement of peak oxygen consumption [15]. Results of this trial are awaited. LIBERTY-HCM, a multicenter international phase II/III trial, evaluated the efficacy of eleclazine—a more specific and more potent late sodium channel inhibitor than ranolazine—to see if compared to placebo, eleclazine improves exercise capacity as measured by peak oxygen consumption during cardiopulmonary exercise testing in patients with symptomatic HCM [16]. Unfortunately, during the time of this writing, the LIBERTY-HCM study was terminated by the sponsoring party for unclear reasons [16]. The termination may be related to the failure of eleclazine to meet its primary endpoint in a concurrent study on its use in patients with type 3 long QT syndrome although this is unknown [17].
Allosteric inhibition of cardiac myosin
There are reported to be hundreds of pathogenic mutations within the seven-component regulated contractile complex of the cardiac sarcomere, which contribute to HCM pathophysiology [44]. These are predominantly missense mutations in the beta myosin heavy chain or the regulatory protein, cMyBP-C, which attenuate the actin-myosin interaction as well as cause marked variability in calcium sensitivity between muscle fibers leading to imbalances in force generation, contractile dysfunction, and myofibril disarray [31, 45]. Cumulatively, these mutations result in characteristic hypercontractility and diastolic dysfunction of HCM cardiac myocytes that often precedes hypertrophy [31, 46, 47]. Interestingly, however, a few reported mutations have been shown to result in myocyte hypocontractility underscoring that HCM pathophysiology is still not entirely understood [18]. While the altered components of the hyperdynamic contractile apparatus in HCM are attractive targets for therapeutic intervention, the varied effects of different mutations complicate pharmacologic development. Inducible pluripotent stem cell HCM models have recently been reported in the literature to help further our understanding of the complex genetic and molecular pathophysiology underlying HCM and may set the stage for future target-based drug development for HCM [19].
The power of sarcomere contraction is determined by the combined force generated by all myosin heads within the sarcomere—the ensemble force—as well as their velocity of movement along actin myofilaments. The ensemble force (Fe) is determined by the total number of myosin heads overlapping with actin filaments (Nt) in the muscle and the duty ratio (ts/tc), which is the proportion of time spent in the strongly bound actin-myosin interaction—the force-generating state—out of total cycle time, Fe = f[(ts/tc)xNt] [46].
MYK-461 is a small-molecule inhibitor of myosin ATPase, which prevents transition into the strongly bound state of the myosin cross-bridge cycle, thereby decreasing the duty ratio and thus the ensemble force, power, and contractility of the sarcomere [48]. In a transgenic mouse model of HCM, introducing human disease-causing mutations into the murine α-cardiac myosin heavy chain gene, Green et al. demonstrated that MYK-461 caused a dose-dependent reduction in fractional shortening of cardiac muscle with no measurable effect on skeletal muscle despite its low affinity demonstrated in rabbit skeletal myosin. Furthermore, MYK-461 was shown to prevent progression to LV hypertrophy in young pre-hypertrophic HCM mice and was also able to cause a significant reduction in LV wall thickness promoting partial regression of hypertrophy in older HCM mice. Pre-hypertrophic mice administered MYK-461 also demonstrated a reduction in fibrosis on histology as well as normalization of pro-fibrotic gene expression in cardiomyocytes. However, no change in histology or gene expression was seen in hypertrophic mice [48]. Although certain mutations in HCM may result in increased myosin ATPase activity, it is unclear if this will provide MYK-461 sufficient selectivity for abnormal cardiac myocytes over normal to avoid deleterious effect on an already compromised cardiac function [47]. MYK-461 is currently undergoing phase II trials. Preliminary data indicate that the drug is well tolerated with dose-dependent pharmacokinetics [49].
Considering the multitude of mutations in HCM involving the contractile apparatus and the sensitivity of these proteins to allosteric effects, there is significant potential for future targeted therapeutic development. Future potential targets involved in the cross-bridging cycle include cMyBP-C to augment its inhibitory effect on contractility as well as cTnC inhibition to damper enhanced calcium sensitivity [39].
Metabolic modulation: energy depletion hypothesis
Inefficient ATP utilization by mutant sarcomeric proteins in HCM, most notably within the gene encoding beta myosin heavy chain (MYH7 gene), leads to increased energy demand within the cardiac myocyte (more ATP is required per unit of force generated) [50, 51]. The increased energetic cost of contraction compromises the ability of the cardiac myocyte to perform other energy-intensive cellular functions, most notably the maintenance of critical subcellular electrochemical gradients by the sarcoplasmic reticulum calcium re-uptake (SERCA2) pump leading to increased cytosolic calcium concentrations [52]. Increases in cytosolic calcium recruit downstream transcriptional processes, which upregulate signaling pathways such as the CaMKII pathway and the mitogen-activated protein kinase (MAPK) pathway whose downstream targets ultimately result in myocyte hypertrophy [38, 53].
Increased ATP consumption results in a decrease in the cellular ATP to ADP ratio, upregulating AMP activated protein kinase (AMPK) signaling whose downstream targets stimulate both fatty acid and carbohydrate metabolism, both highly oxygen consumptive processes [54]. The cardiac phosphocreatine to ATP ratio (PCr/ATP) is a measurement of the energy status of cardiac muscle. In HCM patients, PCr/ATP is reduced by up to 30% irrespective of presence of hypertrophy providing evidence of an energy deficit [55].
