
Abstract
The atomic force microscope (AFM), a member of the scanning probe family of microscopes, generates height maps of sample surfaces with subnanometer resolution. Importantly, AFM offers the opportunity to image samples with little or no treatment and under physiologically-relevant conditions, making it well-suited for investigating the structure of biological samples, including fixed or living cells and tissues. In addition to its high-resolution imaging capability, AFM used in force spectroscopy mode is a sensitive force measuring device, able to detect or exert forces ranging from the pico- to the nanonewton scale. Here we review a broad range of cell biological applications of AFM, including high resolution imaging of adherent cells, measuring cell adhesion down to the single-receptor level and characterizing the mechanical properties of cells. Furthermore, we present recent examples of how the combined use of AFM and advanced light microscopy techniques can provide complementary structural information.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Soon after its invention more then 20 years ago as a high resolution imaging tool for inorganic materials,10 the atomic force microscope (AFM) emerged as a powerful tool to study biological samples.22,46 A vital advancement in applying AFM for biology was the introduction of an optical cantilever deflection system,4,53 allowing imaging at ambient temperature, and the introduction of a fluid cell which permitted imaging samples in solution.16 This offered the possibility to maintain biological samples in a physiological environment and provided a unique high-resolution imaging technique for living cells. Although AFM thus first evolved as an imaging tool, it has subsequently also been used extensively to exert or probe forces on cells, providing insight into their adhesive and mechanical properties.8,14,64 Here we provide an overview of how AFM has been used to study the morphology of substrate-adhering cells. We furthermore introduce AFM techniques that can be used to measure cellular adhesion both with a single molecule force resolution and at the single cell scale. We then present ways of characterizing mechanical properties of cells by indenting cells using the AFM tip. Finally, recent developments complementing AFM with optical microscopy are presented and an outlook is given on future developments and applications of AFM to nano-biological systems.
Principles of AFM and AFM Imaging
In AFM a sharp tip connected to a flexible cantilever (Fig. 1a) is scanned over the sample in a series of horizontal sweeps.10 For imaging cells or cellular compartments, two AFM imaging modes are most frequently employed. In contact mode, the tip is in direct contact with the sample and consequently traces height changes during scanning (Fig. 1b). Tip movement leads to bending (deflection) of the cantilever, which is detected by recording the deflection of a LASER beam reflected off the end of the cantilever with a set of photodiodes.4,53 Using a feedback loop, the deflection signal is used to minimize the force applied to the sample, either by moving the sample (sample scanner) or the tip (head scanner) up and down via a piezo-driven servo system. In this way, the scanning force is maintained constant regardless of the sample topography. The sample topography (height image) is then reconstructed from the piezo movement. At the same time, the cantilever deflection provides information about sample height changes and can be represented as the error or deflection image.

Basic components of an AFM. (a) A laser is used to read on a diode the deflection of a very soft lever equipped with a sharp tip. The sample is moved in the three directions of space with a sub-nm accuracy using piezo-electric ceramics. (b, c) Two main AFM modes are used to image biological samples. (b) Contact mode. The tip is in continuous contact with the sample. The deflection of the lever is kept constant (resulting in a constant force applied to the sample) and the z-motion of the piezo is the read-out of the surface topology. (c) Intermittent contact mode. A secondary piezo is used to vibrate the lever close to the surface, resulting in soft touches. This mode present the advantage of being more gentle with soft objects and to reduce the lateral dragging force created by the raster scans of the surface since the contact is largely reduced compared to contact mode
In intermittent contact mode32,63 (also known as “tapping” mode), the AFM cantilever is oscillated close to its resonance frequency while scanning the sample (Fig. 1c). Upon approaching the sample surface, tip oscillation is progressively dampened and the cantilever amplitude decreases. Again using a feedback system, the full cantilever amplitude can be continuously restored by adjusting the piezo position, hence generating a topographic image of the sample. Because the tip only contacts the sample briefly at the end of its downward move,74 lateral friction forces are reduced in tapping mode, which facilitates imaging weakly immobilized samples. In addition, specific sample surface properties, such as rigidity or friction may shift the cantilevers oscillation frequency (phase shift), an effect used in phase mode imaging. Phase images may be used to visualize stiffness variations on the surface of living cells.57 While contact mode is most frequently employed for scanning fixed or living cells, tapping mode may prove useful when scanning flexible cellular structures, such as filipodia or microvilli or when scanning specific membrane subdomains.43,44,56,63 Nevertheless, the effective forces exerted on the sample during cantilever oscillation remain to be established. In any case, when scanning fragile biological samples, it is crucial to maintain low scanning forces to avoid sample distortion. Scanning in liquid not only provides a physiologically-relevant environment for cells, but also eliminates strong capillary forces between tip and sample occurring in air.78 In liquid scanning forces can be adjusted to 50–100 pN (10−12 N), which is usually sufficient to prevent irreversible deformation of biological samples,54,68 including living cells.46
AFM Force Spectroscopy
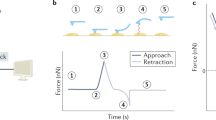
In force spectroscopy mode, the AFM cantilever is stationary in xy-direction but ramped at a given position in z-direction. Measuring cantilever deflection as a function of piezo position compiles force-extension-curves (Fig. 2). For small deflections the AFM cantilever approximately constitutes a Hookean spring and the cantilever deflection is therefore linearly related to the acting force causing the deflection. If the cantilever spring constant is known (for instance through calibration against a reference cantilever or by deduction from its thermal oscillation spectrum12), the cantilever deflection can be converted into a corresponding force. Fragile biological samples require soft cantilevers. Suitable commercially available cantilever feature spring constants as low as 6 pN/nm (Biolever, Olympus) or 10 pN/nm (MLCT, Veeco).

