
Abstract
Toll-like receptors (TLRs) are cellular sensors designed to recognize molecular danger signals associated with exogenous or endogenous threats. Their activation leads to initiation of the host’s immune responses in order to remove or contain the danger. However, one of the most effective methods of defense against invading pathogens and parasites is itch. The perception of itch elicits the rapid defensive action to scratch, which can remove the offending pathogen or parasite before infection can occur. Recent findings show that TLRs such as TLR3, TLR4, and TLR7 are expressed in a subset of pruriceptive/nociceptive neurons in the dorsal root and trigeminal ganglion providing a direct link between TLR activation and itch. Activation of neuronal TLRs can initiate itch sensation by coupling with ion channels. Furthermore, TLRs are expressed in skin cells and glial cells in the spinal cord to regulate inflammation and neuroinflammation in chronic itch. Thus, identification of the role of TLRs in neurons, skin cells, and glial cells may provide new targets for the treatment of chronic itch, a common clinical problem associated with skin diseases, systemic diseases, and metabolic disorders, for which current treatments are far from sufficient.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction to Toll-Like Receptors
Toll-like receptors (TLRs) are pattern recognition receptors that are involved in the initiation of the innate immune response. Firstly, they recognize molecular patterns that are broadly shared by exogenous pathogens but that are distinguishable from host molecules. These are collectively referred to as pathogen-associated molecular patterns (PAMPs). Secondly, a subset of TLRs also recognize endogenous danger-associated molecular patterns (DAMPs) released in response to cellular stress or tissue injury. Thus, TLRs play a key role in initiating the immune responses to both foreign and endogenous threats.
When microbes were first recognized as the cause of infectious diseases, it was immediately clear that organisms must be capable of identifying when infected in order to initiate an immune response. Over a century ago, Richard Pfeiffer discovered “endotoxins” now known as lipopolysaccharide (LPS) which is produced by most Gram-negative bacteria (Westphal et al. 1978). Administration of LPS alone could provoke fever and shock in experimental animals. Additional molecules such as lipopeptides, flagellin, and unmethylated DNA could also provoke host immune responses; however, their receptors remained elusive.
The Toll receptor was first identified in Drosophila melanogaster and was reported for its role in dorsal–ventral patterning in the development (Anderson et al. 1985a, b). In 1996, Hoffmann and colleagues found that Toll also participated in the fly’s immune response to fungal infection (Lemaitre et al. 1996), and in 1997 Charles Janeway and Ruslan Medzhitov showed that activation of human Toll-like receptor 4 (TLR4) could promote an adaptive immune response (Medzhitov et al. 1997). Beutler and colleagues demonstrated that mice that could not respond to LPS had mutations that abolished the function of TLR4 and proved that TLR4 was in fact the receptor for LPS (Poltorak et al. 1998). Notably, Drs. Beutler and Hoffman were awarded the Nobel Prize in Medicine or Physiology for their pioneer work on TLRs in 2011. To date, 10 (TLR 1–10) and 12 (TLR 1–9; TLR 11–13) functional TLRs have been identified in human and mouse, respectively (Kawai and Akira 2010).
1.1 Structure
TLRs are type I transmembrane proteins comprised of an ectodomain responsible for detecting PAMPs or DAMPs, a transmembrane region consisting of a single α-helix, and a cytosolic Toll–interleukin-1 receptor (TIR) domain that activates downstream signaling pathways (Kawai and Akira 2010). The extracellular domain binds either directly to ligands or to cofactors that can modulate their sensitivity to their natural ligand. In the case of TLR4, recognition of LPS requires the presence of MD-2, while cofactors CD14 and LPS-binding protein facilitate the presentation of LPS to MD-2. Upon stimulation, most TLRs form homodimers; however, some TLRs such as TLR1–TLR2 and TLR2–TLR6 also form heterodimers, each with a different ligand specificity (Triantafilou et al. 2007; Oosting et al. 2011).
