
Abstract
Pain and itch are unpleasant sensations that often accompany infections caused by viral, bacterial, parasitic, and fungal pathogens. Recent studies show that sensory neurons are able to directly detect pathogens to mediate pain and itch. Nociceptor and pruriceptor neurons respond to pathogen-associated molecular patterns, including Toll-like receptor ligands, N-formyl peptides, and bacterial toxins. Other pathogens are able to silence neuronal activity to produce analgesia during infection. Pain and itch could lead to neuronal modulation of the immune system or behavioral avoidance of future pathogen exposure. Conversely, pathogens could modulate neuronal signaling to potentiate their pathogenesis and facilitate their spread to other hosts. Defining how pathogens modulate pain and itch has critical implications for sensory neurobiology and our understanding of host-microbe interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pathogenic infections are a major danger to organismic well-being and are often accompanied by pain or itch, behavioral mechanisms that protect mammals from danger. These sensations are mediated by nociceptor and pruriceptor neurons, respectively. The Roman encyclopedist Celsus defined inflammation in the first century A.D. as involving four cardinal signs: Rubor (redness), Calor (heat), Tumor (swelling), and Dolor (Pain). Pain is not only a symptom of inflammation. Nociceptor neurons actively participate in the detection of pathogens by the host and the regulation of inflammation. Pruriceptor neurons are also able to sense pathogenic ligands, and itch may mediate host defense by scratch-mediated pathogen removal. Here, we review recent studies elucidating how pathogens and molecules derived from them are detected by the nervous system to modulate pain or itch.
Nociceptor and pruriceptor neurons encode specific molecular sensors at the nerve terminals to detect noxious/inflammatory stimuli such as temperature, ATP, and reactive chemicals. Nociceptor and pruriceptor neurons also express specific pathogen-recognition mechanisms, including Toll-like receptors (TLRs), formyl peptide receptors (FPRs), and signaling pathways that sensitize transient receptor potential (TRP) channels [1,2,3,4]. Upon activation, antidromic signaling leads to neuropeptide release that can mediate neurogenic inflammation or the activation of innate and adaptive immune cells [5, 6]. These neuron-microbe and neuro-immune interactions could play critical roles in host-pathogen defense.
A survey of major bacterial, viral, fungal, and parasitic pathogens in humans shows that distinct types of infection are characterized by intense pain, while others are characterized by intense itch (Table 1). The molecular and cellular bases for the different sensory modalities during infection are not known. Nociceptors and pruriceptors may possess distinct pathogenic receptors. For example, bacterial pathogens may specifically bind nociceptors to produce pain while parasites bind pruriceptors to produce itch. It is also possible that infectious itch and pain are anatomically encoded: pathogen invasion of the epidermis could lead to itch, while deeper tissue or visceral infections could produce pain.
Here, we discuss the distinct pain, itch, and analgesic mechanisms induced by pathogens. Defining how pathogens and their molecules interact with the sensory nervous system could lead to novel therapeutic approaches to treat pain and itch. For example, botulinum toxins secreted by Clostridium botulinum potently silence neurotransmission and have been successfully applied in the treatment of pain. Many mechanisms of pain and itch remain undefined, and this could be a rich field of future investigation (Table 1). Therefore, this is an emerging field that has basic and translational implications for neurobiology and inflammation.
Pain and Bacterial Infection
Bacterial pathogens often produce pain during host invasion, mediating painful infections of the skin, soft tissues, oral cavity, gastrointestinal (GI) tract, and genitourinary tract (Table 1). Recent work has shown that nociceptor neurons are able to detect gram-positive and gram-negative bacterial ligands through specific molecular pathways including TLRs and the FPR1, TRPA1, and TRPV1 ion channels [3, 7,8,9,10,11,12,13,14].
Lipopolysaccharide (LPS)
LPS, also known as bacterial endotoxin, is a major component of the outer membrane of all gram-negative bacteria. It consists of an extracellular O-antigen, an outer core, inner core components, and a Lipid A moiety. LPS is also a pathogen-associated molecular pattern (PAMP) that allows mammalian host cells to distinguish bacteria from self during infection. Immune cells detect LPS through a pattern-recognition receptor complex consisting of TLR4, CD14 (cluster of differentiation 14), and MD2 (myeloid differentiation factor-2). TLR4 then signals through its cytoplasmic adaptor protein MyD88 (myeloid differentiation primary response gene 88) to activate nuclear factor kappa-B signaling and innate cytokine production.
In one of the first studies to show direct neuronal recognition of pathogens, Hargreaves et al. found that TRPV1-positive trigeminal neurons express both TLR4 and CD14, allowing nociceptor neurons to directly sense LPS during infection [15]. Porphyromonas gingivalis is a gram-negative bacterial pathogen that is often found in ondontogenic abscesses and root canal infections that are associated with pain [16, 17] (Table 1). Neuronal application of LPS derived from P. gingivalis sensitizes nociceptor responses to capsaicin, the prototypic ligand for TRPV1 [7]. LPS sensitizes capsaicin-induced Ca2+ influx and inward currents, as well as the release of the inflammatory neuropeptide CGRP (calcitonin gene-related peptide), and this is abrogated by treatment of neurons with a TLR4 antagonist [7]. Thus, LPS directly sensitizes nociceptors and produces pain sensitivity in tooth and gum decay. Subcutaneous injection of LPS into the footpad of mice also induces significant mechanical allodynia, which is dependent on TLR4 and its downstream signaling adaptor MyD88, but not on TRIF (TIR-domain-containing adapter-inducing interferon-β) [18].