Force mode of the AFM. The schematics are depicting the z-motion of the piezo that leads to a deflection vs. piezo movement curve, a so-called force curve (on the right). (a) The lever is far from the sample and is not deflected. (b) The tip is brought to (“pressed”) and touches the sample, the lever is deflected and follows the movement of the piezo. (c) When the piezo retract (“pulled”), the cantilever can be subjected to an hysteresis due to the interaction with the sample. The height of the force jump when the tip jumps off the contact is called the adhesion force. (d) Similar to (a), but at the end of the cycle. The insert describes how to convert the deflection into the exerted force, by using the system sensitivity and the thermal noise determination of the spring constant of the lever
Imaging Cells and Cellular Compartments
AFM is becoming increasingly popular among cell biologists to investigate the morphology of adherent cells at high resolution. Pioneering studies demonstrated the general possibility to image cell surfaces by AFM with high spatial resolution.31 An important advantage of AFM over other high-resolution imaging techniques, such as electron microscopy or tomography, is the possibility to maintain the sample under physiological conditions. In addition, preparing cells for AFM imaging usually does not require cell labelling or other preparation steps leading to potential structural artefacts, such as detergent extraction, sample drying, coating, or cutting. Importantly, because cells can be maintained in cell culture medium, living cells can be imaged for extended periods of time. Thus, AFM is the only label-free method for high-resolution imaging of living cells and has been successfully employed to image a wide variety of primary cells and cell lines (see Refs. 25,44–46,60 and references therein).
Fixed or Living Adherent Cells
AFM cell imaging requires that cells are firmly attached to a support so that they are not displaced by the probe during scanning. Well-adhering cells, such as fibroblasts or epithelial cells, can be directly imaged on coated or uncoated glass supports. Weakly or non-adherent cells may be immobilized on poly-l-lysine-coated supports, while suspension cells, including yeast and bacteria may be immobilized using microwells or porous membranes.2,17,18 In this case, however, only the top surface of the cell is accessible for imaging since the walls of the microwell supports block access of the AFM tip to lateral cell areas.
Initial concerns that the passing AFM tip would disrupt the cell membrane or cause cell death during scanning proved unfounded. If the scanning parameters are adjusted carefully (low scanning force and speed) and suitable AFM tips are used (non-oxidation sharpened tips), cells can be scanned continuously for hours and remain viable for days after scanning. Experiments using dye-loaded cells demonstrated that under optimized scanning conditions tip penetration of the membrane occurs neither in contact nor tapping mode.36,67 It has been postulated that a repulsive net force between the AFM tip and the cellular glycocalix33 may prevent cell damage despite the direct tip/cell interaction.
Although membraneous material originating from the scanned cells has been observed to accumulate at the AFM tip after prolonged scanning, this does not appear to adversely affect cell behaviour.67 However, the accumulation of cellular debris on the AFM tip will gradually decrease the lateral resolution of the AFM scan.
For live-cell imaging, the softest available cantilevers are commonly used. However, even these soft cantilevers are still at least 10 times stiffer than the plasma membrane of epithelial cells.36 When scanning living cells it is therefore important to keep scanning forces minimal (≤50 pN) to prevent tip penetration of the membrane and the dislodging or dissecting of fine cellular surface structures, such as microvilli and filipodia. In addition, high scanning forces frequently induce cellular retraction out of the scanning path of the AFM tip. Despite the direct contact of the AFM tip, living cells can be imaged for several hours without noticeable changes in cell morphologies.36 Because of the influence of temperature on cell migration, morphology and adhesion, meaningful images of living cells should be acquired at 37 °C using a temperature-controlled sample holder.
Nevertheless, in unfixed cells highly flexible structures, such as microvilli, can frequently not be resolved in contact mode36 because they are moved along the scanning direction by the tip. Chemical fixation with glutaraldehyde, paraformaldehyde or UV-activatable cross linkers can render such flexible structures more rigid, leading to the resolution of small features that remain stable when the tip passes.60
In cell imaging unsharpened AFM tips (radius ∼20–50 nm) frequently yield better images than ultra-sharp tips (radii <10 nm). The reduction in resolution (limited to ∼20–100 nm) is often offset by better control over the tip-cell interaction, improved cell contour tracking, and reduced cell scaring and tip contamination.56 Generally, low scanning speeds (tip velocity as low as 5 μm/s) will improve sample tracking by the tip and consequently provide higher-resolution AFM images of living cells.
The structural detail revealed in AFM images of living cells is also influenced by the scanning force applied. At low scanning forces, the AFM tip will only minimally deform the cell membrane and the scan therefore provides an accurate representation of cell contours. With increasing scan force, the AFM tip will deform the compliant membrane and force it against the much stiffer underlying cortical actin cytoskeleton. Higher scanning force AFM images of living cells are therefore visualization of variations in “surface stiffness” correlating with submembraneous cytoskeletal structures.65,66 The high pliability of the plasma membrane can be exploited to image the underlying cortical cytoskeleton in living cells with high resolution36 when the scanning force is increased. As chemical fixation strongly increases membrane stiffness, the membrane-underlying cytoskeleton in fixed cells contributes less to image contrast generation (Fig. 3).

Imaging cell surface. Living REF-52 fibroblasts imaged in AFM contact mode (a, deflection and b, height image). Stiff actin-containing cytoskeletal fibers become apparent underneath the comparatively soft cell membrane when scanning with slightly increased force. In glutaraldehyde-fixed cells (c, deflection and d, height), cytoskeletal fibers are preserved (e, phase contrast image and f, double fluorescence staining for actin filaments and the focal adhesion marker paxillin) but not revealed in the AFM images because of increased cell membrane stiffness. The full range of the height scale corresponds to 2.4 μm (b) and 4 μm in (d). Each image is 80 × 80 μm2
Observing Dynamic Cell Processes
An exciting application of AFM is the possibility to directly observe morphological changes associated with the dynamic restructuring of the plasma membrane and the underlying cytoskeleton. For example, live-cell time-lapse AFM imaging revealed highly dynamic membrane processes involved in neurite extension.58 In addition, dynamic interactions of cellular protrusions with compartments of the extracellular matrix can be followed with a resolution approaching molecular dimensions.25 Furthermore, it has been possible to monitor granula fusion during platelet activation,27,28 or the formation of membrane pits after receptor-stimulated secretion.72
One of the limitations of time-lapse AFM experiments for studying dynamic processes at the cell membrane are comparatively long image acquisition times of usually around several minutes. If restructuring events of the cell membrane occur at a similar time scale, the image will not provide a snap-shot of the cell but will be blurred comparable to a long-exposure photography shot. Although high-speed (intermittent contact mode) AFM imaging techniques are beginning to become available,59 so far they have not been employed for cell imaging. Scanning of tall cells requires AFMs featuring larger z-ranges (up to 15–20 μm) to be able to track from low membrane structures (filopodia, lamellipodia) to high areas on the cell (for instance above the nucleus). These relatively large height variations along the scan pass require large compensatory z-piezo movements of the cantilever which can exceed the response capabilities of the feedback loop. However, frame acquisition times can be reduced by limiting the scan frame to a cellular subsection and by decreasing the number of pixels per scan line. Higher scan speeds and consequently shorter frame acquisition times (∼60 s) can also be achieved when relatively flat regions of cells are scanned, such as lamellipodial extensions of well-spread cells or the apical region of a cell monolayer.
