
Abstract
Cigarette smoking plays an important role in increased incidence of chronic obstructive pulmonary disease (COPD). The underlying mechanism in which cigarette smoking induced impairment of lung epithelial cells is still unknown. SIRT5 is a nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase, which has been implicated in the regulation of metabolism, stress responses, and aging. Forkhead box O3 (FOXO3) belongs to the O subclass of the forkhead family of transcription factors. It is also involved in protection from oxidative stress by upregulating antioxidants in epithelial cells. Here, we show that cigarette smoke extract (CSE) induces SIRT5 to deacetylate FOXO3 at K271 and K290. Deacetylation of FOXO3 promotes its nuclear localization. Notably, transfection with FOXO3 K271R- or K290R-attenuated CSE-induced apoptosis in SIRT5 knocked down cells, suggesting the protective effects of SIRT5, is mediated by FOXO3. In contrast, CSE stress upregulates SIRT5, which activates FOXO3α leading to rescuing apoptosis. Thus, SIRT5 constitutes a determinant of apoptosis by CSE in lung epithelial cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic obstructive pulmonary disease (COPD), characterized by a progressive and largely irreversible decrement in lung function associated with an abnormal chronic inflammatory response of the lungs to noxious particles, is a lung condition usually caused by smoking (Caramori et al. 2014). Increasing evidence has shown that both endothelial cell and epithelial cell apoptosis is increased in the lung tissue of smokers and patients with emphysematous COPD compared to nonsmokers (Wu et al. 2006). Cigarette smoke extract (CSE) has been considered as an important tool to explore the impact cigarette smoke has on bronchial epithelial cell cultures and facilitates our understanding of crucial intracellular signaling pathways. On one hand, the capacity for CSE to induce a pro-inflammatory response in epithelial cells has been reported (Kode et al. 2006). On the other hand, CSE has been reported to induce apoptosis in primary endothelial cells (Togo et al. 2010) and primary nasal epithelial cells (Lan et al. 2007). However, the molecular basis of smoking-induced cell injury on endothelial cells and epithelial cells remains unclear, and the molecular regulators of CSE-induced cell apoptosis within the lung represent potential determinants of host susceptibility, disease severity, and possible therapeutic targets for intervention.
SIRT5 is an essential member in the sirtuins (SIRTs) family, which are nicotine adenine dinucleotide (NAD+)-dependent enzymes involved in the dynamic regulation of cellular physiology. Like SIRT3 and SIRT4, SIRT5 is found to be located in mitochondria (Verdin et al. 2010). Forkhead box O3 (FOXO3) is the substrate of SIRT3. As a forkhead transcription factor, FOXO3 mediates the expression of multiple genes that govern cellular development, differentiation, survival, apoptosis, stress resistance, metabolism, autophagy, and longevity (vanderHorst et al. 2007; Vogt et al. 2005). Deacetylation of FOXO3 caused by SIRT3 plays an essential role in reducing levels of cellular ROS by upregulating the antioxidant enzymes manganese superoxide dismutase and catalase, which further ameliorates cardiachypertrophy in mice (Sundaresan et al. 2009). However, it is not yet clear whether SIRT5 has a direct role in regulating the physiological role of FOXO3. Here, we provide evidence to suggest a critical role of SIRT5 in the regulation of CSE-induced stress in lung epithelial cells through promoting deacetylation of FOXO3.
Materials and methods
Cell culture
The human type II alveolar epithelial cell line (A549) was purchased from American Type Culture Collection and maintained in continuous culture at 37 °C in a 5 % CO2 atmosphere in RPMI-1640 containing l-glutamine (2 mM), 10 % fetal bovine serum (FBS), penicillin, and streptomycin (1 %).
Preparation of cigarette smoke extract
Preparation of cigarette smoke extract (CSE) was conducted as previously described (Su et al. 1998). Briefly, one commercial cigarette of Marlboro (tar 12 mg and nicotine 0.9 mg) was continuously smoked with a syringe-driven apparatus into 6 ml of culture medium RPMI-1640 at a rate of one cigarette per 4 min. The resulting suspension was adjusted to pH 7.4 and then filtered through a 0.22-μM-pore filter (Millipore, UK). The optical density (OD) was measured at a wavelength that showed maximal absorbance (usually between 270 and 280 nm). CSE was standardized and the concentration was as 100 %. CSE was aliquoted and stored at −80 °C until use.