In addition to increased energy demand by the inefficient HCM contractile apparatus, there is also evidence for compromised energy supply due to mutations causing morphological and functional alterations in HCM mitochondria [56]. Further exacerbating the energy deficit, coronary microvascular dysfunction in HCM hearts limits oxygen delivery to the hypermetabolic tissues and has been shown to contribute to an overall poorer disease prognosis [20, 57,57,59] As such, energy depletion by both supply and demand is a primary process, occurring in pre-hypertrophic patients leading to hypertrophy and not as a result of hypertrophy.
Mismatch between cardiac myocyte energy supply and demand leads to an oxidative cellular state wherein development of reactive oxygen species (ROS) induces S-glutathionylation of cMyBP-C. Functionally, this redox modification augments myofilament calcium sensitivity and depresses cross-bridge cycling kinetics contributing to diastolic dysfunction, thus exacerbating the HCM phenotype [21, 22]. In transgenic animal models of HCM (tropomyosin (Tm-E180G) mice) and beta-myosin heavy-chain (MHC Q403) transgenic rabbits), administration of the glutathione precursor, N-acetylcysteine (NAC), has been shown to reduce levels of glutathionylated myofilaments with reversal of increased myofilament calcium sensitivity, diastolic dysfunction, myocyte hypertrophy, and fibrosis [22, 60]. The phase I HALT trial is currently recruiting participants to evaluate tolerability of an oral form of NAC as well as its ability to alter indices of cardiac mass and function administered over 1 year [61].
Impairment of LV relaxation, a highly energy-consumptive process leading to diastolic dysfunction in HCM, is consistent with the energy depletion hypothesis and contributes to diminished exercise capacity [62, 63]. Perhexiline is a metabolic modulator of myocardial substrate utilization that shifts cellular metabolism to favor more efficient carbohydrate metabolism by inhibiting mitochondrial free fatty acid uptake and utilization, thus improving myocardial energy efficiency [23, 64]. The ability of perhexiline to correct sarcomeric energy depletion was evaluated in a randomized controlled trial comparing perhexiline to placebo in 46 patients with symptomatic non-obstructive cardiomyopathy despite beta-adrenergic or calcium channel blockade over a period of 3–6 months. Those who received perhexiline demonstrated significant improvement in the PCr/ATP as measured by P32 magnetic resonance spectroscopy, reduction in myocardial oxygen consumption, amelioration of diastolic dysfunction, and improvement in heart failure symptoms [65]. It should be noted that there is an association between elevated plasma levels of perhexiline and development of neurotoxicity and hepatotoxicity. However, these adverse effects can be virtually eliminated with routine plasma perhexiline monitoring [24, 66].
A phase III randomized controlled trial was announced in April 2015 to test the efficacy, safety, and tolerability of perhexiline in patients with HCM and moderate to severe heart failure [67]. Recruitment for this trial is pending.
Renin-angiotensin-aldosterone system inhibition
Pathologic hallmarks of HCM include myocyte hypertrophy and interstitial fibrosis [68]. These features are thought to be mediated by elevated cardiac trophic factors including angiotensin II and transforming growth factor beta (TGF-ß) [69]. Their progression over the disease course is associated with worsening diastolic function, increasing LVOT gradient, higher NYHA class, and higher incidence of sudden cardiac death [25, 70,69,72].
In transgenic mouse models of HCM, treatment with losartan or TGF-ß neutralizing antibodies has been shown to prevent development of hypertrophy and fibrosis but was unable to reverse it [73, 74]. Several recent human pilot studies have investigated the effect of angiotensin receptor blockers (ARBs) on progression of hypertrophy, fibrosis, and diastolic dysfunction with divergent results. In a study by Kawano et al., administration of valsartan (n = 23) for 12 months resulted in decreased synthesis of pro-collagen I with no change in LV wall thickness or ejection fraction compared to control [75]. A non-randomized study by Araujo et al. showed improvement in Doppler measured diastolic function after 6 months of losartan administration (n = 20) but no change in wall thickness [76]. Yamazaki et al. (n = 19) and Penicka et al. (n = 24) demonstrated a significant reduction in LV mass measured after 1 year of losartan and candesartan, respectively, compared to placebo control [77, 78]. Shimeda et al. (n = 20) reported a significant increase in late gadolinium enhancement in controls compared to those receiving losartan for 1 year as well as a non-significant change in LV mass [26].
In the subsequent randomized, controlled INHERIT trial (n = 133), investigators found no significant difference in left ventricular mass, LV wall thickness, Doppler measured diastolic function, or late gadolinium enhancement between losartan and placebo groups after 1 year [27]. It is thought that the lack of benefit observed in the INHERIT trial was due to selection of a patient population with more advanced hypertrophy and fibrosis than in previous trials, which based on prior mouse models was irreversible by ARB. Currently, recruitment is ongoing for the phase II VANISH trial, which seeks to determine the efficacy of valsartan in preventing disease progression among HCM mutation carriers with overt hypertrophic disease or pre-clinical non-hypertrophic disease [79].
Structural therapies
Percutaneous mitral valve plication
Approximately 70% of HCM patients develop a resting or provocable dynamic LVOT gradient (HOCM) [5]. A larger gradient is a strong predictor of progression to severe heart failure symptoms or death from heart failure or stroke [80].