AFM and Light Microscopy
The x,y-scan ranges of commercially available AFMs usually range between ∼30 and 150 μm. While the resulting maximum scan frames may contain one or several individual cells, they are frequently too small to provide overview scans of larger groups or colonies of cells. It is therefore useful to combine AFM with optical microscopy by mounting the AFM on top of an inverted light microscope. In this way, optical and AFM images can be directly correlated in the same experimental setup. Potentially interesting cellular regions are first identified by light microscopy. The AFM cantilever is then repositioned and the regions are imaged by AFM at increased resolution.
The combination of AFM with fluorescent microscopy techniques is proving a valuable tool in the investigation of selected cellular compartments.23,29,76 Using immunofluorescence labeling or expression of recombinant fluorescent fusion proteins, interesting regions are first identified on the cell scale. Labeled subregions are then imaged at superior resolution by AFM (Fig. 4). Because the AFM can be easily accommodated on a standard inverted optical microscope, common fluorescent microscopy techniques, such as confocal laser scanning microscopy (CLSM)70 (Fig. 5) or total internal reflection fluorescence (TIRF) microscopy and even single-molecule fluorescent techniques, such as fluorescence correlation spectroscopy (FCS),15 can be readily incorporated into the setup.

Imaging cell substructures. REF-52 cells stably expressing YFP-paxillin as a focal adhesion marker protein were de-roofed by a sonication procedure and fixed. Subsequently, focal adhesion complexes were identified by detecting the YFP signal (b) and staining for actin filaments using TRITC-phalloidin (a). The contact mode AFM images (deflection, c and height, d) reveal ultrastructural detail not available from the corresponding optical images, such as actin filament organization. Insert: height profile corresponding to a cross section taken along the indicated line. The cross section demonstrates that focal adhesions have a corrugated surface formed by the parallel array of actin filaments. The full range of the height scale in (d) corresponds to 500 nm. Each image is 8 × 6 μm2 23

Coupling AFM and laser scanning confocal microscopy. Combined imaging of WM39 melanoma cells. Imaging of melanoma cells (isolated from a vertical growth phase melanoma) using AFM leads to the visualisation of flexible structures on the surface of the melanoma cells (a). Confocal imaging of the same cell stained with FITC-phalloidin shows that these ridges are based on sub-membranous filamentous actin structures (b). An overlay is presented in (c). In addition by comparing TRITC-phalloidin labelled cells (d) with cells in which the beta1 integrin has been labelled (e) shows that the beta1 integrin is localized to these surface ridges. Overlay presented in (f). Adapted from Ref. 60, courtesy of Dr. K. Poole
The integration of fluorescent microscopy is not limited to the identification of specific cellular structures or organelles. By correlating fluorescence intensities with the height information contained in the AFM topographs, information about differential vertical localization of fluorophores can be obtained with higher spatial resolution than optical sectioning techniques provide.23 Furthermore, in living cells expressing fluorescently-labeled proteins, morphological changes can be correlated with changes in the intracellular localization of specific proteins. Fluorescent microscopy can be used as a read-out of mechanical stimulation using the AFM tip.13
Cell Compartments
As a surface-sensitive technique, AFM is most commonly applied for studying the extracellular side of the cell membrane. Nevertheless, AFM has also been successfully employed to visualize intracellular compartments. However, this requires that these structures are made accessible to the AFM tip first. Adherent cells can be opened using hydrodynamic shear, gentle sonification or detergent extraction (references in Franz and Müller23). If the intracellular structures of interest, such as the basal cell membrane43 or matrix adhesion complexes23 remain attached to the support in “de-roofed” cells, they can then be directly scanned. Alternatively, membraneous compartments can be isolated from suspended cells and immobilized on glass or mica support for AFM scanning. Examples include nuclear pore complexes from Xenopus eggs,73 voltage-dependent ion channels from mitochondrial membranes30 or the purification of functional gap junction domains from HeLa cells.55
Measuring Cell Adhesion and Mechanics
Measuring Adhesive Forces
At Single Molecule Scale
AFM force spectroscopy provides powerful means of quantifying the complex inter- and intramolecular interactions that determine the properties of biological molecules and biomaterials.41 In particular, AFM is frequently applied for investigating unbinding forces between single molecular interaction partners using different in vitro systems. Thus, the binding strength of many receptor/ligand pairs could be measured,14,42 including biotin/streptavidin21; antigen/antibody35 and cadherin/cadherin.5,19
Such studies have also been instrumental in validating models describing the physics of bond breakage under an external force, in particular the Kramers/Bell/Evans model20,52). Furthermore, mechanical properties (such as (visco)elasticity) of individual biomolecules11,38,39,50 have been characterized. Importantly, force measurements performed at the single-molecule scale can reveal molecular mechanisms that are leveled out in ensemble experiments. For instance, single-molecule studies have unraveled subtle variations in the unbinding mechanism of some bond types, such as the “catch bond” (bonds that follow different unbinding pathways depending on the level of force they are subjected to48) or shed light on the influence of bond history on unbinding dynamics.49
By feeding experimental results from single-molecule force spectroscopy experiments into sophisticated computer simulations of bond rupture, a quantitative understanding of how molecular composition, architecture, and dynamics govern specific interactions in biology is coming within reach.82,84
At Single Cell Scale