LPS is also an important mediator of pain caused by urinary tract infections (UTIs). Uropathogenic Escherichia coli (UPEC) causes significant pelvic pain during UTI and is associated with bladder cystitis. A series of elegant studies by Rudick et al. have determined the mechanisms of pain in a mouse model of UPEC-induced UTI. They found that a UPEC strain caused pelvic pain, whereas an asymptomatic bacteriuria-inducing strain of E. coli did not. Moreover, UPEC LPS induced pain that was dependent on TLR4 signaling [19]. Infection with a UPEC strain from a chronic prostatitis patient induced persistent pain after bacterial clearance in NOD (non-obese diabetic) mice but not in C57BL/6J mice, indicating potential neuropathic damage and a dependence on genetic background [20]. Further work by the same group showed that the establishment of pain by E. coli depends on TRPV1 [21]. Moreover, they found that the O-antigen moiety of LPS modulates pain [22]. Mutant UPEC bacteria lacking the O-antigen induced chronic pain whereas the wild-type of the same UPEC strain induced acute pain, and the expression of cloned O-antigen gene clusters altered the pain phenotypes [22].
By contrast with the above studies, Meseguer et al. found that LPS induces Ca2+ flux in nociceptor neurons that is independent of TLR4, but depends on the TRPA1 ion channel [8]. LPS from several bacterial pathogens (E. coli, Salmonella typhimurium, Klebsiella pneumoniae, Serratia marcescens, and Pseudomonas aeruginosa) directly induced Ca2+ flux in cultured dorsal root ganglia (DRG) neurons, which was abrogated in TRPA1−/− neurons or by treatment with a TRPA1 antagonist. LPS induced CGRP release and neurogenic inflammation, and the hyperalgesia was lower in TRPA1−/− mice than in wild-type mice [8]. Moreover, they found that the shape of the Lipid A moiety of LPS determined the degree of TRPA1 activation. Therefore, LPS is able to sensitize nociceptor neurons through TLR4-mediated sensitization of the TRPV1 ion channel or through direct gating of the TRPA1 ion channel (Fig. 1). Future work is needed to elucidate whether these molecular mechanisms mediate pain production and hyperalgesia during different types of gram-negative bacterial infections, many of which lead to significant pain (Table 1).

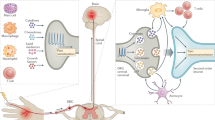
Nociceptor neurons directly detect pathogens and their molecular ligands to mediate pain. Nociceptor sensory nerve terminals directly detect bacterial, fungal, and viral pathogens or their molecular ligands through several mechanisms. Lipopolysaccharide (LPS), a major cell wall component of gram-negative bacteria, binds to neuronal TLR4 to sensitize the TRPV1 ion channel, or directly mediates the opening of the TRPA1 ion channel. Staphylococcus aureus activates nociceptor neurons through bacterial N-formyl peptides that bind to FPR1 or through binding of the pore-forming toxin α-hemolysin to ADAM10. Bacterial flagellin activates TLR5 in Aβ fibers involved in neuropathic pain production. Candida albicans activates nociceptors through the cell-wall component zymosan. Herpesviruses produce significant pain during acute infection, and infection can result in post-herpetic neuralgia characterized by chronic pain. Mycobacterium ulcerans releases a mycolactone that silences pain through signaling downstream of the angiotensin II receptor (ATR2), leading to neuronal hyperpolarization.
Staphylococcus aureus and α-Hemolysin
S. aureus is a gram-positive bacterial coccus and one of the leading causes of human bacterial infections [23, 24]; it commonly causes painful skin and soft-tissue infections in the form of boils, abscesses, and cellulitis. The increased prevalence of antibiotic-resistant bacteria, including both community- and hospital-acquired methicillin-resistant S. aureus strains, highlights the urgent need for better anti-microbial strategies.
In a mouse model of intraplantar infection by S. aureus, we found that mechanical and thermal hyperalgesia closely mirror the bacterial expansion in tissues, but not the tissue swelling or immune cell influx [11]. Pain during infection was thought to be mediated by immune cells and their mediators. However, we found that depletion of key immune components, including neutrophils, monocytes, T cells, B cells, natural killer cells, and TLR signaling pathways, does not reduce S. aureus-induced pain. Rather, S. aureus directly induces Ca2+ flux and action potential generation in nociceptor neurons to cause pain.
Pain during S. aureus infection depends on at least two mechanisms: neuronal recognition of N-formyl peptides through FPR1, and neuronal activation by the bacterial pore-forming toxin α-hemolysin (Fig. 1). Alpha-hemolysin is a beta-barrel pore-forming toxin secreted by S. aureus that binds to its host receptor ADAM10 [25]. Upon binding, α-hemolysin inserts into target-cell membranes, self-oligomerizing into heptameric pores that allow the influx of cations [23]. We found that nociceptor neurons express ADAM10, and that S. aureus α-hemolysin induces neuronal action potential generation and Ca2+ flux. Intraplantar injection of α-hemolysin in mice produces dose-dependent acute pain, as well as thermal and mechanical hyperalgesia [11]. S. aureus deficient in α-hemolysin induces significantly less pain behavior than wild-type bacteria, indicating a key role for this bacterial toxin in infectious pain [11]. We also found that S. aureus mediates pain during infection through bacterial N-formyl peptides, as discussed below.
What is the role of pain in S. aureus host defense? Mice deficient in Nav1.8-lineage nociceptor neurons show increased neutrophil and monocyte infiltration, and greater lymph node hypertrophy during infection. S. aureus induces nociceptor neurons to release the neuropeptide CGRP, which suppresses the production of TNF-α by macrophages and lymph node hypertrophy [11]. Thus, S. aureus may gain a selective advantage by activating nociceptor neurons to suppress innate immunity.