Structurally, TLRs may be classified into two groups. The first has structural transitions that divide the proteins into three subdomains: N terminal, central, and C terminal (Jin et al. 2007; Kim et al. 2007; Kang et al. 2009; Kang and Lee 2011). The ligand-binding pockets of TLR1, TLR2, and TLR6 are located close to the transition site between N terminal and central (Jin et al. 2007; Kang et al. 2009; Kang and Lee 2011), and the MD-2-binding pocket of TLR4 is located between the transition from central to C terminal (Kim et al. 2007). The second group has a single-domain configuration such as TLR3 (Choe et al. 2005; Bell et al. 2006). While the structures of TLR5, TLR7, TLR8, TLR9, and TLR10 have not yet been reported, sequence analysis suggests that the first four should belong to the single-domain group, while TLR10 should belong to the three-domain group (Kang and Lee 2011). These structural observations agree with functional analyses that have demonstrated that the three-domain TLRs interact with lipid-containing molecules such as LPS and lipoproteins. Accordingly, all three-domain TLRs (1, 2, 6, and 10) are expressed on the cell membrane for the detection of microbial membrane components. Conversely, the single-domain TLRs interact with hydrophilic ligands, such as vial or endogenous nucleic acids, and are expressed intracellularly on endosomes and the endoplasmic reticulum (TLR3, TLR7/TLR8, and TLR9). However, some TLRs (TLR3 and TLR7) are localized both on membrane and in intracellular compartments (Akira et al. 2006) (Table 1).
1.2 Ligands
Each type of TLR detects distinct molecular patterns specific to pathogenic threats to the organism such as viruses, bacteria, mycobacteria, fungi, and parasites. For example, TLR4 recognizes LPS (Poltorak et al. 1998; Shimazu et al. 1999), a component of Gram-negative bacterial cell membranes; TLR5 detects flagellin, a protein in bacterial flagella (Hayashi et al. 2001); and TLR11 senses profilin-like protein (Yarovinsky et al. 2005). TLR2 heterodimers bind to specific lipopeptides, another component of bacterial cell membranes. The ligand specificity of TLR2 is modulated by its heterodimeric partner. The TLR1–TLR2 complex binds to triacyl lipopeptides with only a weak affinity for diacyl lipopeptides, while the TLR2–TLR6 complex interacts with diacyl lipopeptides but not to triacyl lipopeptides (Takeuchi et al. 2001, 2002; Alexopoulou et al. 2002; Yamamoto et al. 2002; Jin et al. 2007; Kang et al. 2009). TLR3 senses double-stranded RNAs and TLR7/TLR8 single-stranded RNAs, present in retroviruses and viruses, respectively (Alexopoulou et al. 2001; Diebold et al. 2004; Heil et al. 2004; Town et al. 2006). TLR9 senses unmethylated CpG DNA (Hemmi et al. 2000; Krieg 2002). Importantly, TLRs also recognize endogenous markers of cell necrosis and tissue injury, termed DAMPs. Activation of TLRs by DAMPs induces sterile inflammatory responses (Okamura et al. 2001; Biragyn et al. 2002; Vabulas et al. 2002; Kariko et al. 2004; Jiang et al. 2005; Tian et al. 2007; Imai et al. 2008; Midwood et al. 2009; West et al. 2010). Thus, TLRs can recognize both pathogen invasion through their recognition of PAMPs and tissue injury through their recognition of endogenous DAMPs (summarized in Table 1).
1.3 Signaling Pathways
Activation of TLR signaling leads to a tightly regulated intracellular signaling pathway that initiates the production and secretion of proinflammatory mediators. The intracellular signaling domains of TLRs have a high sequence similarity with the interleukin-1 receptor and are termed Toll/IL-1R (TIR) homology domains. Signaling adaptor proteins MyD88, TRIF, and TRAM also contain TIR domains and interact with the TIR domains of the TLR receptors through heterotypic TIR–TIR interactions (Watters et al. 2007; Kenny and O’Neill 2008). TRIF and TRAM are also referred to as TICAM-1 and TICAM-2, respectively. All TLRs, with the exception of TLR3, signal through MyD88. TLR3 signals through a TRIF-dependent pathway discussed later. TLR4 is also capable of signaling in a MyD88-independent manner by recruiting TRAM (Yamamoto et al. 2003). TLR1/TLR2, TLR4, and TLR6 recruit the additional adapter protein TIRAP (Akira and Takeda 2004). Upon activation of the MyD88-dependent pathway, MyD88 recruits the IL-1R-associated kinases (IRAKs), which interact with tumor necrosis factor receptor-associated factor 6 (TRAF6), leading to the phosphorylation and degradation of the inhibitor of nuclear factor-κB (NF-kB) IκB. Degradation of IκB releases NF-κB which translocates to promote transcription of proinflammatory genes. The MyD88-dependent pathway also activates the mitogen-activated protein kinase (MAPK) signaling pathways, such as p38, and c-Jun N-terminal kinase (JNK), leading to the activation of AP-1 and interferon regulatory factors (IRFs) (see Fig. 1).