AFM-based force spectroscopy can be used to characterize molecular mechanisms underlying cell adhesion of living cells and has been adapted to study cell–substrate62,83 and cell–cell interactions.7,62 For single-cell force spectroscopy (SCFS), a living cell is gently attached to a tipless, lectin-functionalized cantilever.34 Alternatively, cells can be cultured directly on the cantilever for several hours or days before adhesion measurements.8
The cantilever-attached cell is approached to the substrate (or another cell) until a preset contact force is achieved. After a defined contact time, the cell is removed and a force-distance curve is recorded. These curves provide information about the detachment work and the maximal detachment force (Fig. 6). Although other force spectroscopy techniques, such as the biomembrane force probe52 or optical and magnetic tweezers47 offer equal or superior force resolution, the maximum forces that can be detected or applied are frequently below the detachment forces required to remove well-adhering cells. With AFM-based SCFS, cell detachment forces can be recorded over a range of roughly 4 orders of magnitude, limited at the upper end by the maximum strength the cantilever/cell lectin-coupling can sustain (<100 nN (10−9N)) and at the lower end by the thermal noise of the cantilever (>10 pN).34,75

Quantifying cell adhesion at single cell scale. The schematics depicts a cell/cell adhesion experiment using zebrafish primary mesendodermal cells. The two cells are brought in contact (a, b) and let to interact for a given period of time, and then separated with a controlled speed (c, d). The experimental force curve shows typical large and small jumps preceded (grey arrows, tethers) or not (black arrows) by a plateau region. This has been obtained in calcium containing buffer, with a contact time of 10 s and approaching/retracting speeds of 10 μm/s. The large force jump decreases when calcium is removed from the solution40
Unbinding force curves frequently contain a series of small force jumps corresponding to small discrete rupture events. The smallest detectable rupture events correspond to the unbinding of single receptor-ligand pairs,7,26,75,80 demonstrating the high force-resolution of the setup. To address the contribution of individual cell surface receptors to cell adhesion, SCFS can be performed with single-molecule sensitivity. When contact time (<1 s) and contact force (<500 pN) are minimized, cell-substrate interactions are limited to sporadically occurring discrete adhesion events. By probing these interactions over a range of loading rates (pulling speeds), a dynamic force spectrum (DFS) can be generated from which bond parameters, such as bond strength, bond life time or the width of the potential barrier can be determined according to the Bell/Evans theory of bond rupture under force.6,20 An important requirement for ensuring that predominantly single-molecule interactions is detected are a low binding frequency.24
As substrates pure preparations of one type of extracellular adhesion molecule should be used to which the tested cell type binds using a single class of adhesion receptor. Furthermore, the specificity of the interaction has to be demonstrated by using blocking antibodies or excess free ligand/receptor or by chelating extracellular ions coordinating the adhesive interaction.24,80 Under these conditions, SCFS can be applied to elucidate the contribution of individual cell adhesion molecules on the cell surface, irregardless of the heterogeneity of the cell surface and the complex molecular mechanisms determining overall cell adhesion.
Because of the excellent temporal and spatial control of the piezo-driven cantilever approach mechanism, the cell/substrate contact time can be precisely controlled even for short contact times (<1 s). SCFS is therefore well-suited for studying early events involved in adhesion formation. For instance, the transgression from single to cooperative receptor-mediated adhesion could be monitored during the establishment of integrin-mediated adhesion to collagen type I.75
In contrast to bulk adhesion assays which measure the mean behavior of the entire cell population, with SCFS the adhesive properties of different cell subpopulations can be distinguished.75 A further advantage of SFCS is that the effect of inhibitors or enhancers of adhesion can be monitored directly on an individual cell. For instance, inhibitors can be added and flushed out using perfusion chambers without dislocating the cell from the cantilever. AFM-based force spectroscopy is also especially useful when bulk adhesion assays are not practicable because the number of cells available for adhesion measurements is limited. For example, during embryonal development, the specific adhesive properties of different cell lineages are thought to play an important role in driving tissue formation but collectively probing adhesion of these cells is difficult due to their relatively small number in the developing embryo. Using differentiation markers, a limited number of lineage-specific cells can be isolated at defined developmental stages and homo- or heterotypic adhesion of cell pairs can be measured.40,62,76
SCFS measurements can be performed using standard AFM force spectroscopy setups but should be conducted using a temperature-controlled sample-holder if mammalian cells are tested. In addition, as different cell types tend to deform strongly under load along the pulling direction, complete cell/substrate or cell/cell separation may require an AFM featuring an extended z-range pulling range (>80 μm).61
In conclusion, the ability to measure forces with high resolution over a wide range makes AFM a valuable tool to study cellular adhesion forces across dimensions from the single-molecule level to that of the entire cell in the same experimental setup.61 SCFS promises to become a valuable tool for quantitating molecular contributions to adhesion forces in living cells in a wide range of applications.34
Characterizing Cell Mechanics
Eukaryotic cells are mechanically complex, heterogenous structures exhibiting both elastic and viscous characteristics. The cell membrane, intracellular organelles, structural proteins forming the cytoskeleton as well as a viscous cytosol all contribute to the overall mechanical properties of the cell. Furthermore, the mechanical properties of living cells are not static but may undergo drastic spatial and temporal changes, for instance during cytokinesis, differentiation, extravasion or migration. Characterizing changes in cell mechanics during these processes provides important insight into the underlying mechanisms regulating cell morphology and function.