Assessment of apoptosis and DNA fragmentation by Hoechst staining
After indicated treatment, cells were fixed with 4 % paraformaldehyde for 15 min at room temperature, followed by stained with 0.2 ml of Hoechst 33258 staining solution (Sigma) for 5 min. After washed for three times in PBS, fluorescence signals were examined under a fluorescence microscope.
Immunoblotting
Whole cell lysates were prepared by using cell lysis buffer (Cell signaling, USA) as described previously (Sheng et al. 2012). Protein extracts were subjected to 10 % SDS-PAGE and then transferred to hydrophobic polyvinylidene fluoride (PVDF) membranes. The membranes were blocked in TBST buffer (20 mM Tris–HCl, 150 mM sodium chloride, 0.1 % Tween-20) containing 5 % non-fat dry milk (Santa Cruz, USA) for 1 h at RT, followed by sequentially incubated with primary antibodies and with corresponding secondary antibodies. Immunoreactive bands were visualized by using Immobilon Western Chemiluminescent HRP substrate.
Statistical analysis
Values were expressed as mean ± SD. Differences between groups were examined for statistical significance by one-way analysis of variance (ANOVA) using SPSS software. P < 0.05 was considered as the presence of a statistically significant difference.
Results
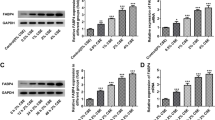
To determine whether members of the SIRT gene family are critical in regulating CSE toxicity, we first performed quantitative real-time PCR analysis for SIRT1–7 in A549 cells after stimulation with 1 % CSE for 24 h. And the results indicated that treatment with 1 % CSE differentially regulated the expression of several SIRT family members. The expression of SIRT5 was particularly noteworthy, as it was greatly increased by CSE treatment (Fig. 1). Then we examined whether the expression of aging-related SIRT5 is modulated by CSE in A549 cells in a dose-dependent manner. When A549 cells were treated with 1 or 2 % CSE for 12 h, the levels of SIRT5 increased in a dose-dependent manner in contrast to the untreated controls (Figs. 2a, b). We then examined whether the acetylation of FOXO3 is also modulated by CSE treatment. CSE treatment caused a significant increase in FOXO3 acetylation (Fig. 3a). However, levels of FOXO3 acetylation were significantly reduced when SIRT5 was overexpressed but increased when SIRT5 was knocked down (Fig. 3b). These results suggest that CSE-induced SIRT5 deacetylates FOXO3.

mRNA levels of SIRT family members in lung epithelial A549 cells after cigarette smoke extract (CSE) treatment. Cells were stimulated with 1 % CSE for 24 h, and mRNA levels of SIRT1-SIRT7 were determined by real-time PCR (*P < 0.01 vs. non-treated control, n = 4–5)

Expression of SIRT5 in A549 cells after cigarette smoke extract (CSE) treatment. A549 cells were stimulated with 1 or 2 % CSE for 24 h. a mRNA levels of SIRT5 were determined by real-time PCR; b Protein levels of SIRT5 were determined by Western blot analysis (*P < 0.01 vs. non-treated control, n = 4)

Effects of cigarette smoke extract (CSE) and SIRT5 on FOXO3 acetylation. a 2 % CSE treatment increases FOXO3 acetylation; b Overexpressing SIRT5 inhibits the effects of CSE on FOXO3 acetylation; c Knockdown of SIRT5 exacerbates the effects of CSE on FOXO3 acetylation
Three lysines in the protein of FOXO3 are involved in acetylation of FOXO3, including K259, K271, and K290. K259 of FOXO3 is acetylated by p300/CBP, whereas K271 and K290 of FOXO3 are targeted by SIRT1 (Calnan and Brunet 2008). To illustrate which lysine residue of FOXO3 is modulated by SIRT5, mimetic lysine-to-arginine (KR) mutants of FOXO3 were generated at K259, K271, and K290. SIRT5 siRNA together with various FOXO3 mutants were cotransfected into A549 cells. And the result indicated that knockdown of SIRT5 significantly increased the acetylation of FOXO3 (Fig. 4a). Knockdown of SIRT1 by transfection with siSIRT1 was used as a positive control. Importantly, there was no increment in FOXO3 acetylation when the KR mutation was introduced at either K271 or K290. In contrast, SIRT5 knockdown enhanced the level of FOXO3 K259R acetylation (Fig. 4b). All the data suggest that K271 and K290 rather than K259 of FOXO3 are targeted by SIRT5.