Conventional signaling of TLRs. Activation of extracellular TLRs (e.g., TLR4) and intracellular TLRs (e.g., TLR3 and TLR7) initiates canonical TRIF- and MYD88-dependent signaling pathways resulting in the transcription of proinflammatory genes. Specifically, the activation of the transcriptional factors IRF3/IRF7 and NF-κB induces the production of inflammatory mediators such as cytokines (e.g., IL-1β, interferons), chemokines (e.g., CCL2), PGE2 via the enzyme cyclooxygenase 2 (COX-2), and NO via the enzyme inducible nitric oxide synthase (iNOS). These inflammatory mediators act in concert to initiate the host’s innate immune response
TLR3 signals through a TRIF-dependent pathway which activates two signaling pathways. First, TRIF recruits TRAF3 to activate IRF3 and IRF7 and initiate the production of type I interferons (e.g., IFN-α/IFN-β), which are the first line of defense produced by the innate immune system. Second, TRIF interacts with receptor-interacting protein 1 (RIP-1) and TRAF6 to activate NF-κB and/or MAPK pathways (Fig. 1). This is involved in the late-phase induction of proinflammatory genes.
The signaling molecules that comprise each TLR signaling pathway are also subject to a high degree of regulation through physical interactions, conformational changes, phosphorylation, ubiquitination, and proteasome-mediated degradation (Carpenter and O’Neill 2009). Some miRNAs are capable of regulating TLR signaling through targeting the 3′ untranslated regions of mRNAs encoding components of the TLR signaling system (O’Neill et al. 2011). Activation of TLR signaling produces a wide array of proinflammatory mediators, such as cytokines, chemokines, and reactive oxygen/nitrogen intermediates including nitric oxide (Takeda and Akira 2004). Upregulated activity of TLR signaling may promote chronic itch (Liu and Ji 2013).
2 Introduction to Itch
Itch, or pruritus, is defined as an unpleasant sensation that elicits the desire or reflex to scratch. While acute itch is an adaptive mechanism to warn an organism of potential chemical or parasitic danger (Ikoma et al. 2006), chronic itch is a common clinical problem associated with skin diseases (Reich and Szepietowski 2007; Bieber 2008), liver and kidney diseases (Cassano et al. 2010; Kremer et al. 2010), and metabolism disorders (Yamaoka et al. 2010). Additionally, itching itself leads to scratching which can cause inflammation, skin degradation, and secondary infection.
While some itch-sensing circuitry may overlap with existing nociceptive circuits, there are features that have recently been shown that are specific to itch. Itch sensation arises from MrgprA3+ fibers, derived from a specific subset of primary sensory neurons that reside in the skin, but not in deeper tissues, muscles, organs, or bones where itch sensation does not occur (Han et al. 2013). Their cell bodies are located in the dorsal root ganglia (DRG) or trigeminal ganglia, while their peripheral terminals, free nerve endings, reach to the stratum granulosum of the epidermis, and their central projections terminate in the superficial horn of the spinal cord. Itch-specific neurons terminate in lamina I–II and release glutamate as well as gastrin-releasing peptide (GRP), a neuropeptide known to elicit itch sensation via activation of GRP receptors (GRPRs) on superficial dorsal horn neurons (Sun and Chen 2007; Liu et al. 2009; Sun et al. 2009). A major source of GRP in the spinal cord may arise from interneurons (Alemi et al. 2013; Mishra and Hoon 2013). In addition, the neuropeptide natriuretic polypeptide b (Nppb) and substance P are also implicated in spinal cord itch transmission (Akiyama and Carstens 2013; Mishra and Hoon 2013). In the dorsal horn, itch signals can be processed and modulated before ascending to the brain where activation of specific brain regions then results in the perception of itch. Notably, in acute conditions, pain can suppress itch sensation via spinal cord inhibitory neurons (Liu et al. 2010b; Ross et al. 2010; Liu and Ji 2013).