Bacterial N-formyl Peptides
N-formyl peptides are molecular signatures of all bacteria, due to their unique expression of a formylase enzyme, which appends formyl groups during protein synthesis. In nature, this enzyme is only present within bacteria and mitochondria [26]. Mammalian immune cells express formyl peptide receptors (FPRs), a family of G-protein-coupled receptors that specifically recognizes N-formyl peptides as a signature of bacterial infection or mitochondrial injury [26]. Neutrophils and monocytes express FPR1 and FPR2, which critically mediate their chemotaxis to sites of bacterial infection [26]. FPRs have recently been found to be expressed in the sensory nervous system. Olfactory chemosensory neurons in the mouse vomeronasal organ express several FPRs (Fpr-rs1, -rs3, -rs4, -rs6, and -rs7), though their ligand specificity may differ from immune FPRs [27,28,29]. We found that nociceptor DRG neurons express FPR1 and FPR2 [11], which are also the receptors expressed by the innate immune system [26]. We found that fMIFL, a formyl peptide from S. aureus, and fMLF, a formyl peptide from E. coli, induce Ca2+ influx in a subset of nociceptor neurons that also respond to capsaicin and mustard oil. FPR1−/− mice show decreased mechanical allodynia induced by subcutaneous injection of N-formyl peptides or of heat-killed S. aureus [11]. Therefore, nociceptor neurons express FPRs, which are able to detect N-formyl peptides to induce mechanical hyperalgesia (Fig. 1).
Bacterial Flagellin
Flagellin is the globular protein core component that forms the filament of the bacterial flagellum, and is expressed by many types of bacterial pathogen. Mammalian host cells are able to specifically recognize bacterial flagellin as a PAMP through TLR5. Ji et al. recently found that NF200-positive Aβ-fiber sensory neurons that mediate chronic neuropathic pain specifically express TLR5 [10] (Fig. 1). Interestingly, concurrent injection of bacterial flagellin together with the charged lidocaine derivative QX-314 produces significant pain blockade in mouse models of pain due to chemotherapy, nerve injury, and diabetic neuropathy [10]. This analgesic delivery is specifically mediated by TLR5-induced neuronal entry, as it does not occur in TLR5−/− mice. Though the authors did not address it specifically in their study, it would be interesting to determine whether flagellated bacteria induce neuronal TLR5 activation and neuropathic pain-like symptoms by the action of flagellin on A-fibers during live pathogen infection.
Citrobacter rodentium and Post-infectious Irritable Bowel Syndrome (IBS)
GI infections caused by major human pathogens including Clostridium difficile, enterohemorrhagic E. coli, Helicobacter pylori, and Salmonella enterica produce intense acute pain during infection (Table 1). Such infections can also lead to post-infectious IBS, characterized by chronic visceral pain hypersensitivity and irritability. Understanding the mechanisms of pain caused by GI pathogens could lead to better treatments for IBS and other painful GI conditions.
C. rodentium is a rodent-specific pathogen that causes GI pathology and colitis that resembles enterohemorrhagic E. coli infection in humans. Vanner et al. showed that C. rodentium infection in mice causes post-infectious visceral pain sensitivity [30]. This infection significantly increases the rheobase and action potential discharge in colonic nociceptive DRG neurons as assessed by patch-clamp analysis. Electrophysiological studies have shown that transient outward (I(A)) and delayed rectifier (I(K)) currents are suppressed, and this could be a key contributor to neuronal hypersensitivity [30]. They further showed that exposure to water-avoidance stress compounds the DRG excitability following C. rodentium infection in mice, indicating that psychological stress contributes to visceral pain mechanisms in post-infectious IBS [31].
Meningitis and Pain
Bacterial pathogens can invade the meninges to produce acute inflammation or meningitis, which is associated with migraine-like headaches [32, 33]. S. pneumoniae and L. monocytogenes are major causes of bacterial meningitis (Table 1). Though the mechanisms underlying pain during meningitis are not fully defined, the molecular mechanisms underlying migraine such as CGRP-induced nociceptor sensitization may also be at play.
Immune-Mediated Mechanisms of Pain
The immune system plays an important role in pain sensitization during tissue inflammation. Upon activation by pathogen-derived ligands, immune cells secrete cytokines such as IL-1β, IL-6, and TNF-α, which are able to decrease the threshold for firing action potentials [6, 34, 35]. Cytokines act via mitogen-activated protein kinase and other signaling mechanisms that lead to the phosphorylation and gating of voltage-gated Na+ channels (e.g. Nav1.8) or TRP channels [36,37,38,39]. Immune cells are also a major source of histamine and bradykinin as well as inflammatory lipids including prostaglandins (e.g. PGE2) and leukotrienes that are all potent sensitizers of nociceptor neurons and inflammatory pain. We refer the reader to reviews on the topics of immune cells, their communication with nociceptor neurons, and the immune modulation of pain (Ji et al. [34, 35] and Pinho-Ribeiro et al. [6]). Neurons have also been found to express molecular mediators of innate immunity, including TLRs and their signaling molecules [Sarm1 (sterile alpha and TIR motif-containing 1), MyD88, and Trif (TIR-domain-containing adapter-inducing interferon-β)], and neurons also produce cytokines that can play a role in infection, which is well-reviewed by Liu et al. [40]. Microglia and astrocytes, glial cells in the central nervous system, also play key roles in neuropathic and inflammatory pain by signaling to nociceptor neurons to mediate central sensitization (for comprehensive reviews, see Berta et al. [41] and Ji et al. [42]). Because the area of live pathogen-induced pain and itch is still novel, further investigations are needed to determine whether immune cells or glial cells play a key role in pain induction in each infectious context. This is a rich area for future investigation.