Multiple hemodynamic studies utilizing a variety of imaging modalities have demonstrated that in a majority of HOCM patients, systolic anterior motion (SAM) of an anatomically altered mitral valve apparatus leads to apposition of the anterior mitral leaflet and an asymmetrically hypertrophied septum causing an LVOT gradient [81,80,81,82,85]. Mitral regurgitation (MR) results secondary to SAM with a predominantly posteriorly directed jet when the posterior leaflet is not mobile enough to participate in SAM and anterior leaflet coaptation. As such, the severity of MR for comparable degrees of SAM is dependent on the geometry of the posterior leaflet [83, 86,85,88]. A correlation is also established between the severity of MR and the magnitude of the LVOT gradient [89].
While surgical septal myectomy is currently the gold standard for symptomatic patients with HOCM that is refractory to optimal medical management, there is controversy about whether mitral valve abnormalities should be corrected at the time of myectomy as there has never been a randomized controlled trial comparing the results of isolated myectomy versus myectomy plus mitral repair [90]. Additionally, as the complex anatomy and pathophysiology of the LVOT and obstruction are highly diverse, surgical approach must be tailored to individual patients. Pre-operative assessment of septal thickness, mitral leaflet length, and anterolateral papillary muscle displacement determine the extent of myectomy, leaflet plication, and need for papillary muscle release, respectively [85]. The so called “resect-plicate-release” procedure, which combines myectomy, mitral repair, and papillary muscle release, has been safe and effective in the treatment of symptomatic refractory obstructive HCM [91, 92]. Use of an Alfieri stitch at the time of myectomy has also been used effectively for reduction of SAM and improvement in MR [93]. Recently, Ferrazzi et al. demonstrated the favorable results of transaortic cutting of fibrotic secondary chordae with associated shallow myectomy in 39 patients with a large LVOT gradient and mild septal hypertrophy in whom septal hypertrophy is inadequate for classic extended myectomy [94]. In these patients, alterations in the mitral valve apparatus play a central role in LVOT gradient production. By displacing the mitral valve coaptation point away from the LVOT to a more posterior normal position within the LV cavity, secondary chordal cutting resulted in abolished or substantial reduction in resting LVOT gradient from 82 ± 43 to 9 ± 5 mmHg (p < 0.001) and improvement in NYHA class from 2.9 ± 0.5 to 1.1 ± 1.1 at median 24-month follow-up. Regardless of surgical approach, however, there is currently an inadequate number of surgeons proficient in myectomy, which unfortunately has become a major barrier to optimal management of these complex patients [95].
Percutaneous mitral valve plication by MitraClip (Abbot Vascular) implantation—essentially a percutaneous Alfieri stitch—is an established treatment for patients with MR and prohibitive surgical risk [28, 96, 97]. In a recent study by Soraja et al., five patients with pharmacologic refractory symptoms due to HOCM underwent MitraClip implantation [98]. SAM, MR, and LVOT gradients were virtually eliminated immediately post-procedure. At average 15-month follow-up however, while symptoms remained improved by at least one NYHA class in all participants, significant LVOT gradients persisted in three of five participants despite continued abolition of SAM. This suggests that the symptomatic benefit achieved may have been due to reduction of MR and not of SAM or the LVOT gradient. The subset of obstructive HCM patients most likely to respond favorably to MitraClip implantation are those with limited septal thickness, where elongation and alteration of the mitral apparatus are central to gradient production, much like the patient subset selected by Ferrazzi et al.
Simultaneous echocardiography and invasive hemodynamics performed on one study participant in the Soraja study revealed a threefold higher Doppler-derived LVOT velocity (64 mmHg) compared to that measured on cardiac catheterization (22 mmHg). A high-velocity LVOT gradient on continuous wave Doppler with no true pressure gradient seen on catheterization has been described previously [99]. These findings may be explained by the following: (1) cavity obliteration by a hypercontractile HCM left ventricle may generate high intraventricular velocities without true impedance to flow [100,99,102]; (2) a “pressure recovery” phenomenon from a long tubular narrowing of the midventricular region may lead to overestimation of pressure gradients measured by Doppler compared to catheterization [103]; and (3) there is significant variation in LVOT pressure gradients with different pharmacologic and physiologic provocations that alter ventricular loading and contractility even during a single hemodynamic assessment, which may result in misclassification of patients [104,103,106]. Taken together, the hemodynamic effect of percutaneous mitral valve plication in obstructive HCM patients with severe MR is not entirely understood, although preliminary findings are intriguing and warrant further study.
Endocardial radiofrequency ablation of septal hypertrophy
Percutaneous transcoronary alcohol septal ablation (ASA) is an alternative to surgical myectomy in patients with pharmacologic refractory HOCM, which provides comparable improvement in functional status and similar long-term mortality to surgical myectomy when performed at experienced centers [8]. However, ASA is a very different intervention from myectomy as it causes a large, oftentimes transmural septal infarct occupying about 10% of the LV by instilling absolute alcohol into the first major septal perforator artery. As such, alcohol ablation has greater than twice the risk of permanent pacemaker implantation (10 versus 4%) and five times the risk of need for additional septal reduction therapy compared to myectomy [9, 10]. Despite this, after refinement of procedural technique over the last 20 years since its introduction, ASA has a relatively low post-procedural mortality rate of 1% per year, making it a valued option in places where a surgeon proficient in myectomy is not available [107, 108]. A major limitation of ASA however is its reliance on suitable coronary anatomy to provide access to target ablation, and as such, number of patients may be ineligible for ASA based on depth and distribution of the first septal perforator [29, 72, 109].