The most characterized mediator of itch is histamine. The released histamine from local mast cells binds to H1/H4 receptors on nerve terminals (Shim and Oh 2008) which is followed by activation of PLCbeta3 and transient receptor potential vanilloid subtype 1 (TRPV1) (Han et al. 2006; Imamachi et al. 2009). TRPV1-positive C-fibers are required for both histamine-dependent and histamine-independent itch (Imamachi et al. 2009). Histamine-independent itch can be induced by the activation of transient receptor potential cation channel, subfamily A, member 1 (TRPA1), chloroquine (an antimalarial drug and MrgprA3 agonist), BAM8–22 (an endogenous MrgprC11 agonist), and oxidative stress (Liu et al. 2009; Wilson et al. 2011; Liu and Ji 2012), respectively.
3 Control of Itch by Peripheral TLRs
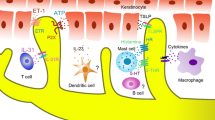
The skin is the body’s largest organ and first line of defense against microbial and parasitic invaders. The skin expresses every type of known TLR receptor; however, each cell type has a unique expression pattern and distinct contribution to the skin’s immune response (Ermertcan et al. 2011). In particular, two TLR-expressing cell type keratinocytes and mast cells have been implicated in chronic itch (Ikoma et al. 2006) (see Fig. 2).

Involvement of peripherally expressed TLRs in itch sensation via different mechanisms. Cells residing in the skin such as keratinocytes and mast cells express TLRs. Their activation by exogenous ligands (PAMP) and endogenous ligands (DAMP) results in the release of multiple pruritogens including NGF, ET-1, 5HT, tryptase, and histamine, all of which can activate G protein coupled receptors (GPCRs) or TrkA receptor (in the case of NGF) on pruriceptive neuronal terminals to increase neuronal excitation and trigger itch sensation. TLRs such as TLR3 and TLR7 are also expressed by pruriceptor terminals, which can be activated by DAMP such as double-strand and single-strand RNAs. Activation of TLR3 and TLR7 elicits rapid inward currents and action potentials, due to the direct coupling with ion channels in pruriceptors, leading to increased neuronal excitation and itch sensation. A second late-phase response to the activation of TLRs may also involve the increased transcription of proinflammatory mediators which can maintain neuronal hyperexcitability in chronic itch conditions
Epidermal keratinocytes express TLRs 1–6, 9, and 10, which are upregulated in pruritic skin diseases, such as psoriasis and atopic dermatitis (Baker et al. 2003; Ermertcan et al. 2011). Mast cells, predominantly in the dermis, express TLRs 1–7 and 9 and play a key role in IgE-mediated allergic inflammation. Activation of these TLRs results in the synthesis and release of cytokines and chemokines to recruit immune cells from the circulation and mount an adaptive immune response. In particular, keratinocytes and mast cells are the major sources of nerve growth factor (NGF) (Ikoma et al. 2006). Intradermal administration of NGF enhances itch sensation in humans (Rukwied et al. 2013), and NGF is upregulated in dry skin models of itch in mice (Tominaga et al. 2007). Of interest, this increase is suppressed in Tlr3 knockout animals (Liu et al. 2012). Therefore, TLR3-mediated release of NGF from skin cells may contribute to peripheral sensitization in chronic itch conditions.
While TLRs have mainly been studied on immune cells, we have recently demonstrated the expression of TLR3 and TLR7 on a subset of primary sensory neurons that are responsible for itch. Single-cell Real-time PCR (RT-PCR) and immunohistochemistry demonstrated that TLR7 is expressed in small TRPV1-expressing DRG neurons (Liu et al. 2010a, 2012) and is completely colocalized with MrgprA3+. TLR3 partially colocalizes with TLR7 and is also expressed in TRPV1-positive DRG neurons as shown by in situ hybridization, immunocytochemistry, single-cell RT-PCR, and electrophysiology (Liu et al. 2012).
TLR7 responds to synthetic ligands such as imidazoquinoline derivatives [e.g., imiquimod and resiquimod (R848)] and guanine analogs (e.g., loxoribine) (Hemmi et al. 2002). Intradermal injection of these synthetic compounds produces scratching behavior in wild-type mice, which is reduced in Tlr7 −/− mice, suggesting that these responses are, at least partially, TLR7 dependent (Liu et al. 2010a). Tlr7 −/− mice also show a significant reduction of scratching behavior in response to non-histaminergic pruritogens such as chloroquine, endothelin-1 (ET-1), and SLIGRL-NH2, an agonist of protease-activated receptor 2 (PAR2) (Liu et al. 2010a). Interestingly, Tlr7 −/− mice exhibited normal thermal and mechanical pain sensitivity. These findings show that TLR7 may serve an important role as a non-histaminergic itch receptor.