Pain Induced by Viral and Fungal Pathogens
Viral Infection and Pain
Herpesviruses consist of a family of DNA viruses that cause significant human infections. Of note, 3 major types are neurotropic and neuroinvasive: Herpes Simplex Virus 1 (HSV1), Herpes Simplex Virus 2, and Varicella Zoster Virus (VZV). During primary infection, these viruses specifically infect peripheral sensory neurons including nociceptors and pruriceptors. Subsequently, the viruses are retrogradely transported to neuronal cell bodies in the DRG and trigeminal ganglia where they establish viral latency. Upon reactivation, which can occur due to compromise of the immune system, Herpesviruses are transported back to the skin where they establish painful infections. For example, HSV1 reactivation causes painful “cold sores” during reactivation. VZV, the causative agent of shingles, produces significant acute pain during primary infection and can also induce neuropathic pain leading to post-herpetic neuralgia, a chronic, intractable pain [43,44,45]. Post-herpetic neuralgia is a significant complication in people over the age of 60, necessitating vaccination against VZV in geriatric populations [46]. Although herpesviruses cause pain during infection and reactivation, the molecular mechanisms of neuronal activation are not well understood (Fig. 1). Infections of the GI tract by noroviruses or rotaviruses are characterized by marked visceral pain and discomfort (Table 1). However, the cellular and molecular mechanisms underlying pain during norovirus and rotavirus infection remain undefined.
Infection of the respiratory tract by influenza virus leads to fever, significant myalgia (muscle pain), and peripheral tissue pain. In a recent study, Huang et al. found that acute infection with the Influenza A virus leads to acute peripheral mechanical allodynia that is dependent on the enzyme indoleamine 2,3 dioxygenase (IDO) [47]. In parallel, they found that chronic intravenous infection with murine leukemia retrovirus leads to chronic IDO-dependent pain. IDO is induced in splenic CD19-positive dendritic cells during infection, and this mediates the production of kynurenine that mediates pain hypersensitivity during viral infection [47]. Therefore, pain during viral infection can be caused by peripheral immune activation that leads to sensitization via kynurenine (Table 1).
TLR7 is a pathogen-recognition receptor that plays a key role in immune system responses to viruses by binding to single-stranded viral RNA. Ji et al. found that nociceptor neurons express TLR7, which serves as a receptor for microRNAs in pain [14]. This study showed that stimulation of DRG neurons induces the activity-dependent release of extracellular microRNAs such as let-7b, which then induce neuronal firing through TLR7 signaling [14]. This nociceptor activation depends on molecular coupling of TLR7 with the ion channel TRPA1. Pain induced by intraplantar injection of let-7b is abolished in TRPA1−/− mice [14]. It will be interesting to determine whether live viruses also induce the activation of neuronal TLR7 and TRPA1 to contribute to pain during infection.
Fungal Infection and Pain
Fungal pathogens such as the dimorphic fungus Candida albicans or the yeast Saccharomyces cerevisiae induce both painful and itchy infections of the skin and genitourinary tract (Table 1), but the molecular mechanisms are not fully defined. C, albicans commonly causes infections of the female genital tract that are accompanied by acute pain; it can also lead to tissue damage and chronic urogenital pain. Mogil et al. found that repeated acute exposure of the vaginal tract of mice to C. albicans or an extended type of C. albicans infection leads to the development of mechanical allodynia [48]. Injections of S. cerevisiae zymosan also induce chronic mechanical vulval allodynia [48]. They found that the vulva shows increased peptidergic nociceptor and sympathetic innervation following C. albicans infection, which may mediate these changes in pain sensitivity.
Zymosan is a major component of fungal cell walls and consists of a heterogeneous mixture of glucans. Intraplantar injection of zymosan results in significant edema as well as mechanical and thermal pain-like sensitivity; thus, it is widely used as a mouse model of inflammatory pain [49,50,51,52]. Pain during fungal infection is mediated by TLR signaling, as Ferreira et al. showed that zymosan induces joint hyperalgesia and this depends on TLR2 and MyD88, but not on TLR4 in mice [49].
Kashem et al. recently found that DRG nociceptor neurons show Ca2+ influx upon direct application of zymosan or heat-killed C. albicans [53] (Fig. 1). In their study, chemical ablation of TRPV1-positive neurons led to significantly greater proliferation of C. albicans in a mouse model of cutaneous candidiasis. They found that CGRP released by neurons plays a role in signaling to CD301b-positive dermal dendritic cells to release IL-23, a key mediator of downstream γδ-T cell activation and the host defense against C. albicans infection [53]. Therefore, nociceptor neurons are able to directly detect C. albicans to signal pain and to modulate the downstream immune response.
Itch and Infection
Parasites
Two major types of parasite affect human health: ectoparasites and endoparasites. Ectoparasites live outside of the host and feed on mammals; these include insects such as mosquitoes, sand flies, and black flies. Itch-induced scratching may be a behavioral response to protect mammals from ectoparasites [54]. Mosquito bites lead to allergic responses to insect salivary gland secretions and mast cell release of histamine and leukotrienes [55]. These inflammatory mediators specifically activate pruriceptor neurons, leading to the induction of acute itch [56].