Operating an AFM in force spectroscopy mode and using the AFM tip as a microindentor can be employed to probe cell mechanics on defined points of the cell surface. Depending on the size and the shape of the probe, cell mechanics can be tested on a local or global scale. Usually, either unsharpened AFM tips (tip radius >20 nm) or cantilevers carrying polymeric or glass beads are used as microindentors (Fig. 7a). Beads can be of micrometer size (i.e. be smaller than the cell) or measure several tens of micrometers of diameter (i.e. be larger than the cell). The resolution of such AFM measurements is determined both by the probe size and the range of elastic deformation along the membrane. Beads provide a defined contact geometry but their comparatively large diameter limits the spatial resolution of the elasticity measurements. Instead, using the unmodified AFM tip as a conical indentor, measurements can be performed with a higher local resolution of ∼50–500 nm.64–66

Quantifying cell mechanical properties. The use of different indenter shapes and sizes allow to probe different regimes/scales of cell mechanics (see text). (a) Using the blunt pyramidal tip of the lever implies to use the Hertz model for a cone. It gives a local description of the cell elasticity and allow its mapping. (b, c) Using a glued bead implies to use the Hertz model for a sphere and the different diameters allow to probe either a larger local zone as in (a), or the whole cell elasticity. (d) Typical force curve recorded while pressing on a SAOS cell in the situation (a) fitted with the relevant Hertz model. This allows to extract the local Young modulus of the cell, here E ∼ 1220 Pa (courtesy A. Taubenberger)
From force-distance curves recorded during indentation, information about the local elastic or Young’s modulus (E) can be extracted, most commonly by applying the Sneddon or Hertz model for elastic indentation.64 However, using such a model requires knowing the exact tip shape, Poisson ratio (directional compressibility under strain) of the cell and indentation depth. Because a soft cell will start deforming before a deflection of the stiffer cantilever can be detected, determining the exact cell/probe contact point is usually difficult and results in erroneous indentation depths and consequently, in E moduli. Further errors may result from setting the Poisson ratio of living cells to 0.5 (i.e. that of an entirely incompressible body), and by deviations of the tip geometry. Unsurprisingly, E moduli measured for different cell types vary greatly (from 1 kPa (103 Pa) to several 100 kPa). It has been discussed whether absolute values for E moduli obtained by AFM indentation measurements provide meaningful information, given the arguments mentioned above and the cells’ structurally heterogeneity.1 In contrast, relative variations in elasticity across an individual cell or cells in a tissue may yield relevant information. FIEL (force integration to equal limits) provides such relative elasticity data by determining the indentation work required to obtain a predefined indentation force. Importantly, FIEL does not require knowledge of probe geometry, Poisson ratio or the precise position of tip/cell contact.
Because of viscous contributions to cell mechanics, the time scale of indentation measurements has to considered. Generally, short (subsecond) indentation times will probe the visco-elastic response of the cell, while longer indentation times allow viscous relaxation, reducing the cellular response predominantly to its elastic component. Transitions between different elasticity regimes can be analyzed in more detail by oscillating the AFM tip over a range of frequencies.3,71 Plastic deformation of cells has also been evaluated in some cases.81 Since temperature strongly affects viscous relaxation times, performing indentation assays at physiological temperature is critical.1
When force curves are recorded while the tip is raster-scanned over the cell in x,y-direction, 2-dimensional elasticity maps can be obtained.64 Comparing the resulting “force-map” with the corresponding height image allows correlating elastic properties with structural features. “Force-maps” usually feature arrays of 32 × 32 or 64 × 64 force curves. This typically require acquisition times of between 20 min and 2 h, which complicates force mapping of highly-dynamic processes, such as cell division. In these cases, elasticity measurements can be accelerated by restricting the data acquisition to discrete points or lines.51
Outlook
AFM is unique as an imaging tool for cell biology because it provides high-resolution topographic images of cellular surfaces and compartments under physiological conditions. However, native cellular membranes are heterogeneous and elastic, causing image degradation due to the dislocation of flexible structures (e.g. villis, ridges) during scanning and blurring by tip convolution. Nevertheless, under optimized scanning conditions, resolutions down to 10 nm have been reported on cell surfaces.46 In such high-resolution scans, however, unequivocally assigning structural features to specific membrane components is often problematic since the topographic image contains no information about the chemical make-up of the sample. Hence, molecular or even submolecular resolution of specific membrane components is usually only obtained on reconstituted membranes containing homogenous distributions of purified transmembrane proteins. A current challenge in AFM cell imaging is therefore the integration of high-resolution fluorescence microscopy techniques, such as photo-activated localization microscopy (PALM),9,37 stimulated emission depletion (STED) microscopy79 or structured illumination. These powerful new observation methods allow localizing fluorophores well below the limits of optical diffraction. In combination with AFM, such light microscopy implementations could be used to correlate structural features with the localization of specific cellular components. Likewise, single-molecule fluorescence techniques are beginning to be integrated into AFM setups. For instance, FCS and AFM have already been combined to study single-molecule diffusion processes in artificial lipid bilayers.15 Similar techniques could be used to follow receptor aggregation within the plasma membrane together with nm-changes in membrane topography.
AFM-based SCFS provides a powerful setup to study cell adhesion from the single-molecule level to overall cell adhesion in the same experimental setup. However, so far SCFS has been mainly used to study cell adhesion to homogeneously decorated substrates. In contrast, the native cellular environment is formed from a complex, flexible scaffold of highly-structured macromolecules. Transferring experimental results from SCFS adhesion measurements onto physiologically-relevant systems will benefit from using flexible and structured cell adhesion substrates. Through recent advances in surface functionalization techniques, such as microcontact printing or laserlithography have reproducible micro- and nano-structured supports are now available.69 Determining the impact of the structure of the presented substrate on cell adhesion may lead to a better understanding of the molecular and geometric cues that drive adhesion formation. Furthermore, performing SCFS simultaneously with TIRFM or Interference Microscopy (RICM) may allow correlating the breaking of adhesive structures (such as focal adhesion sites) with the rupture force information contained in AFM force curves. However, due to the stability of the AFM/optical set-ups, contact times are limited to a few minutes, limiting the investigation to early adhesion events.
In a single technique, AFM collects two biologically crucial types of data: Cells shape from the micrometer down to the nanometer scale, and cell adhesion, from single-molecule-mediated adhesion events to dynamically arranged adhesive super-structures. This combination presents an unique opportunity to link cellular structure and function in adhesion studies.
References
E. A-Hassan, W. F. Heinz, M. D. Antonik, N. P. D’Costa, S. Nageswaran, C. A. Schoenenberger, and J. H. Hoh. Relative microelastic mapping of living cells by atomic force microscopy. Biophys J, 74(3):1564–1578, Mar 1998.
Fraçois Ahimou, Ahmed Touhami, and Yves F Dufrêne. Real-time imaging of the surface topography of living yeast cells by atomic force microscopy. Yeast, 20(1):25–30, Jan 2003.
Jordi Alcaraz, Lara Buscemi, Mireia Grabulosa, Xavier Trepat, Ben Fabry, Ramon Farré, and Daniel Navajas. Microrheology of human lung epithelial cells measured by atomic force microscopy. Biophys J, 84(3):2071–2079, Mar 2003.