Although still in its beginning of its development with a relative paucity of data, endocardial radiofrequency ablation of septal hypertrophy (ERASH) is a new and reasonable alternative for septal reduction in patients who are not candidates for myectomy or ASA. The concept of ERASH in HOCM was first demonstrated by Lawrenz and Kuhn in 2004 on a 45-year-old man [30]. In this case report, ERASH resulted in sub-aortic septal hypokinesia, enlargement of the LVOT, reduction in septal thickness, and improved performance on 6-min walk testing at 7 days post-procedure. In 2011, the same group reported the efficacy of ERASH in 19 patients [109]. Septal tissue was targeted with the use of the CARTO (Biosense Webster) electroanatomical mapping system and transesophageal echocardiography. A retrograde left ventricular approach was taken in 9 patients and a right ventricular approach in 10 patients with no significant difference in procedural outcome. Ablation resulted in a significant post-procedural reduction of resting and provoked LVOT gradients by 62 and 60%, respectively. At 6-month follow-up, patients demonstrated improvement in exercise capacity and NYHA functional class from 3.0 ± 0.0 to 1.6 ± 0.7. Complete AV block occurred in four patients (21%) necessitating permanent pacemaker placement and one patient suffered acute cardiac tamponade from RV perforation requiring surgical repair.
Sreeram et al. demonstrated encouraging results using ERASH in 32 children with HOCM during a median follow-up of 48 months showing sustained beneficial effect [110]. Three subsequent smaller studies in adults with a composite of 23 patients and follow-up ranging from 6 to 12 months have shown similar favorable outcomes [111,110,113]. Use of the CARTOSound System in these more recent studies, which incorporates intracardiac echocardiography, allows for more accurate definition of the ablation target minimizing damage to the conduction system such that only 2 of these 23 patients (8.7%) required permanent pacemaker placement.
ERASH is a promising therapeutic option for patients with pharmacologic refractory HOCM in whom myectomy and ASA are not suitable, providing reduction in the LVOT gradient and symptomatic benefit. Further large-scale studies are needed however before any conclusions may be drawn.
Conclusion
HCM is transitioning into an exciting phase of novel, target-based pharmacologic discovery and modern minimally invasive structural intervention. While many of these therapies hold significant promise to better treat HCM patients, it is unclear how they will ultimately change or fit into clinical practice. As HCM has heterogeneous molecular pathophysiology and hemodynamics, it is important to recognize the patient selection criteria for each therapy studied in this review. Looking toward the future, a more standardized incorporation of genetic testing in HCM patients may prove to enhance the patient selection and drug discovery process.
References
Maron BJ, Gardin JM, Flack JM et al (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Circulation 92:785–789. https://doi.org/10.1161/01.CIR.92.4.785
Maron BJ, Olivotto I, Spirito P et al (2000) Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 102:858–864. https://doi.org/10.1161/01.CIR.102.8.858
Maron BJ, McKenna WJ, Danielson GK et al (2003) American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on hypertrophic cardiomyopathy. J Am Coll Cardiol 42:1687–1713. https://doi.org/10.1016/S0735-1097(03)00941-0
Sen-Chowdhry S, Jacoby D, Moon JC, McKenna WJ (2016) Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat Rev Cardiol 13:651–675. https://doi.org/10.1038/nrcardio.2016.140
Maron MS, Olivotto I, Zenovich AG et al (2006) Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 114:2232–2239. https://doi.org/10.1161/CIRCULATIONAHA.106.644682
Spoladore R, Maron MS, D’Amato R et al (2012) Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 33:1724–1733. https://doi.org/10.1093/eurheartj/ehs150
Ammirati E, Contri R, Coppini R et al (2016) Pharmacological treatment of hypertrophic cardiomyopathy: current practice and novel perspectives. Eur J Heart Fail 18:1106–1118. https://doi.org/10.1002/ejhf.541
Agarwal S, Tuzcu EM, Desai MY et al (2010) Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 55:823–834. https://doi.org/10.1016/j.jacc.2009.09.047
Valeti US, Nishimura RA, Holmes DR et al (2007) Comparison of surgical septal myectomy and alcohol septal ablation with cardiac magnetic resonance imaging in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol 49:350–357. https://doi.org/10.1016/j.jacc.2006.08.055
Liebregts M, Vriesendorp PA, Mahmoodi BK et al (2015) A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Hear Fail 3:896–905. https://doi.org/10.1016/j.jchf.2015.06.011
Semsarian C, Ahmad I, Giewat M et al (2002) The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest 109:1013–1020. https://doi.org/10.1172/JCI200214677
Ho CY, Lakdawala NK, Cirino AL et al (2015) Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Hear Fail 3:180–188. https://doi.org/10.1016/j.jchf.2014.08.003