Endoparasites live inside hosts for part of their infectious life-cycle and include helminths, schistosomes, pinworms, and tapeworms. Itch also often accompanies infections caused by endoparasites, but the underlying mechanisms are not well understood (Table 1). Onchocerca volvulus is a parasitic worm that causes river blindness and infections are characterized by extreme itch. It infects humans through bites by its insect vector the black fly, subsequently spreading through the skin and bloodstream, eventually migrating to the cornea to cause sclerosing keratitis. Schistosomes, commonly known as blood flukes, also cause acute itch during infection. They attach to and burrow into the skin of humans from water in aquatic habitats, causing what is known as “swimmer’s itch”. Pinworms reside in the GI tract during infection. Female pinworms lay eggs in the skin around the anus, which causes intense itch. The resulting scratching behavior facilitates their spread to other hosts via the fingernails. Therefore, itch may play an important role in the life-cycle of the parasitic pinworm.
It is also conceivable that scratching benefits endoparasites by allowing them to break into and access subcutaneous tissues. By contrast, itch can also be protective for host defenses against endoparasites in some infections by augmenting skin inflammation and allergic immunity at the site of infection. Future studies establishing animal models of parasite infection with concurrent studies of itch behavior could begin to unravel the role of itch in parasite infections and host defense.
Fungal Pathogens
Fungal pathogens often cause skin infections characterized by itch, though the mechanisms remain undefined (Table 1). For example, Tinea pedis or “athlete’s foot”, is a fungal infection of the feet characterized by itching and burning sensations. T. cruris, or “jock itch”, is a fungal infection of the groin area commonly found in young men. Tinea, also known as ringworm, generally refers to dermatophyte-type infections caused by the fungal Trichophyton genus. The fungal pathogen C. albicans can also cause itch during infection. Candida intertrigo is caused by C. albicans infection of the folds of hair-bearing skin and is intensely itchy. Despite this close association of skin fungal infections with itch, the molecular and cellular mechanisms of pruriceptor neuron activation are still mostly unknown.
Bacterial and Viral Pathogens
Bacterial and viral infections can also result in itch-inducing infections. For example, the bacterial pathogen S. aureus is found in 90% of the skin lesions of patients with atopic dermatitis, an inflammatory skin disease characterized by chronic itch [57]. One potential mechanism of itch during S. aureus infection is mast cell-mediated pruriceptor activation. Nunez et al. found that S. aureus releases delta-toxin, an amphipathic peptide that induces mast cell degranulation and mediates skin pathology in atopic dermatitis [58].
VZV, the causative agent of chickenpox, is known to cause acute pruritus in children during infection and this can be mediated by histamine [59]. Above, we discussed how both S. aureus and VZV also cause pain during infection. Therefore, the same pathogens are able to produce pain or itch in different infectious contexts. It remains to be determined what mediates these distinct sensory modalities, though the anatomical site of bacterial or viral infection may play an important role. Epicutaneous or superficial skin infections may activate pruriceptor neurons to produce itch, while deeper tissue infections could activate nociceptor neurons to produce pain.
Itch and Toll-Like Receptors
Recent work by Ji, Liu, and others have demonstrated that TLRs play an important role in pruriceptor activation and itch (for reviews on this topic, see Taves and Ji [60] and Liu and Ji [4]). Pruriceptor neurons express TLR3, which recognizes double-stranded RNA, and TLR7, which recognizes single-stranded RNA [12, 13]. The TLR7 ligand imiquimod induces inward currents in TRPV1-positive neurons and induces itch when injected into the cheek of mice [13]. Imiquimod responses are abrogated when TRPV1-positive neurons are ablated by treatment with resiniferatoxin, and in Tlr7−/− mice. Non-histaminergic itch induced by the pruritogens chloroquine, SLIGRL-NH2 (a peptide sequence), serotonin, or endothelin-1 is reduced in Tlr7−/− mice [13]. In a subsequent study, Liu et al. showed that the TLR3 agonist Poly I:C induces the firing of action potentials in DRG neurons and acute itch when injected into mice, and this is abrogated in Tlr3−/− mice [12]. Tlr3−/− mice show decreased acute itch induced by the mast cell activator 48/80 and the pruritogen chloroquine. Tlr3−/− mice also show decreased pruritus induced in a dry skin model of itch [12]. Therefore, both TLR3 and TLR7 are able to induce pruriception.
TLR4 also plays a role in pruriceptor neuron activation and chronic itch [61, 62]. Liu et al. found that Tlr4−/− mice show reduced scratching in models of dry-skin itch induced by acetone and diethylether followed by water, contact dermatitis, and allergic contact dermatitis [61]. Intrathecal injection of TLR4 antagonists decreases chronic itch, and this is partially mediated by their action on astrocytes. Min et al. found that TLR4 expressed by pruriceptor neurons potentiates itch by the sensitization of TRPV1 and histamine responses [62]. It is still unknown whether these TLR-mediated itch mechanisms play a role in live pathogen infection.
Analgesic Infections
Some pathogenic infections are characterized by a complete lack of pain or are even analgesic in nature (Table 1). Indeed, pathogens can silence pain for their selective advantage. For example, syphilis, caused by the spirochete bacterium Treponema pallidum, induces large genital lesions that are characteristically painless. The mechanism of pain blockade is unknown, but the painless nature of syphilis could facilitate the spread of this sexually-transmitted disease. Below, we discuss the mechanisms of pain blockade induced by the bacterial pathogens C. botulinum and M. ulcerans, mechanisms that could be harnessed as novel analgesic approaches.