S. Alexander, L. Hellemans, O. Marti, J. Schneir, V. Elings, P. K. Hansma, Matt Longmire, and John Gurley. An atomic-resolution atomic-force microscope implemented using an optical lever. J. Appl. Phys., 65:164, 1989.
W. Baumgartner, P. Hinterdorfer, W. Ness, A. Raab, D. Vestweber, H. Schindler, and D. Drenckhahn. Cadherin interaction probed by atomic force microscopy. Proc Natl Acad Sci U S A, 97(8):4005–4010, Apr 2000.
G. I. Bell. Models for the specific adhesion of cells to cells. Science, 200(4342):618–627, May 1978.
M. Benoit, D. Gabriel, G. Gerisch, and H. E. Gaub. Discrete interactions in cell adhesion measured by single-molecule force spectroscopy. Nat Cell Biol, 2(6):313–317, Jun 2000.
Martin Benoit and Hermann E Gaub. Measuring cell adhesion forces with the atomic force microscope at the molecular level. Cells Tissues Organs, 172(3):174–189, 2002.
Eric Betzig, George H Patterson, Rachid Sougrat, O. Wolf Lindwasser, Scott Olenych, Juan S Bonifacino, Michael W Davidson, Jennifer Lippincott-Schwartz, and Harald F Hess. Imaging intracellular fluorescent proteins at nanometer resolution. Science, 313(5793):1642–1645, Sep 2006.
Binnig, Quate, and Gerber. Atomic force microscope. Phys. Rev. Lett. 56(9):930–933, 1986.
Christian A Bippes, Andrew D L Humphris, Martin Stark, Daniel J Müller, and Harald Janovjak Direct measurement of single-molecule visco-elasticity in atomic force microscope force-extension experiments. Eur Biophys J 35(3), 287–292, 2006.
H.J. Butt, M. Jaschke Calculation of thermal noise in atomic force microscopy. Nanotechnology, 6:1–7, 1995.
G. T. Charras, P. P. Lehenkari, and M. A. Horton. Atomic force microscopy can be used to mechanically stimulate osteoblasts and evaluate cellular strain distributions. Ultramicroscopy, 86(1-2):85–95, Jan 2001.
Aileen Chen and Vincent T Moy. Single-molecule force measurements.Methods Cell Biol, 68:301–309, 2002.
Salvatore Chiantia, Jonas Ries, Nicoletta Kahya, and Petra Schwille. Combined afm and two-focus sfcs study of raft-exhibiting model membranes. Chemphyschem, 7(11):2409–2418, Nov 2006.
B. Drake, C.B. Prater, A.L. Weisenhorn, S.A. Gould, T.R. Albrecht, C.F. Quate, D.S. Cannell, H.G. Hansma, and P.K. Hansma. Imaging crystals, polymers, and processes in water with the atomic force microscope.Science, 243(4898):1586–1589, Mar 1989.
Y. F. Dufrêne. Surface morphology and mechanical properties of mdck monolayers by atomic force microscopy. J Cell Sci, 107 (Pt 5):1105–1114, May 2008.
Y. F. Dufrêne, C. J. Boonaert, P. A. Gerin, M. Asther, and P. G. Rouxhet. Direct probing of the surface ultrastructure and molecular interactions of dormant and germinating spores of phanerochaete chrysosporium. J Bacteriol, 181(17):5350–5354, Sep 1999.
O. Duroure, A. Buguin, H. Feracci, and P. Silberzan. Homophilic interactions between cadherin fragments at the single molecule level: an afm study. Langmuir, 22(10):4680–4684, May 2006.
E. Evans and K. Ritchie Dynamic strength of molecular adhesion bonds. Biophys J 72(4), 1541–1555, 1997.
E. L. Florin, V. T. Moy, and H. E. Gaub. Adhesion forces between individual ligand-receptor pairs. Science, 264(5157):415–417, Apr 1994.
Dimitrios Fotiadis, Simon Scheuring, Shirley A Müller, Andreas Engel, and Daniel J Müller. Imaging and manipulation of biological structures with the afm. Micron, 33(4):385–397, 2002.
Clemens M Franz and Daniel J Müller. Analyzing focal adhesion structure by atomic force microscopy. J Cell Sci, 118(Pt 22):5315–5323, Nov 2005.
Clemens M Franz, Anna Taubenberger, Pierre-Henri Puech, and Daniel J Muller. Studying integrin-mediated cell adhesion at the single-molecule level using afm force spectroscopy. Sci STKE, 2007(406):pl5, Oct 2007.
Jens Friedrichs, Anna Taubenberger, Clemens M Franz, and Daniel J Muller. Cellular remodelling of individual collagen fibrils visualized by time-lapse afm. J Mol Biol, 372(3), 594–607 2007.
Jens Friedrichs, Juha M Torkko, Jonne Helenius, Terhi P Teráváinen, Joachim Füllekrug, Daniel J Muller, Kai Simons, and Aki Manninen. Contributions of galectin-3 and −9 to epithelial cell adhesion analyzed by single cell force spectroscopy. J Biol Chem, 282(40):29375–29383, Oct 2007.
M. Fritz, M. Radmacher, and H. E. Gaub. In vitro activation of human platelets triggered and probed by atomic force microscopy. Exp Cell Res, 205(1):187–190, Mar 1993.
M. Fritz, M. Radmacher, and H. E. Gaub. Granula motion and membrane spreading during activation of human platelets imaged by atomic force microscopy. Biophys J, 66(5):1328–1334, May 1994.
G. Fuhr, E. Richter, H. Zimmermann, H. Hitzler, H. Niehus, and R. Hagedorn. Cell traces – footprints of individual cells during locomotion and adhesion. Biol. Chem. 379(8–9):1161–1173, 1998.
Gonçalves, R., N. Buzhysnskyy, and S. Scheuring. Mini review on the structure and supramolecular assembly of vdac. J. Bioenerg. Biomembr. 40(3):133–138, 2008.
W. Häberle, JK Hörber, F Ohnesorge, DP Smith, and G Binnig. In situ investigations of single living cells infected by viruses. Ultramicroscopy, 42-44:1161, 1992.
H. G. Hansma and J. H. Hoh. Biomolecular imaging with the atomic force microscope.Annu Rev Biophys Biomol Struct, 23:115–139, 1994.