Activation of TLR7 in immune cells initiates signaling cascades leading to a variety of transcriptional changes to promote inflammation. However, in small DRG neurons, application of TLR7 agonists elicits rapid inward currents and action potentials, which are abolished in Tlr7 −/− knockout mice (Liu et al. 2010a). This immediate increase in neuronal excitability suggests that TLR7 may be coupled to ion channel activation in primary sensory neurons. Our recent study shows that TLR7 is functionally coupled to TRPA1 but not TRPV1 (Park et al. 2014).
TLR3 responds to dsRNA as well as its synthetic analog, polyinosinic–polycytidylic acid (poly I:C), in which one strand of RNA is replaced by a polymer of inosinic acid. Activation of TLR3 by its ligands poly I:C or purified total RNAs also elicits rapid inward currents and the generation of action potentials from DRG neurons in wild-type but not Tlr3 −/− mice (Liu et al. 2012). Furthermore, intradermal application of poly I:C produced dose-dependent scratching behavior in wild-type mice which was abolished in Tlr3 −/− mice (Liu et al. 2012). Similar to TLR7, TLR3 also seems to serve as an itch receptor/co-receptor on pruriceptive neurons. In contrast to Tlr7 −/− mice, which showed a partial reduction in histamine-independent itch, Tlr3 −/− mice displayed significant reductions in both histamine-dependent and histamine-independent itch (Liu et al. 2012). Knockdown of TLR3 by intrathecal injection of Tlr3 antisense oligonucleotides significantly reduced TLR3 expression in the DRG and reduced both histamine-dependent and histamine-independent itch in wild-type mice (Liu et al. 2012). This corroborates the results found using Tlr3 knockout animals.
TLR3 and TLR7 expression by primary sensory neurons seems to serve as itch receptors to detect foreign pathogens and endogenous ligands (e.g., ds- and ssRNA, respectively), leading to a rapid defensive response: scratching. As neuronal excitation occurs within minutes of agonist application, the neuronal signaling pathway must have a distinct component from the traditional TLR signaling pathway in immune cells; however, the details remain to be determined. We postulate that TLR3 and TLR7 are expressed on the cell surface and are coupled to unidentified ion channels capable of inducing inward currents and action potentials (see Fig. 2). Of note, it was also demonstrated that the activation of TLRs, including TLR3, TLR7, and TLR9, in DRG neurons by their respective ligands may indirectly influence the excitability of DRG neurons by inducing the expression of proinflammatory mediators such as prostaglandin E2 (PGE2), calcitonin gene-related peptide (CGRP), and interleukin-1beta (IL-1β) (Qi et al. 2011) (see Table 2). Thus, TLRs expressed by DRG neurons may regulate neuronal excitability by both transcriptional and non-transcriptional mechanisms.
4 Control of Itch by Central TLRs
While both TLRs play a role in eliciting itch sensation from peripheral stimuli, only TLR3 was found to play a critical role in spinal synaptic transmission and the central sensitization underlying itch processing in the spinal cord (Liu et al. 2012). Tlr3 −/− mice displayed decreased spontaneous excitatory postsynaptic currents (sEPSCs) in spinal lamina II neurons, while activation of TLR3 by poly I:C resulted in an increase in sEPSCs in wild-type animals (Liu et al. 2012). Furthermore, knockout of Tlr3 abolished spinal long-term potentiation (LTP) following tetanic stimulation of the sciatic nerve (Liu et al. 2012). In contrast, both sEPSC activity and LTP remained intact in Tlr7 −/− mice (Liu et al. 2012). Given the important role of spinal synaptic plasticity in itch hypersensitivity (Ross et al. 2010), impairments in central sensitization are likely to contribute to the profound itch deficit in Tlr3 −/− mice.
Within the spinal cord, glial cells also express TLRs. Microglia, the resident immune cell of the central nervous system (CNS), express almost all known members of the TLR family, while astrocyte expression of TLRs is limited (Farina et al. 2007) (see Table 2). Similar to the activation of TLRs on peripheral immune cells, the activation of TLRs on glia results in the production and secretion of proinflammatory mediators including cytokines (e.g., TNF-α), chemokines [e.g., chemokine ligand 2 (CCL2)], and enzymes [e.g., cyclooxygenase 2 (COX-2)], as well as other inflammatory mediators (e.g., prostaglandins) (Basbaum et al. 2009; van Noort and Bsibsi 2009; Lehnardt 2010; Nicotra et al. 2011). While these mediators are known to generate central sensitization and pain hypersensitivity, their role in chronic itch is unknown.