Clostridium botulinum
C. botulinum is the causative bacterial agent of infant botulism, characterized by loss of facial expression, swallowing problems, and progressive paralysis due to inhibition of neurotransmission. C. botulinum and related species produce neurotoxins termed botulinum toxins (BoNTs), which mediate the neuronal blockade. BoNTs are classified into seven serotypes based on their ability to react to immune serum (BoNT/A-BoNT/G) [63, 64]. BoNTs consist of heavy-chain (HC) and light-chain (LC) moieties linked by a disulfide bridge. The BoNT HCs mediate binding to mammalian cell membranes through their recognition of membrane polysialogangliosides, and to the synaptic vesicle proteins SV2 or synaptotagmin [65,66,67,68]. Subsequently, BoNTs insert into the cell membrane and deliver the LC moieties into the cytosol. BoNT LCs then serve as metalloproteinases that cleave components of the SNARE complex, thus blocking synaptic vesicle fusion with the plasma membrane and effectively silencing neurotransmission. Specifically, BoNT/A and BoNT/E cleave SNAP25 within the SNARE complex, while BoNT/B, BoNT/D, BoNT/F, and BoNT/G cleave synaptobrevin [63]. BoNTs potently silence peripheral sensory and motor functions during infection through this mechanism. Importantly, BoNTs have been successfully used for the treatment of muscle spasticity and the cosmetic smoothening of facial wrinkles. Moreover, BoNTs have recently been applied in the treatment of chronic neck, shoulder, back, and migraine-associated pain [69,70,71].
Recent work has shown that BoNT/A also targets TRPV1 structurally and functionally to block pain [72,73,74]. BoNT/A treatment significantly decreases TRPV1 expression in trigeminal neurons, and this is mediated by inhibition of TRPV1 membrane trafficking and the induction of proteasome-mediated degradation [74]. Similarly, intraplantar treatment with BoNT/A induces loss of TRPV1 in the DRG in a rat model of neuropathic pain [73]. BoNT/A also co-localizes with TRPV1, SNAP-25, and synaptic vesicle glycoprotein 2A when applied to DRG cultures, and co-immunoprecipitates with TRPV1, suggesting structural interactions [72].
Mycobacterium ulcerans
M. ulcerans is a bacterial pathogen that causes Buruli ulcer in humans, an infectious disease characterized by large, painless skin ulcers. M. ulcerans induces Schwann cell death and nerve damage during late stages of infection, and this was originally hypothesized to be a mechanism of analgesia during infection [75]. Marion et al. recently found that there may also be a direct molecular interaction between microbe and nociceptor neuron that leads to the silencing of pain [76]. They found that M. ulcerans produces a specific mycolactone, a polyketide-derived macrolide, that hyperpolarizes neurons through activation of the angiotensin II type 2 receptor (Fig. 1). Downstream signaling through phospholipase A2 leads to the activation of TWIK-related arachidonic acid-stimulated K+ channels and induces analgesia in mice [76]. Thus, defining specific microbial mechanisms of neuronal modulation could lead to novel therapeutic applications in pain.
Future Work and Conclusions
Pain and itch can play an important role in host defense against pathogens. Pain induces behavioral avoidance of placing pressure on the infection site, which could facilitate the immune response and subsequent wound healing [53]. Pain also leads to social avoidance and sickness behaviors that could decrease pathogenic spread from infected individuals to healthy people. Itch-scratch behavior can also be protective, by the removal of insects that carry disease and by inducing a local allergic immune responses that could contain parasitic or fungal pathogens. Pathogens may also modulate pain or itch to their advantage. For example, sensory neurons release neuropeptides that suppress innate immunity, and could thus facilitate survival within the host [11]. The beneficial or harmful nature of pain and itch in host-pathogen interactions may depend on the specific type of infection, the neuronal subtype involved, and the type of host immune response.
As highlighted in our review, mechanisms of infection-induced pain or itch can be direct through the neuronal recognition of PAMPs. Neuronal activation may also be indirect, in particular through immune cells that are activated during infection. These cells release cytokines, lipids, and other inflammatory mediators in response to pathogens that then sensitize neurons. Pathogenic infections may also indirectly cause pain or itch through other mechanisms such as the induction of oxidative stress. Recent work has shown that anti-oxidants are potent modulators of acute and chronic itch, and thus could be useful in the treatment of infectious pain or itch [3, 77].
There is a need for the development of animal models of parasite, viral, bacterial, and fungal infections in which pain or itch mechanisms can be studied in detail. Despite recent advances, the molecular and cellular bases of pain or itch during most infections are unknown (Table 1). Such interdisciplinary studies also require increased scientific collaboration between microbiologists, immunologists, and neurobiologists. Treatment of pain or itch could use or block specific pathogenic products that regulate neuronal activation. In conclusion, determining sensory neuron-microbe interactions in pain and itch is an emerging, rich field of investigation.
References
Baral P, Mills K, Pinho-Ribeiro FA, Chiu IM. Pain and itch: beneficial or harmful to antimicrobial defense? Cell Host Microbe 2016, 19: 755–759.
Chiu IM, Pinho-Ribeiro FA, Woolf CJ. Pain and infection: pathogen detection by nociceptors. Pain 2016, 157: 1192–1193.
Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull 2012, 28: 131–144.
Liu T, Ji RR. Toll-like receptors and itch. In: Carstens E, Akiyama T (Eds). Itch: Mechanisms and Treatment. Boca Raton (FL): CRC Press/Taylor & Francis, 2014.
Ji RR. Neuroimmune interactions in itch: do chronic itch, chronic pain, and chronic cough share similar mechanisms? Pulm Pharmacol Ther 2015, 35: 81–86.
Pinho-Ribeiro FA, Verri WA, Jr., Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol 2017, 38: 5–19.
Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res 2011, 90: 759–764.
Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 2014, 5: 3125.