SIRT5 deacetylates FOXO3 at K271 and K290. a Levels of FOXO3 acetylation in A549 cells were increased in SIRT5 knockdown cells; b Cells were overexpressed with EGFP-tagged FOXO3WT, FOXO3 K259R, FOXO3 K271R, or FOXO3 K290R. Whole-cell lysates (WCL) were immunoprecipitated with anti-EGFP and analyzed by immunoblotting with antibodies against the proteins shown on the right (Ac-K, acetyllysine)
Nuclear localization of FOXO family members plays a critical role in its transcriptional activity. Posttranslational modifications such as deacetylation regulate the nuclear localization of FOXOs. For example, a recent study has reported that SIRT1-dependent deacetylation renders FOXO1 immobile within the nucleus, thereby promoting the transcription of FOXO1-dependent genes (Frescas et al. 2005). Cells were overexpressed with FOXO3 K271/290R or FOXO3 K271/290Q (acetylation mimetic mutant). After 48 h, cells were fractionated into nuclear, mitochondrial, and cytosolic components. We found that the two mutants exhibited distinct distributions in nuclear, mitochondrial, and cytosolic components. As shown in Fig. 5a, b, FOXO3 K271/290R showed a more prominent localization in nucleus. However, FOXO3 K271/290Q was preferentially localized in the cytosol and mitochondria than did FOXO3 K271/290R. These results indicate that deacetylation of FOXO3 promotes its nuclear localization.

Deacetylation of FOXO3 promotes its nuclear localization. a Fractionated cellular extracts (cytosolic, nuclear, and mitochondrial fractions) were prepared from A549 cells that overexpressed FOXO3 K271/290R or FOXO3 K271/290Q and subjected to immunoblotting analysis. EGFP shows both FOXO3 K271/290R and FOXO3 K271/290Q; tubulin was used as the internal control of cytosolic enrichment; lamin A was used as the internal control of nuclear enrichment; VDAC1 was used as the internal control of mitochondrial enrichment
Exposure to CSE meaningfully increased the levels of apoptosis in A549 cells (Fig. 6b). Knockdown of SIRT5 further augmented the levels of apoptosis in A549 cells in response to CSE exposure (Fig.6c). Importantly, transfection with FOXO3 K271R (Fig. 6d) or K290R (Fig. 6e) ameliorated CSE-induced apoptosis in SIRT5 knockdown cells. Quantitative analysis of apoptosis was shown in Fig. 6f. These data suggested that the protective effects of SIRT5 against CSE-induced apoptosis are mediated by FOXO3.

Protective effects of SIRT5 against cigarette smoke extract (CSE)-induced apoptosis are mediated by FOXO3. Cells were treated with 2 % CSE for 24 h after transfection with FOXO3 K271/290R and siSIRT5. Cell apoptosis was determined by Hoechst 33258. a Untreated control; b CSE (2 %, 24 h)-treated group; c CSE (2 %, 24 h) + siSIRT5 group; d CSE (2 %, 24 h) + siSIRT5 + FOXO3 K271R group; e CSE (2 %, 24 h) + siSIRT5+ FOXO3 K290R group; e quantitative analysis (*, #, and $ indicate P < 0.01 vs. previous group)
Discussion
Sirtuins are a class of nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylases found in mammalian cells with various subcellular localizations. In mammals, seven sirtuins were found to modulate distinct metabolic pathways, diverse stress responses, and other biological activities in aging, metabolism, and other diseases (Michishita et al. 2005). SIRT5 localizes in mitochondrial matrix (Schlicker et al. 2008), which exhibits demalonylase and desuccinylase activities in addition to its deacetylase activity (Du et al. 2011). FOXO3 is a member of the FOXO transcription factor family and modulates diverse cellular and physiological processes, including metabolism, development, tumor suppression, and longevity (Calnan and Brunet 2008). Increasing evidence has shown that deacetylation of FOXO3 reduces levels of cellular ROS by upregulating the antioxidant enzymes manganese superoxide dismutase and catalase, which further ameliorates cardiac hypertrophy in mice (Sundaresan et al. 2009).