5 Clinical Significance and Future Perspectives
Chronic itch is a common and significant clinical problem. Chronic itch may involve the entire skin or be localized to a specific area or dermatome. It is more common in women than in men (Ständer et al. 2013), and its incidence increases with age (Weisshaar and Dalgard 2009; Ständer et al. 2010). It can be broadly categorized into four major etiologies: dermatologic causes (e.g., atopic eczema, psoriasis, or scabies), systemic causes (e.g., liver and kidney disease or metabolism disorders), neuropathic causes (such as spinal nerve impingement), and psychogenic causes (Yosipovitch and Bernhard 2013). However, regardless of the cause, it is associated with a marked reduction in the quality of life. In fact, a recent study showed that chronic itch was as debilitating as chronic pain (Kini et al. 2011). Clinically, the current treatments for chronic itch are far from sufficient (Yosipovitch and Bernhard 2013); however, targeting TLRs may offer new therapy options for treating debilitating itch-related problems.
TLRs are emerging as important players in the regulation of acute and chronic itch. They are expressed by many components of the itch signaling pathway including the cells of the skin, resident and infiltrating immune cells, peripheral and central neurons, as well as central glia. However, the precise contribution of TLR signaling in each cell type remains to be determined. Additionally, our work has raised the exciting possibility that TLR-mediated neuronal excitation occurs in a non-transcriptional manner. Again, much work remains to be done in order to elucidate the details of this pathway. These distinctions may provide targets for developing new therapies that block the detrimental effects of persistent unregulated TLR signaling, while leaving their beneficial effects intact.
Abbreviations
- 5HT:
-
Serotonin
- BDNF:
-
Brain-derived neurotrophic factor
- CCL2:
-
Chemokine ligand 2 (MCP-1)
- CGRP:
-
Calcitonin gene-related peptide
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase 2
- CXCL:
-
Chemokine (C-X-C motif) ligand
- DAMPs:
-
Danger-associated molecular patterns
- DRG:
-
Dorsal root ganglia
- dsRNA:
-
Double-stranded RNA
- ERK:
-
Extracellular signal-regulated kinase
- ET-1:
-
Endothelin-1
- GRP:
-
Gastrin-releasing peptide
- GRPR:
-
Gastrin-releasing peptide receptor
- IL-1β:
-
Interleukin-1beta
- IRAKs:
-
IL-1R-associated kinases
- IRFs:
-
Interferon regulatory factors
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- LTP:
-
Long-term potentiation
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemoattractant protein 1 (CCL2)
- miRNA:
-
Microribonucleic acid
- mRNA:
-
Messenger ribonucleic acid
- NF-kB:
-
Nuclear factor-kappa B
- NGF:
-
Nerve growth factor
- NO:
-
Nitric oxide
- Nppb:
-
Natriuretic polypeptide b
- PAMPs:
-
Pathogen-associated molecular patterns
- PAR2:
-
Protease-activated receptor 2
- PGE2:
-
Prostaglandin E2
- Poly I:C:
-
Polyinosinic:polycytidylic acid
- RIP-1:
-
Receptor-interacting protein 1
- sEPSCs:
-
Spontaneous excitatory postsynaptic currents
- ssRNA:
-
Single-stranded RNA
- TIR:
-
Toll–interleukin-1 receptor
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor alpha
- TRAF6:
-
Tumor necrosis factor receptor-associated factor 6
- TRPV1:
-
Transient receptor potential vanilloid subtype 1
References
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Akiyama T, Carstens E (2013) Neural processing of itch. Neuroscience 250:697–714
Alemi F, Kwon E, Poole DP, Lieu T, Lyo V, Cattaruzza F, Cevikbas F, Steinhoff M, Nassini R, Materazzi S, Guerrero-Alba R, Valdez-Morales E, Cottrell GS, Schoonjans K, Geppetti P, Vanner SJ, Bunnett NW, Corvera CU (2013) The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest 123:1513–1530
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, Medzhitov R, Fikrig E, Flavell RA (2002) Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat Med 8:878–884
Anderson KV, Bokla L, Nusslein-Volhard C (1985a) Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42:791–798
Anderson KV, Jurgens G, Nusslein-Volhard C (1985b) Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42:779–789
Baker BS, Ovigne JM, Powles AV, Corcoran S, Fry L (2003) Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: modulation of TLR expression in chronic plaque psoriasis. Br J Dermatol 148:670–679
Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139:267–284
Bell JK, Askins J, Hall PR, Davies DR, Segal DM (2006) The dsRNA binding site of human Toll-like receptor 3. Proc Natl Acad Sci USA 103:8792–8797