Coppini R, Ferrantini C, Yao L et al (2013) Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127:575–584. https://doi.org/10.1161/CIRCULATIONAHA.112.134932
Gentry JL, Mentz RJ, Hurdle M, Wang A (2016) Ranolazine for treatment of angina or dyspnea in hypertrophic cardiomyopathy patients (RHYME). J Am Coll Cardiol 68:1815–1817. https://doi.org/10.1016/j.jacc.2016.07.758
Bemporad D (2016) Ranolazine in patients with symptomatic hypertrophic cardiomyopathy: a pilot study assessing the effects on exercise capacity, diastolic function and symptomatic status. EU Clin Trials Regist. https://www.clinicaltrialsregister.eu/ctr-search/trial/2011-004507-20/DE. Accessed August 1, 2016
Gilead Sciences (2017) Effect of eleclazine (GS-6615) on exercise capacity in subjects with symptomatic hypertrophic cardiomyopathy (LIBERTY-HCM). US National Library of Medicine. https://clinicaltrials.gov/ct2/show/NCT02291237. Accessed February 17, 2017
Gilead Sciences (2017) Effect of eleclazine on shortening of the QT interval, safety, and tolerability in adults with long QT syndrome type 3. US National Library of Medicine. https://clinicaltrials.gov/ct2/show/NCT02300558?te. Accessed February 17, 2017
Nag S, Sommese RF, Ujfalusi Z et al (2015) Contractility parameters of human beta-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv 1:1–16. https://doi.org/10.1126/sciadv.1500511
Kamdar F, Klaassen Kamdar A, Koyano-Nakagawa N et al (2015) Cardiomyopathy in a dish: using human inducible pluripotent stem cells to model inherited cardiomyopathies. J Card Fail 21:761–770. https://doi.org/10.1016/j.cardfail.2015.04.010
Maslov MY, Chacko VP, Stuber M et al (2007) Altered high-energy phosphate metabolism predicts contractile dysfunction and subsequent ventricular remodeling in pressure-overload hypertrophy mice. Am J Physiol Heart Circ Physiol 292:H387–H391. https://doi.org/10.1152/ajpheart.00737.2006
Ferrantini C, Belus A, Piroddi N et al (2009) Mechanical and energetic consequences of HCM-causing mutations. J Cardiovasc Transl Res 2:441–451. https://doi.org/10.1007/s12265-009-9131-8
Wilder T, Ryba DM, Wieczorek DF et al (2015) N-acetylcysteine reverses diastolic dysfunction and hypertrophy in familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. doi:https://doi.org/10.1152/ajpheart.00339.2015
Lee L, Campbell R, Scheuermann-Freestone M et al (2005) Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation 112:3280–3288. https://doi.org/10.1161/CIRCULATIONAHA.105.551457
Horowitz JD, Sia STB, Macdonald PS et al (1986) Perhexiline maleate treatment for severe angina pectoris—correlations with pharmacokinetics. Int J Cardiol 13:219–229. https://doi.org/10.1016/0167-5273(86)90146-4
Olivotto I, Gistri R, Petrone P et al (2003) Maximum left ventricular thickness and risk of sudden death in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 41:315–321. https://doi.org/10.1016/S0735-1097(02)02713-4
Shimada YJ, Passeri JJ, Baggish AL et al (2013) Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Hear Fail 1:480–487. https://doi.org/10.1016/j.jchf.2013.09.001
Axelsson A, Iversen K, Vejlstrup N et al (2015) Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 3:123–131. https://doi.org/10.1016/S2213-8587(14)70241-4
Lim DS, Reynolds MR, Feldman T et al (2014) Improved functional status and quality of life in prohibitive surgical risk patients with degenerative mitral regurgitation after transcatheter mitral valve repair. J Am Coll Cardiol 64:182–192. https://doi.org/10.1016/j.jacc.2013.10.021
Maron BJ, Dearani JA, Ommen SR et al (2004) The case for surgery in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 44:2044–2053. https://doi.org/10.1016/j.jacc.2004.04.063
Lawrenz T, Kuhn HJ (2004) Endocardial radiofrequency ablation of septal hypertrophy: a new catheter-based modality of gradient reduction in hypertrophic obstructive cardiomyopathy. Z Kardiol 93:493–499
Pohlmann L, Kröger I, Vignier N et al (2007) Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res 101:928–938. https://doi.org/10.1161/CIRCRESAHA.107.158774
Iorga B, Blaudeck N, Solzin J et al (2008) Lys184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc Res 77:676–686. https://doi.org/10.1093/cvr/cvm113
Huke S, Knollmann BC (2010) Increased myofilament Ca2+ sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 48:824–833. https://doi.org/10.1016/j.yjmcc.2010.01.011
Baudenbacher F, Schober T, Pinto JR et al (2008) Myofilament Ca sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118:3845–3903. https://doi.org/10.1172/JCI36642
Ho CY, Sweitzer NK, McDonough B et al (2002) Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 105:2992–2997. https://doi.org/10.1161/01.CIR.0000019070.70491.6D
Nagueh SF, Bachinski LL, Meyer D et al (2001) Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation 104:128–130. https://doi.org/10.1161/01.CIR.104.2.128
Michele D, Albayya FP, Metzger JM (1999) Direct, convergent hypersensitivity of calcium-activated force generation produced by hypertrophic cardiomyopathy mutant alpha-tropomyosins in adult cardiac myocytes. Nat Med 5:1413–1417. https://doi.org/10.1038/70990