FOXO3 is a target of SIRT family. It has been shown that FOXO3 can be deacetylated by SIRT1 to induce cell cycle arrest and elicit cellular resistance to oxidative stress. Notably, the SIRT1-mediated deacetylation of FOXO3 inhibits FOXO3-mediated cell death (Brunet et al. 2004). Moreover, FOXO3 can be deacetylated by SIRT2 under oxidative stress to elevate the expression of p27, MnSOD, and Bim, which suppresses cell death (Wang et al. 2007). It was also reported that SIRT3 upregulate MnSOD and catalase to detoxify ROS which further blocks cardiac hypertrophy in mice through deacetylating FOXO3 (Jacobs, et al. 2008). To date, little is known about SIRT5 function in the regulation of FOXO3 and cell death. In this study, we found that SIRT3 was induced by CSE to deacetylate FOXO3 at K271 and K290, suggesting an important function of SIRT5 in regulating FOXO3 signaling.
With regard to the protective effects of SIRT5 against COPD, little information has been provided in previous studies. But increasing evidence has shown that SIRT1 and SIRT6 are considered to have protective effects against COPD. In the lungs, SIRT1 plays pivotal roles in inhibiting autophagy, cellular senescence, fibrosis, and inflammation by deacetylation of target proteins using NAD (+) as co-substrate and is therefore linked to the redox state. In addition to SIRT1, SIRT6 have also been shown to improve or slow down COPD by inhibiting cellular senescence and fibrosis (Chun. 2015). Similar with our current findings, a recent study showed that SIRT1 protects against CSE-induced oxidative stress mediated by FOXO3 but not Nrf2 (Yao et al. 2014).
References
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27:2276–2288
Caramori G, Adcock IM, Di Stefano A, Chung KF (2014) Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis 9:397–412
Chun P (2015) Role of sirtuins in chronic obstructive pulmonary disease. Arch Pharm Res 38(1):1–10
Du J, Zhou Y, Su X, Yu JJ, Khan S et al (2011) Sirt5 is a NAD-dependent protein lysinedemalonylase and desuccinylase. Science 334(6057):806–9
Frescas D, Valenti L, Accili D (2005) Nuclear trapping of the forkhead transcription factor FoxO1via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 280:20589–20595
Jacobs KM, Pennington JD, Bisht KS, Aykin-Burns N, Kim HS, Mishra M et al (2008) SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci 4:291–299
Kode A, Yang SR, Rahman I (2006) Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res 7:132
Lan MY, Ho CY, Lee TC et al (2007) Cigarette smoke extract induces cytotoxicity on human nasal epithelial cells. Am J Rhinol 21:218–223
Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRTproteins. Mol Biol Cell 16(10):4623–35
Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C (2008) Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 382(3):790–801
Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120(3):419–29
Su Y, Han W, Giraldo C, De Li Y, Block ER (1998) Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 19:819–825
Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP (2009) Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 119:2758–2771
Togo S, Sugiura H, Nelson A et al (2010) Hepatic growth factor (HGF) inhibits cigarette smoke extract induced apoptosis in human bronchial epithelial cells. Exp Cell Res 316:3501–3511
Van der Horst A, Burgering BM (2007) Stressing the role of FoxO proteins in life span and disease. Nat Rev Mol Cell Biol 8:440–450
Verdin E, Hirschey MD, Finley LWS, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675
Vogt PK, Jiang H, Aoki M (2005) Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 4:908–913
Wang F, Nguyen M, Qin FX, Tong Q (2007) SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6:505–514
Wu CH, Lin HH, Yan FP, Wu CH, Wang CJ (2006) Immunohistochemical detection of apoptotic proteins, p53/Bax and JNK/FasL cascade, in the lung of rats exposed to cigarette smoke. Arch Toxicol 80:328–336
Yao H, Sundar IK, Ahmad T, Lerner C, Gerloff J, Friedman AE, Phipps RP, Sime PJ, McBurney MW, Guarente L, Rahman I (2014) SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 306(9):L816–28
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Y., Zhu, Y., Xing, S. et al. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3 . Cell Stress and Chaperones 20, 805–810 (2015). https://doi.org/10.1007/s12192-015-0599-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-015-0599-7