Qi J, Buzas K, Fan H, Cohen JI, Wang K, Mont E, et al. Painful pathways induced by TLR stimulation of dorsal root ganglion neurons. J Immunol 2011, 186: 6417–6426.
Xu ZZ, Kim YH, Bang S, Zhang Y, Berta T, Wang F, et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med 2015, 21: 1326–1331.
Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501: 52–57.
Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, et al. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest 2012, 122: 2195–2207.
Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci 2010, 13: 1460–1462.
Park CK, Xu ZZ, Berta T, Han Q, Chen G, Liu XJ, et al. Extracellular microRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron 2014, 82: 47–54.
Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res 2006, 85: 49–53.
Jacinto RC, Gomes BP, Shah HN, Ferraz CC, Zaia AA, Souza-Filho FJ. Incidence and antimicrobial susceptibility of Porphyromonas gingivalis isolated from mixed endodontic infections. Int Endod J 2006, 39: 62–70.
Siqueira JF, Jr., Rocas IN, Silva MG. Prevalence and clonal analysis of Porphyromonas gingivalis in primary endodontic infections. J Endod 2008, 34: 1332–1336.
Calil IL, Zarpelon AC, Guerrero AT, Alves-Filho JC, Ferreira SH, Cunha FQ, et al. Lipopolysaccharide induces inflammatory hyperalgesia triggering a TLR4/MyD88-dependent cytokine cascade in the mice paw. PLoS One 2014, 9: e90013.
Rudick CN, Billips BK, Pavlov VI, Yaggie RE, Schaeffer AJ, Klumpp DJ. Host-pathogen interactions mediating pain of urinary tract infection. J Infect Dis 2010, 201: 1240–1249.
Rudick CN, Berry RE, Johnson JR, Johnston B, Klumpp DJ, Schaeffer AJ, et al. Uropathogenic Escherichia coli induces chronic pelvic pain. Infect Immun 2011, 79: 628–635.
Rosen JM, Klumpp DJ. Mechanisms of pain from urinary tract infection. Int J Urol 2014, 21 Suppl 1: 26–32.
Rudick CN, Jiang M, Yaggie RE, Pavlov VI, Done J, Heckman CJ, et al. O-antigen modulates infection-induced pain states. PLoS One 2012, 7: e41273.
Berube BJ, Bubeck Wardenburg J. Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel) 2013, 5: 1140–1166.
Mistry RD. Skin and soft tissue infections. Pediatr Clin North Am 2013, 60: 1063–1082.
Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A 2010, 107: 13473–13478.
Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol 2002, 23: 541–548.
Liberles SD, Horowitz LF, Kuang D, Contos JJ, Wilson KL, Siltberg-Liberles J, et al. Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc Natl Acad Sci U S A 2009, 106: 9842–9847.
Bufe B, Schumann T, Zufall F. Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J Biol Chem 2012, 287: 33644–33655.
Riviere S, Challet L, Fluegge D, Spehr M, Rodriguez I. Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature 2009, 459: 574–577.
Ibeakanma C, Miranda-Morales M, Richards M, Bautista-Cruz F, Martin N, Hurlbut D, et al. Citrobacter rodentium colitis evokes post-infectious hyperexcitability of mouse nociceptive colonic dorsal root ganglion neurons. J Physiol 2009, 587: 3505–3521.
Spreadbury I, Ochoa-Cortes F, Ibeakanma C, Martin N, Hurlbut D, Vanner SJ. Concurrent psychological stress and infectious colitis is key to sustaining enhanced peripheral sensory signaling. Neurogastroenterol Motil 2015, 27: 347–355.
Davis LE, Katzman JG. Chronic daily headache: when to suspect meningitis. Curr Pain Headache Rep 2008, 12: 50–55.
Lampl C, Yazdi K, Buzath A, Klingler D. Migraine-like headache in bacterial meningitis. Cephalalgia 2000, 20: 738–739.
Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354: 572–577.
Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 2014, 13: 533–548.
Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, et al. Nociceptors are interleukin-1 beta sensors. J Neurosci 2008, 28: 14062–14073.
Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005, 114: 149–159.
Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 2002, 36: 57–68.
Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci 2002, 22: 478–485.
Liu HY, Chen CY, Hsueh YP. Innate immune responses regulate morphogenesis and degeneration: roles of Toll-like receptors and Sarm1 in neurons. Neurosci Bull 2014, 30: 645–654.
Berta T, Qadri YJ, Chen G, Ji RR. Microglial signaling in chronic pain with a special focus on caspase 6, p38 MAP kinase, and sex dependence. J Dent Res 2016, 95: 1124–1131.
Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain 2013, 154 Suppl 1: S10–28.
Mallick-Searle T, Snodgrass B, Brant JM. Postherpetic neuralgia: epidemiology, pathophysiology, and pain management pharmacology. J Multidiscip Healthc 2016, 9: 447–454.
Lee EG, Lee HJ, Hyun DJ, Min K, Kim DH, Yoon MS. Efficacy of low dose gabapentin in acute herpes zoster for preventing postherpetic neuralgia: a prospective controlled study. Dermatol Ther 2016, 29: 184–190.
Forbes HJ, Bhaskaran K, Thomas SL, Smeeth L, Clayton T, Mansfield K, et al. Quantification of risk factors for postherpetic neuralgia in herpes zoster patients: A cohort study. Neurology 2016, 87: 94–102.
Nagel MA, Gilden D. Neurological complications of varicella zoster virus reactivation. Curr Opin Neurol 2014, 27: 356–360.
Huang L, Ou R, Rabelo de Souza G, Cunha TM, Lemos H, Mohamed E, et al. Virus Infections Incite Pain Hypersensitivity by Inducing Indoleamine 2,3 Dioxygenase. PLoS Pathog 2016, 12: e1005615.