P. G. Haydon, R. Lartius, V. Parpura, and S. P. Marchese-Ragona. Membrane deformation of living glial cells using atomic force microscopy.J Microsc, 182(Pt 2):114–120, May 1996.
Jonne Helenius, Carl-Philipp Heisenberg, Hermann E Gaub, and Daniel J Muller. Single-cell force spectroscopy. J Cell Sci, 121(Pt 11):1785–1791, Jun 2008.
P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher, and H. Schindler. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc Natl Acad Sci U S A, 93(8):3477–3481, Apr 1996.
J. H. Hoh and C. A. Schoenenberger. Surface morphology and mechanical properties of mdck monolayers by atomic force microscopy. J Cell Sci, 107 (Pt 5):1105–1114, May 1994.
Marko Kaksonen and David G Drubin. Palm reading: Seeing the future of cell biology at higher resolution. Dev Cell, 11(4):438–439, Oct 2006.
Masaru Kawakami, Katherine Byrne, David J Brockwell, Sheena E Radford, and D. Alastair Smith. Viscoelastic study of the mechanical unfolding of a protein by afm. Biophys J, 91(2):L16–L18, Jul 2006.
Masaru Kawakami, Katherine Byrne, Bhavin Khatri, Tom C B McLeish, Sheena E Radford, and D. Alastair Smith. Viscoelastic properties of single polysaccharide molecules determined by analysis of thermally driven oscillations of an atomic force microscope cantilever. Langmuir, 20(21):9299–9303, Oct 2004.
M. Krieg, Y. Arboleda-Estudillo, P-H. Puech, J. Káfer, F. Graner, D. J. Müller, and C-P. Heisenberg. Tensile forces govern germ-layer organization in zebrafish. Nat Cell Biol, 10(4):429–436, Apr 2008.
D. Leckband (2004) Force as a probe of membrane protein structure and function. Curr Opin Struct Biol 11(4), 433–439, 2001.
Deborah Leckband. Nanomechanics of adhesion proteins. Curr Opin Struct Biol, 14(5):524–530, Oct 2004.
C. Legrimellec, E. Lesniewska, M. C. Giocondi, E. Finot, V. Vié, and J. P. Goudonnet.Imaging of the surface of living cells by low-force contact-mode atomic force microscopy. Biophys J, 75(2):695–703, Aug 1995.
C. Legrimellec, E. Lesniewska, M. C. Giocondi, E. Finot, and J. P. Goudonnet. Simultaneous imaging of the surface and the submembraneous cytoskeleton in living cells by tapping mode atomic force microscopy. C R Acad Sci III, 320(8):637–643, Aug 1997.
C. Legrimellec, E. Lesniewska, M. C. Giocondi, E. Finot, V. Vié, and J. P. Goudonnet.Imaging of the surface of living cells by low-force contact-mode atomic force microscopy. Biophys J, 75(2):695–703, Aug 1998.
Eric Lesniewska, Pierre Emmanuel Milhiet, Marie-Cécile Giocondi, and Christian Legrimellec. Atomic force microscope imaging of cells and membranes.Methods Cell Biol, 68:51–65, 2002.
Rustem I Litvinov, Gaston Vilaire, Henry Shuman, Joel S Bennett, and John W Weisel. Quantitative analysis of platelet alpha v beta 3 binding to osteopontin using laser tweezers. J Biol Chem 278(51):51285–51290, 2003.
Bryan T Marshall, Mian Long, James W Piper, Tadayuki Yago, Rodger P McEver, and Cheng Zhu. Direct observation of catch bonds involving cell-adhesion molecules. Nature, 423(6936):190–193, May 2003.
Bryan T Marshall, Krishna K Sarangapani, Jizhong Lou, Rodger P McEver, and Cheng Zhu. Force history dependence of receptor-ligand dissociation. Biophys J, 88(2):1458–1466, Feb 2005.
Bryan T Marshall, Krishna K Sarangapani, Jianhua Wu, Michael B Lawrence, Rodger P McEver, and Cheng Zhu (2006). Measuring molecular elasticity by atomic force microscope cantilever fluctuations. Biophys J 90(2), 681–692.
R. Matzke, K. Jacobson, and M. Radmacher. Direct, high-resolution measurement of furrow stiffening during division of adherent cells. Nat Cell Biol, 3(6):607–610, Jun 2001.
R. Merkel, P. Nassoy, A. Leung, K. Ritchie, and E. Evans. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature, 397(6714):50–53, Jan 1999.
Gerhard Meyer and Nabil M. Amer. Novel optical approach to atomic force microscopy. Novel optical approach to atomic force microscopy, 53:1045, 1988.
D. J. Müller, D. Fotiadis, S. Scheuring, S. A. Müller, and A. Engel. Electrostatically balanced subnanometer imaging of biological specimens by atomic force microscope. Biophys J, 76(2):1101–1111, Feb 1999.
Daniel J Müller, Galen M Hand, Andreas Engel, and Gina E Sosinsky. Conformational changes in surface structures of isolated connexin 26 gap junctions. EMBO J, 21(14):3598–3607, Jul 2002.
E. Nagao and J.A. Dvorak. An integrated approach to the study of living cells by atomic force microscopy. J Microsc, 191(Pt 1):8–19, Jul 1998.
E. Nagao and J.A. Dvorak. Phase imaging by atomic force microscopy: analysis of living homoiothermic vertebrate cells. Biophys J, 76(6):3289–3297, Jun 1999.
V. Parpura, P.G. Haydon, and E. Henderson Three-dimensional imaging of living neurons and glia with the atomic force microscope. J Cell Sci 104(2), 427–432 1993.
LM Picco and L Bozec. Analyzing focal adhesion structure by atomic force microscopy. J Cell Sci, 118(Pt 22):5315–5323, Nov 2007.
K. Poole and D. Müller. Flexible, actin-based ridges colocalise with the beta1 integrin on the surface of melanoma cells. Br J Cancer, 92(8):1499–1505, Apr 2005.
Pierre-Henri Puech, Kate Poole, Detlef Knebel, and Daniel J Muller. A new technical approach to quantify cell-cell adhesion forces by afm.Ultramicroscopy, 106(8-9):637–644, 2006.