Bieber T (2008) Atopic dermatitis. N Engl J Med 358:1483–1494
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW (2002) Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science 298:1025–1029
Carpenter S, O’Neill LA (2009) Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem J 422:1–10
Cassano N, Tessari G, Vena GA, Girolomoni G (2010) Chronic pruritus in the absence of specific skin disease: an update on pathophysiology, diagnosis, and therapy. Am J Clin Dermatol 11:399–411
Choe J, Kelker MS, Wilson IA (2005) Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 309:581–585
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531
Ermertcan AT, Ozturk F, Gunduz K (2011) Toll-like receptors and skin. J Eur Acad Dermatol Venereol 25:997–1006
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28:138–145
Han SK, Mancino V, Simon MI (2006) Phospholipase Cbeta 3 mediates the scratching response activated by the histamine H1 receptor on C-fiber nociceptive neurons. Neuron 52:691–703
Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, Kim Y, Nie H, Qu L, Patel KN, Li Z, McNeil B, He S, Guan Y, Xiao B, Lamotte RH, Dong X (2013) A subpopulation of nociceptors specifically linked to itch. Nat Neurosci 16:174–182
Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 3:196–200
Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M (2006) The neurobiology of itch. Nat Rev Neurosci 7:535–547
Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249
Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, Han SK (2009) TRPV1-expressing primary afferents generate behavioral responses to pruritogens via multiple mechanisms. Proc Natl Acad Sci USA 106:11330–11335
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11:1173–1179
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082
Kang JY, Lee JO (2011) Structural biology of the Toll-like receptor family. Annu Rev Biochem 80:917–941
Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279:12542–12550
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Kenny EF, O’Neill LA (2008) Signalling adaptors used by Toll-like receptors: an update. Cytokine 43:342–349
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130:906–917
Kini SP, DeLong LK, Veledar E, McKenzie-Brown AM, Schaufele M, Chen SC (2011) The impact of pruritus on quality of life: the skin equivalent of pain. Arch Dermatol 147:1153–1156
Kremer AE, Martens JJ, Kulik W, Rueff F, Kuiper EM, van Buuren HR, van Erpecum KJ, Kondrackiene J, Prieto J, Rust C, Geenes VL, Williamson C, Moolenaar WH, Beuers U, Oude Elferink RP (2010) Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 139(1008–18):1018
Krieg AM (2002) CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20:709–760
Lehnardt S (2010) Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58:253–263
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983
Liu T, Ji RR (2012) Oxidative stress induces itch via activation of transient receptor potential subtype ankyrin 1 in mice. Neurosci Bull 28:145–154
Liu T, Ji RR (2013) New insights into the mechanisms of itch: are pain and itch controlled by distinct mechanisms? Pflugers Arch 465:1671–1685
Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, Ru F, Guan Y, Weng HJ, Geng Y, Undem BJ, Kollarik M, Chen ZF, Anderson DJ, Dong X (2009) Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 139:1353–1365
Liu T, Xu ZZ, Park CK, Berta T, Ji RR (2010a) Toll-like receptor 7 mediates pruritus. Nat Neurosci 13:1460–1462
Liu Y, Abdel SO, Zhang L, Duan B, Tong Q, Lopes C, Ji RR, Lowell BB, Ma Q (2010b) VGLUT2-dependent glutamate release from nociceptors is required to sense pain and suppress itch. Neuron 68:543–556
Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, Liu Q, Liu Y, Gao YJ, Liu YC, Ma Q, Dong X, Ji RR (2012) TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest 122:2195–2207
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397
Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F, Foxwell B (2009) Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15:774–780
Mishra SK, Hoon MA (2013) The cells and circuitry for itch responses in mice. Science 340:968–971
Nicotra L, Loram LC, Watkins LR, Hutchinson MR (2011) Toll-like receptors in chronic pain. Exp Neurol 234:316–329
O’Neill LA, Sheedy FJ, McCoy CE (2011) MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 11:163–175
Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF III (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 276:10229–10233
Oosting M, Ter HH, Sturm P, Adema GJ, Kullberg BJ, van der Meer JW, Netea MG, Joosten LA (2011) TLR1/TLR2 heterodimers play an important role in the recognition of Borrelia spirochetes. PLoS One 6:e25998