Farmer MA, Taylor AM, Bailey AL, Tuttle AH, MacIntyre LC, Milagrosa ZE, et al. Repeated vulvovaginal fungal infections cause persistent pain in a mouse model of vulvodynia. Sci Transl Med 2011, 3: 101ra191.
Guerrero AT, Cunha TM, Verri WA, Jr., Gazzinelli RT, Teixeira MM, Cunha FQ, et al. Toll-like receptor 2/MyD88 signaling mediates zymosan-induced joint hypernociception in mice: participation of TNF-alpha, IL-1beta and CXCL1/KC. Eur J Pharmacol 2012, 674: 51–57.
Vale ML, Cunha FQ, Brito GA, Benevides VM, Ferreira SH, Poole S, et al. Anti-nociceptive effect of thalidomide on zymosan-induced experimental articular incapacitation. Eur J Pharmacol 2006, 536: 309–317.
Yoon SY, Kwon YB, Kim HW, Roh DH, Kang SY, Kim CY, et al. Intrathecal neostigmine reduces the zymosan-induced inflammatory response in a mouse air pouch model via adrenomedullary activity: involvement of spinal muscarinic type 2 receptors. Neuropharmacology 2005, 49: 275–282.
Meller ST, Gebhart GF. Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain 1997, 1: 43–52.
Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 2015, 43: 515–526.
Green D, Dong X. The cell biology of acute itch. J Cell Biol 2016, 213: 155–161.
Horsmanheimo L, Harvima IT, Harvima RJ, Brummer-Korvenkontio H, Francois G, Reunala T. Histamine and leukotriene C4 release in cutaneous mosquito-bite reactions. J Allergy Clin Immunol 1996, 98: 408–411.
Han L, Dong X. Itch mechanisms and circuits. Annu Rev Biophys 2014, 43: 331–355.
Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet 1998, 351: 1715–1721.
Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503: 397–401.
Tebruegge M, Kuruvilla M, Margarson I. Does the use of calamine or antihistamine provide symptomatic relief from pruritus in children with varicella zoster infection? Arch Dis Child 2006, 91: 1035–1036.
Taves S, Ji RR. Itch control by Toll-like receptors. Handb Exp Pharmacol 2015, 226: 135–150.
Liu T, Han Q, Chen G, Huang Y, Zhao LX, Berta T, et al. Toll-like receptor 4 contributes to chronic itch, alloknesis and spinal astrocyte activation in male mice. Pain 2016, 157: 806–817..
Min H, Lee H, Lim H, Jang YH, Chung SJ, Lee CJ, et al. TLR4 enhances histamine-mediated pruritus by potentiating TRPV1 activity. Mol Brain 2014, 7: 59.
Rossetto O, Megighian A, Scorzeto M, Montecucco C. Botulinum neurotoxins. Toxicon 2013, 67: 31–36.
Rossetto O, Pirazzini M, Montecucco C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol 2014, 12: 535–549.
Dong M, Liu H, Tepp WH, Johnson EA, Janz R, Chapman ER. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol Biol Cell 2008, 19: 5226–5237.
Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, et al. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312: 592–596.
Peng L, Berntsson RP, Tepp WH, Pitkin RM, Johnson EA, Stenmark P, et al. Botulinum neurotoxin D-C uses synaptotagmin I and II as receptors, and human synaptotagmin II is not an effective receptor for type B, D-C and G toxins. J Cell Sci 2012, 125: 3233–3242.
Peng L, Tepp WH, Johnson EA, Dong M. Botulinum neurotoxin D uses synaptic vesicle protein SV2 and gangliosides as receptors. PLoS Pathog 2011, 7: e1002008.
Guyer BM. Mechanism of botulinum toxin in the relief of chronic pain. Curr Rev Pain 1999, 3: 427–431.
Royal MA. Botulinum toxins in pain management. Phys Med Rehabil Clin N Am 2003, 14: 805–820.
Wheeler AH, Goolkasian P, Gretz SS. Botulinum toxin A for the treatment of chronic neck pain. Pain 2001, 94: 255–260.
Li X, Coffield JA. Structural and functional interactions between transient receptor potential vanilloid subfamily 1 and botulinum neurotoxin serotype A. PLoS One 2016, 11: e0143024.
Xiao L, Cheng J, Zhuang Y, Qu W, Muir J, Liang H, et al. Botulinum toxin type A reduces hyperalgesia and TRPV1 expression in rats with neuropathic pain. Pain Med 2013, 14: 276–286.
Shimizu T, Shibata M, Toriumi H, Iwashita T, Funakubo M, Sato H, et al. Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol Dis 2012, 48: 367–378.
Goto M, Nakanaga K, Aung T, Hamada T, Yamada N, Nomoto M, et al. Nerve damage in Mycobacterium ulcerans-infected mice: probable cause of painlessness in buruli ulcer. Am J Pathol 2006, 168: 805–811.
Marion E, Song OR, Christophe T, Babonneau J, Fenistein D, Eyer J, et al. Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 2014, 157: 1565–1576.
Zhou FM, Cheng RX, Wang S, Huang Y, Gao YJ, Zhou Y, et al. Antioxidants attenuate acute and chronic itch: peripheral and central mechanisms of oxidative stress in pruritus. Neurosci Bull 2016. doi:10.1007/s12264-016-0076-z.
Acknowledgements
We thank Felipe Pinho-Ribeiro for help with drawing the figure. This review was supported by funding from the National Institutes of Health (NCCIH DP2AT009499 and NIAID K22AI114810), USA.