Pierre-Henri Puech, Anna Taubenberger, Florian Ulrich, Michael Krieg, Daniel J Muller, and Carl-Philipp Heisenberg.Measuring cell adhesion forces of primary gastrulating cells from zebrafish using atomic force microscopy. J Cell Sci, 118(18), 4199–4206, 2005.
C. A. Putman, K. O. van der Werf, B.G. de Grooth, N.F. van Hulst, and J.Greve. Viscoelasticity of living cells allows high resolution imaging by tapping mode atomic force microscopy. Biophys J, 67(4):1749–1753, Oct 1994.
Manfred Radmacher. Measuring the elastic properties of living cells by the atomic force microscope. Methods Cell Biol, 68:67–90, 2002.
C. Rotsch, F. Braet, E. Wisse, and M. Radmacher. Afm imaging and elasticity measurements on living rat liver macrophages.Cell Biol Int, 21(11):685–696, Nov 1997.
C. Rotsch and M. Radmacher. Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J, 78(1):520–535, Jan 2000.
S. S. Schaus and E. R. Henderson. Cell viability and probe-cell membrane interactions of xr1 glial cells imaged by atomic force microscopy. Biophys J, 73(3):1205–1214, Sep 1997.
S. Scheuring, P. Ringler, M. Borgnia, H. Stahlberg, D.J. Müller, P. Agre, and A. Engel (1999) High resolution afm topographs of the escherichia coli water channel aquaporin z. EMBO J 18(18):4981–4987.
Selhuber-Unkel, C., M. López-García, H. Kessler, and J. P. Spatz. Cooperativity in adhesion cluster formation during initial cell adhesion. Biophys. J. 95(11):5424–5431, 2008.
Amita Sharma, Kurt I Anderson, and Daniel J Müller Actin microridges characterized by laser scanning confocal and atomic force microscopy. FEBS Lett 579(9), 2001–2008 2005.
S. G. Shroff, D. R. Saner, and R. Lal. Dynamic micromechanical properties of cultured rat atrial myocytes measured by atomic force microscopy. Am J Physiol, 269(1 Pt 1):C286–C292, Jul 1995.
A. Spudich and D. Braunstein. Large secretory structures at the cell surface imaged with scanning force microscopy. Proc Natl Acad Sci U S A, 92(15):6976–6980, Jul 1995.
D. Stoffler, K. N. Goldie, B. Feja, and U. Aebi. Calcium-mediated structural changes of native nuclear pore complexes monitored by time-lapse atomic force microscopy. J Mol Biol, 287(4):741–752, Apr 1999.
J Tamayo and R Garcia. Deformation, contact time, and phase contrast in tapping mode scanning force microscopy. Langmuir, 12:4430–4435, 1996.
Anna Taubenberger, David A Cisneros, Jens Friedrichs, Pierre-Henri Puech, Daniel J Muller, and Clemens M Franz. Revealing early steps of alpha2beta1 integrin-mediated adhesion to collagen type i by using single-cell force spectroscopy. Mol Biol Cell, 18(5):1634–1644, May 2007.
A. Trache and G. A. Meininger. Atomic force multi optical imaging integrated microscope for monitoring molecular dynamics in live cells. J. Biomed. Opt. 10(6):064023, 2005.
Florian Ulrich, Michael Krieg, Eva-Maria Schötz, Vinzenz Link, Irinka Castanon, Viktor Schnabel, Anna Taubenberger, Daniel Mueller, Pierre-Henri Puech, and Carl-Philipp Heisenberg. Wnt11 functions in gastrulation by controlling cell cohesion through rab5c and e-cadherin. Dev Cell, 9(4):555–564, Oct 2005.
A. L. Weisenhorn, B. Drake, C. B. Prater, S. A. Gould, P. K. Hansma, F. Ohnesorge, M. Egger, S. P. Heyn, and H. E. Gaub. Immobilized proteins in buffer imaged at molecular resolution by atomic force microscopy. Biophys J, 58(5):1251–1258, Nov 1990.
Katrin I Willig, Robert R Kellner, Rebecca Medda, Birka Hein, Stefan Jakobs, and Stefan W Hell.Nanoscale resolution in gfp-based microscopy. Nat Methods, 3(9):721–723, Sep 2006.
Ewa P Wojcikiewicz, Midhat H Abdulreda, Xiaohui Zhang, and Vincent T Moy. Force spectroscopy of lfa-1 and its ligands, icam-1 and icam-2. Biomacromolecules, 7(11):3188–3195, Nov 2006.
H. W. Wu, T. Kuhn, and V. T. Moy. Mechanical properties of l929 cells measured by atomic force microscopy: effects of anticytoskeletal drugs and membrane crosslinking. Scanning, 20(5):389–397, Aug 1998.
Tadayuki Yago, Jizhong Lou, Tao Wu, Jun Yang, Jonathan J Miner, Leslie Coburn, Josó A López, Miguel A Cruz, Jing-Fei Dong, Larry V McIntire, Rodger P McEver, and Cheng Zhu. Platelet glycoprotein ibalpha forms catch bonds with human wt vwf but not with type 2b von willebrand disease vwf. J Clin Invest, 118(9):3195–3207, Sep 2008.
Xiaohui Zhang, Ewa Wojcikiewicz, and Vincent T Moy. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys J, 83(4):2270–2279, Oct 2002.
Cheng Zhu, Jizhong Lou, and Rodger P McEver. Catch bonds: physical models, structural bases, biological function and rheological relevance. Biorheology, 42(6):443–462, 2005.
Acknowledgments
The authors want to thank Pr. D.J. Müller for his invaluable help and for introducing them to AFM. A. Taubenberger, M. Krieg, J. Friedrich, Dr. J. Helenius and Dr. K. Poole are thanked for their valuable input in the described experiments. JPK Instruments’s continuous support is greatly acknowledged. This work was supported by the DFG Research Center for Functional Nanostructures, Karlsruhe, Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Franz, C.M., Puech, PH. Atomic Force Microscopy: A Versatile Tool for Studying Cell Morphology, Adhesion and Mechanics. Cel. Mol. Bioeng. 1, 289–300 (2008). https://doi.org/10.1007/s12195-008-0037-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-008-0037-3