Frey N, McKinsey TA, Olson EN (2000) Decoding calcium signals involved in cardiac growth and function. Nat Med 6:1221–1227. https://doi.org/10.1038/81321
Tardiff JC, Carrier L, Bers DM et al (2015) Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res 105:457–470. https://doi.org/10.1093/cvr/cvv023
Robinson P, Griffiths PJ, Watkins H, Redwood CS (2007) Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 101:1266–1273. https://doi.org/10.1161/CIRCRESAHA.107.156380
Fatkin D, Mcconnell BK, Mudd JO et al (2000) An abnormal Ca2+ response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest 106:1351–1359. https://doi.org/10.1172/JCI11093
Lovelock JD, Monasky MM, Jeong EM et al (2012) Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res 110:841–850. https://doi.org/10.1161/CIRCRESAHA.111.258251
Flenner F, Friedrich FW, Ungeheuer N et al (2016) Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc Res 109:90–102. https://doi.org/10.1093/cvr/cvv247
Buvoli M, Hamady M, Leinwand LA, Knight R (2008) Bioinformatics assessment of β-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc Med 18:141–149. https://doi.org/10.1016/j.tcm.2008.04.001
Kirschner SE, Becker E, Antognozzi M et al (2004) Hypertrophic cardiomyopathy-related-myosin mutations cause highly variable calcium sensitivity with functional imbalances among individual muscle cells. AJP Hear Circ Physiol 288:H1242–H1251. https://doi.org/10.1152/ajpheart.00686.2004
Spudich JA (2014) Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J 106:1236–1249. https://doi.org/10.1016/j.bpj.2014.02.011
Walsh R, Rutland C, Thomas R, Loughna S (2009) Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115:49–60. https://doi.org/10.1159/000252808
Green EM, Wakimoto H, Anderson RL et al (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351(80):617–621
MyoKardia Inc (2015) MyoKardia provides update on two phase 1 trials of MYK-461 for the treatment of hypertrophic cardiomyopathy. In: http://investors.myokardia.com/phoenix.zhtml?c=254211&p=irol-newsArticle&ID=2097088
Witjas-Paalberends ER, Güclü A, Germans T et al (2014) Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res 103:248–257. https://doi.org/10.1093/cvr/cvu127
Witjas-Paalberends ER, Ferrara C, Scellini B et al (2014) Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol 592:3257–3272. https://doi.org/10.1113/jphysiol.2014.274571
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19:263–268. https://doi.org/10.1016/S0168-9525(03)00081-7
Frey N, Olson EN (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65:45–79. https://doi.org/10.1146/annurev.physiol.65.092101.142243
Blair E, Redwood C, Ashrafian H et al (2001) Mutations in the γ 2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet 10:1215–1220. https://doi.org/10.1093/hmg/10.11.1215
Crilley JG, Boehm EA, Blair E et al (2003) Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 41:1776–1782. https://doi.org/10.1016/S0735-1097(02)03009-7
Unno K, Isobe S, Izawa H et al (2009) Relation of functional and morphological changes in mitochondria to myocardial contractile and relaxation reserves in asymptomatic to mildly symptomatic patients with hypertrophic cardiomyopathy. Eur Heart J 30:1853–1862. https://doi.org/10.1093/eurheartj/ehp184
Camici P, Chiriatti G, Lorenzoni R et al (1991) Coronary vasodilation is impaired in both hypertrophied and nonhypertrophied myocardium of patients with hypertrophic cardiomyopathy: a study with nitrogen-13 ammonia and positron emission tomography. J Am Coll Cardiol 17:879–886. https://doi.org/10.1016/0735-1097(91)90869-B
Cecchi F, Olivotto I, Gistri R et al (2003) Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med 349:1027–1035
Petersen SE, Jerosch-Herold M, Hudsmith LE et al (2007) Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation 115:2418–2425. https://doi.org/10.1161/CIRCULATIONAHA.106.657023
Lombardi R, Rodriguez G, Chen SN et al (2009) Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation 119:1398–1407. https://doi.org/10.1161/CIRCULATIONAHA.108.790501.Resolution
Marian AJ (2015) Hypertrophic regression with N-Acetylcysteine in HCM (HALT). US National Library of Medicine. https://clinicaltrials.gov/ct2/show/NCT01537926?. Accessed February 17, 2017
Lele SS, Thomson HL, Seo H et al (1995) Exercise capacity in hypertrophic cardiomyopathy. Circulation 92:2886–2894
Phan TT, Shivu GN, Abozguia K et al (2010) Impaired heart rate recovery and chronotropic incompetence in patients with heart failure with preserved ejection fraction. Circ Hear Fail 3:29–34. https://doi.org/10.1161/CIRCHEARTFAILURE.109.877720
Jeffrey FMH, Alvarez L, Diczku V et al (1995) Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate. J Cardiovasc Pharmacol 25:469–472
Abozguia K, Elliott P, McKenna W et al (2010) Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 122:1562–1569. https://doi.org/10.1161/CIRCULATIONAHA.109.934059