Park CK, Xu ZZ, Berta T, Han QJ, Liu XJ, Ji RR (2014) Extracellular miRNAs activate nociceptor neurons to elicit pain via TLR7 and TRPA1. Neuron 82:47–54
Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088
Qi J, Buzas K, Fan H, Cohen JI, Wang K, Mont E, Klinman D, Oppenheim JJ, Howard OM (2011) Painful pathways induced by TLR stimulation of dorsal root ganglion neurons. J Immunol 186:6417–6426
Reich A, Szepietowski JC (2007) Mediators of pruritus in psoriasis. Mediators Inflamm 2007:1–6
Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, Tolias C, Corfas R, Chen S, Inquimbert P, Xu Y, McInnes RR, Rice FL, Corfas G, Ma Q, Woolf CJ, Greenberg ME (2010) Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron 65:886–898
Rukwied RR, Main M, Weinkauf B, Schmelz M (2013) NGF sensitizes nociceptors for cowhage- but not histamine-induced itch in human skin. J Invest Dermatol 133:268–270
Shim WS, Oh U (2008) Histamine-induced itch and its relationship with pain. Mol Pain 4:29
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782
Ständer S, Schafer I, Phan NQ, Blome C, Herberger K, Heigel H, Augustin M (2010) Prevalence of chronic pruritus in Germany: results of a cross-sectional study in a sample working population of 11,730. Dermatology 221:229–235
Ständer S, Stumpf A, Osada N, Wilp S, Chatzigeorgakidis E, Pfleiderer B (2013) Gender differences in chronic pruritus: women present different morbidity, more scratch lesions and higher burden. Br J Dermatol 168:1273–1280
Sun YG, Chen ZF (2007) A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature 448:700–703
Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF (2009) Cellular basis of itch sensation. Science 325:1531–1534
Takeda K, Akira S (2004) TLR signaling pathways. Semin Immunol 16:3–9
Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S (2001) Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol 13:933–940
Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 169:10–14
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La RG, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Tominaga M, Ozawa S, Tengara S, Ogawa H, Takamori K (2007) Intraepidermal nerve fibers increase in dry skin of acetone-treated mice. J Dermatol Sci 48:103–111
Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA (2006) Microglia recognize double-stranded RNA via TLR3. J Immunol 176:3804–3812
Triantafilou M, Uddin A, Maher S, Charalambous N, Hamm TS, Alsumaiti A, Triantafilou K (2007) Anthrax toxin evades Toll-like receptor recognition, whereas its cell wall components trigger activation via TLR2/6 heterodimers. Cell Microbiol 9:2880–2892
Vabulas RM, Wagner H, Schild H (2002) Heat shock proteins as ligands of toll-like receptors. Curr Top Microbiol Immunol 270:169–184
van Noort JM, Bsibsi M (2009) Toll-like receptors in the CNS: implications for neurodegeneration and repair. Prog Brain Res 175:139–148
Watters TM, Kenny EF, O’Neill LA (2007) Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol 85:411–419
Weisshaar E, Dalgard F (2009) Epidemiology of itch: adding to the burden of skin morbidity. Acta Derm Venereol 89:339–350
West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467:972–976
Westphal O, Westphal U, Sommer T (1978) The history of pyrogen research. In: Schlessinger D (ed) Microbiology. American Society for Microbiology, Washington, DC, pp 221–238
Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, Bautista DM (2011) TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci 14:595–602
Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 169:6668–6672
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643
Yamaoka H, Sasaki H, Yamasaki H, Ogawa K, Ohta T, Furuta H, Nishi M, Nanjo K (2010) Truncal pruritus of unknown origin may be a symptom of diabetic polyneuropathy. Diabetes Care 33:150–155
Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629
Yosipovitch G, Bernhard JD (2013) Clinical practice. Chronic pruritus. N Engl J Med 368:1625–1634
Acknowledgements
This work is supported by NIH grants R01DE17794, R01DE22743, and R01NS89479.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Taves, S., Ji, RR. (2015). Itch Control by Toll-Like Receptors. In: Cowan, A., Yosipovitch, G. (eds) Pharmacology of Itch. Handbook of Experimental Pharmacology, vol 226. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44605-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-662-44605-8_7
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44604-1
Online ISBN: 978-3-662-44605-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)