Cole PL, Beamer AD, Mcgowan N et al (1990) Efficacy and safet of perhexiline maleate refractory angina a double-blind placebo-controlled clinical trial of a novel. Circulation 81:1260–1270
Heart Metabolics Limited (2015) Efficacy, safety, and tolerability of perhexiline in subjects with hypertrophic cardiomyopathy and heart failure. US National Library of Medicine. https://clinicaltrials.gov/ct2/show/NCT02431221. Accessed February 17, 2017
Varnava AM, Elliott PM, Sharma S et al (2000) Hypertrophic cardiomyopathy: the interrelation of disarray, fibrosis, and small vessel disease. Heart 84:476–482. https://doi.org/10.1136/heart.84.5.476
Marian AJ (2000) Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet 355:58–60. https://doi.org/10.1016/S0140-6736(99)06187-5
Spirito P, Chiarella F, Carratino L et al (1989) Clinical course and prognosis of hypertrophic cardiomyopathy in an outpatient population. N Engl J Med 320:749–755. https://doi.org/10.1056/NEJM198603273141302
Elliott PM, Gimeno Blanes JR, Mahon NG et al (2001) Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 357:420–424. https://doi.org/10.1016/S0140-6736(00)04005-8
Maron BJ, Casey SA, Hauser RG, Aeppli DM (2003) Clinical course of hypertrophic cardiomyopathy with survival to advanced age. J Am Coll Cardiol 42:882–888. https://doi.org/10.1016/S0735-1097(03)00855-6
Teekakirikul P, Eminaga S, Toka O et al (2010) Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires TGF-β. J Clin Invest 120:3520–3529. https://doi.org/10.1172/JCI42028DS1
Lim D, Lutucuta S, Bachireddy P et al (2001) Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 103:789–792
Kawano H, Toda G, Nakamizo R et al (2005) Valsartan decreases type I collagen synthesis in patients with hypertrophic cardiomyopathy. Circ J 69:1244–1248
Araujo AQ, Arteaga E, Ianni BM et al (2005) Effect of losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am J Cardiol 96:1563–1567. https://doi.org/10.1016/j.amjcard.2005.07.065
Yamazaki T, Suzuki J-I, Shimamoto R et al (2007) A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an angiotensin II receptor blocker. Int Heart J 48:715–724. https://doi.org/10.1536/ihj.48.715
Penicka M, Gregor P, Kerekes R et al (2009) The effects of candesartan on left ventricular hypertrophy and function in nonobstructive hypertrophic cardiomyopathy. J Mol Diagnostics 11:35–41. https://doi.org/10.2353/jmoldx.2009.080082
US National Library of Medicine (2016) clinicaltrials.gov https://clinicaltrials.gov/ct2/show/NCT01912534
Maron MS, Olivotto I, Betocchi S et al (2003) Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 348:295–303. https://doi.org/10.1056/NEJMoa021332\r348/4/295
Wigle ED, Marquis Y, Auger P (1967) Muscular subaortic stenosis: initial left ventricular inflow tract pressure in the assessment of intraventricular pressures differences in man. Circulation 36:1100–1117
Ross J, Braunwald E, Gault JH et al (1966) The mechanism of the intraventricular pressure gradient in idiopathic hypertrophic subaortic stenosis. Circulation 34:558–578
Shah PM, Gramiak R, Kramer D (1969) Ultrasound localization of left ventricular outflow obstruction in hypertrophic obstructive cardiomyopathy. Circulation 40:3–11. https://doi.org/10.1161/01.CIR.40.1.3
Cape EG, Simons D, Jimoh A et al (1989) Chordal geometry determines the shape and extent of systolic anterior mitral motion: in vitro studies. J Am Coll Cardiol 13:1438–1448. https://doi.org/10.1016/0735-1097(89)90326-4
Sherrid MV, Balaram S, Kim B et al (2016) The mitral valve in obstructive hypertrophic cardiomyopathy: a test in context. J Am Coll Cardiol 67:1846–1858. https://doi.org/10.1016/j.jacc.2016.01.071
Spirito P, Maron BJ (1984) Patterns of systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy: assessment by two-dimensional echocardiography. Am J Cardiol 54:1039–1046. https://doi.org/10.1016/S0002-9149(84)80141-1
Grigg LE, Wigle ED, Williams WG et al (1992) Transesophageal Doppler echocardiography in obstructive hypertrophic cardiomyopathy: clarification of pathophysiology and importance in intraoperative decision making. J Am Coll Cardiol 20:42–52. https://doi.org/10.1016/0735-1097(92)90135-A
Schwammenthal E, Nakatani S, He S et al (1998) Mechanism of mitral regurgitation in hypertrophic cardiomyopathy: mismatch of posterior to anterior leaflet length and mobility. Circulation 98:856–865
Yu EHC, Omran AS, Wigle ED et al (2000) Mitral regurgitation in hypertrophic obstructive cardiomyopathy: relationship to obstruction and relief with myectomy. J Am Coll Cardiol 36:2219–2225. https://doi.org/10.1016/S0735-1097(00)01019-6
Smedira NG, Lytle BW, Lever HM et al (2008) Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 85:127–133. https://doi.org/10.1016/j.athoracsur.2007.07.063
Balaram SK, Tyrie L, Sherrid MV et al (2008) Resection-plication-release for hypertrophic cardiomyopathy: clinical and echocardiographic follow-up. Ann Thorac Surg 86:1539–1545. https://doi.org/10.1016/j.athoracsur.2008.07.048
Balaram SK, Ross RE, Sherrid MV et al (2012) Role of mitral valve plication in the surgical management of hypertrophic cardiomyopathy. Ann Thorac Surg 94:1990–1998. https://doi.org/10.1016/j.athoracsur.2012.06.008
Shah AA, Glower DD, Gaca JG (2016) Trans-aortic Alfieri stitch at the time of septal myectomy for hypertrophic obstructive cardiomyopathy. J Card Surg 31:503–506. https://doi.org/10.1111/jocs.12804
Ferrazzi P, Spirito P, Iacovoni A et al (2015) Transaortic chordal cutting mitral valve repair for obstructive hypertrophic cardiomyopathy with mild septal hypertrophy. J Am Coll Cardiol 66:1687–1696. https://doi.org/10.1016/j.jacc.2015.07.069
Maron BJ, Dearani JA, Maron MS, et al (2017) Why we need more septal myectomy surgeons: an emerging recognition. J Thorac Cardiovasc Surg 1–6. doi:https://doi.org/10.1016/j.jtcvs.2016.12.038
Feldman T, Foster E, Glower DD et al (2011) Percutaneous repair or surgery for mitral regurgitation. N Engl J Med 364:1395–1406
Mauri L, Foster E, Glower DD et al (2013) 4-year results of a randomized controlled trial of percutaneous repair versus surgery for mitral regurgitation. J Am Coll Cardiol 62:317–328. https://doi.org/10.1016/j.jacc.2013.04.030
Sorajja P, Pedersen W, Bae R et al (2016) First experience with percutaneous mitral valve plication as primary therapy for symptomatic obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 67:1516. https://doi.org/10.1016/S0735-1097(16)31517-0
Jaber WA, Nishimura RA, Ommen SR (2007) Not all systolic velocities indicate obstruction in hypertrophic cardiomyopathy: a simultaneous Doppler catheterization study. J Am Soc Echocardiogr 20:5–7. https://doi.org/10.1016/j.echo.2007.01.015
Criley JM, Siegel RJ (1986) Obstruction is unimportant in the pathophysiology of hypertrophic cardiomyopathy. Postgrad Med J 62:515–529. https://doi.org/10.1136/pgmj.62.728.515
Murgo JP, Alter BR, Dorethy JF et al (1980) Dynamics of left ventricular ejection in obstructive and nonobstructive hypertrophic cardiomyopathy. J Clin Invest 66:1369–1382. https://doi.org/10.1172/JCI109990
Criley JM (1997) Unobstructed thinking (and terminology) is called for in the understanding and management of hypertrophic cardiomyopathy. J Am Coll Cardiol 29:741–743. https://doi.org/10.1016/S0735-1097(96)00590-6
Sahn DJ, Yoganathan AP, Editors G et al (1989) Pressure recovery distal to a stenosis: potential cause of a gradient “overestimation” by Doppler echocardiography. J Am Coll Cardiol 13:706–715
Braunwald E, Lambrew CT, Rockoff SD et al (1964) Idiopathic hypertrophic subaortic stenosis: I. A description of the disease based upon an analysis of 64 patients. Circulation 29:IV-3–IV-119. https://doi.org/10.1161/01.CIR.29.5S4.IV-3
Geske JB, Sorajja P, Ommen SR, Nishimura RA (2009) Left ventricular outflow tract gradient variability in hypertrophic cardiomyopathy. Clin Cardiol 32:397–402. https://doi.org/10.1002/clc.20594
Geske JB, Sorajja P, Ommen SR, Nishimura RA (2011) Variability of left ventricular outflow tract gradient during cardiac catheterization in patients with hypertrophic cardiomyopathy. JACC Cardiovasc Interv 4:704–709. https://doi.org/10.1016/j.jcin.2011.02.014
Veselka J, Jensen MK, Liebregts M et al (2016) Long-term clinical outcome after alcohol septal ablation for obstructive hypertrophic cardiomyopathy: results from the Euro-ASA registry. Eur Heart J 37:1517–1523. https://doi.org/10.1093/eurheartj/ehv693
Veselka J, Anavekar NS, Charron P (2017) Hypertrophic obstructive cardiomyopathy. Lancet 389:1253–1267. https://doi.org/10.1016/S0140-6736(16)31321-6
Lawrenz T, Borchert B, Leuner C et al (2011) Endocardial radiofrequency ablation for hypertrophic obstructive cardiomyopathy: acute results and 6 months’ follow-up in 19 patients. J Am Coll Cardiol 57:572–576. https://doi.org/10.1016/j.jacc.2010.07.055
Sreeram N, Emmel M, De Giovanni JV (2011) Percutaneous radiofrequency septal reduction for hypertrophic obstructive cardiomyopathy in children. J Am Coll Cardiol 58:2501–2510. https://doi.org/10.1016/j.jacc.2011.09.020
Cooper RM, Shahzad A, Hasleton J et al (2015) Radiofrequency ablation of the interventricular septum to treat outflow tract gradients in hypertrophic obstructive cardiomyopathy: a novel use of CARTOSound® technology to guide ablation. Eur Eur pacing, arrhythmias, Card Electrophysiol J Work groups Card pacing, arrhythmias, Card Cell Electrophysiol Eur Soc Cardiol 18:113–120. https://doi.org/10.1093/europace/euv302
Crossen K, Jones M, Erikson C (2016) Radiofrequency septal reduction in symptomatic hypertrophic obstructive cardiomyopathy. Hear Rhythm 13:1885–1890. https://doi.org/10.1016/j.hrthm.2016.04.018
Shelke AB, Menon R, Kapadiya A et al (2016) A novel approach in the use of radiofrequency catheter ablation of septal hypertrophy in hypertrophic obstructive cardiomyopathy. Indian Heart J 68:618–623. https://doi.org/10.1016/j.ihj.2016.02.007
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Philipson, D.J., DePasquale, E.C., Yang, E.H. et al. Emerging pharmacologic and structural therapies for hypertrophic cardiomyopathy. Heart Fail Rev 22, 879–888 (2017). https://doi.org/10.1007/s10741-017-9648-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-017-9648-x