
Abstract
Lung disease is the leading and second-leading cause of death in women and men in Taiwan, respectively. Epidemiological studies conducted in Taiwan have shown that cigarette smoking is the principal risk factor of lung disease, but little is known about the association between apoptosis and cigarette smoke (CS)-induced lung pathogenesis. We designed an animal exposure system to study signal proteins involved in the process of apoptosis induced by smoking in rat terminal bronchiole. Rats were exposed to CS in doses of 5, 10, and 15 cigarettes, respectively, and the exposure lasted for 30 min, twice a day, 6 days a week for 1 month. Following which the rats were sacrificed and the lung tissues were analyzed by histopathological methods. The terminal bronchioles revealed mild to severe inflammation according to the doses of CS and marked lipid peroxidation, lymphocyte infiltration, congestion, and epithelial emphysema of alveolar spaces were also noted. Using an in situ cell death detection kit (TA300), the association of CS with apoptosis was determined in a concentration-dependent manner. Immunohistochemical evaluation showed that CS treatment produced an increase in the cellular levels of Bax, t-Bid, cleaved caspase-3, phospho-p53, phospho-JNK, and FasL but a decline in Bcl-2 and Mcl-1 (p<0.001 for all) in rat terminal bronchioles. The results provided evidences suggesting that exposure to CS not only induced apoptosis, but also involved p53/Bax and JNK/FasL cascade pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cigarette smoke (CS) is a major risk factor which causes the development of many cancers, including cancers of the larynx, oral cavity, pharynx, esophagus, pancreas, kidney, bladder, and lung (Czekaj et al. 2002; Shishodia and Aggarwal 2004). Smokers have a high incidence of lung cancer, which is the most common cause of cancer in Western countries, accounting for more deaths than those caused by prostate, breast, and colorectal cancers combined (Greenlee et al. 2001; Villeneuve and Morrison 1995). Recent estimates indicate that CS causes approximately 80–90% of lung cancer in Taiwan (Ko et al. 1997). However, the mechanisms by which CS induces lung cancer remain unclear, and among the 3,800 compounds identified in CS, a large number of them exert mutagenic and carcinogenic activity (Vineis and Caporaso 1995).
Recent studies have found that exposure to CS results in oxidative stress stimulation of inducible NO synthase (iNOS) and c-fos via regulation of protein tyrosine phosphorylation and MAP kinase, which in turn may promote lung pathogenesis (Chang et al. 2001). CS has been reported to have the ability to induce DNA single strand breaks in cultured human and rodent cells (Fielding et al. 1989; Leanderson and Tagesson 1992; Nakayama et al. 1984; Spencer et al. 1995; Stone et al. 1994, 1995). It was previously reported that a positive correlation exists between the number of cigarettes smoked per day and both the 8-hydroxyguanosine (8-OH-G) levels and their repair activities in human peripheral leukocytes and lung tissue (Asami et al. 1996, 1997). Peroxynitrite was identified in aqueous CS fraction (Muller et al. 1997), and this has been shown to react with guanine and DNA in vitro to form 8-nitroguanine (8-NO2-G) (Yermilov et al. 1995a, b). Moreover, it has recently been found that gaseous NOx can induce 8-NO2-G formation in human lung fibroblast cells and in tobacco cigarette smokers (Hsieh et al. 2001) and smoke-exposed Wistar rats (Hsieh et al. 2002). Although, detailed mechanisms of CS have been established in oxidative stress, DNA damage and aberrant proliferation, CS-regulated apoptosis remains unclear in vivo (De Flora et al. 2003).
Inappropriate regulation of apoptosis is associated with a variety of diseases, ranging from neurodegenerative disorders to malignancy (Thompson 1995). Past studies suggest that apoptosis might influence the malignant phenotype and promote tumor progression (Lowe and Lin 2000). Apoptosis involves two main pathways: death receptor and mitochondria death pathway (Herr and Debatin 2001). The apoptotic death receptor pathway is induced by members of the death receptor superfamily such as Fas (CD95). The Fas ligand (FasL) bonds to its receptor, forming a death-inducing complex with the adaptor molecule Fas-associated death domain (FADD) and finally caspase cascade activation is required for apoptosis. The mitochondrial death pathway is controlled by members of the Bcl-2 family, including the anti-apoptotic Bcl-2 and Bcl-XL proteins and the proapoptotic Bax and Bid proteins. Death signals stimulated cytochrome c, Apaf-1, and other possible factors released from the mitochondria. The two-apoptotic death pathways converge at activation of caspase-3 (Luo et al. 1998; Li et al. 1998).
In addition to caspases, the members of the mitogen-activated protein kinases family (MAPK) are also pathways that mediate apoptosis. The MAPK superfamily of serine/threonine kinases is activated by numerous extracellular stimuli and is involved in signal transduction cascades that play an important regulatory role in cell growth, differentiation, and apoptosis (Cobb and Goldsmith 1995; Chan-Hui and Weaver 1998). Three major mammalian MAPKs have been described: the extracellular signal regulating kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK. However, the diverse MAPK members are activated in response to different extracellular stimuli and have distinct downstream targets, thus serving different roles in cellular responses. It is worth mentioning that JNK is activated by UV irradiation, DNA damage, hydrogen peroxide, heat, and osmotic shock, resulting in apoptotic cell death (Ichijo 1999).
In the past two decades, significant progress has been made in understanding the molecular and cellular pathogenesis of lung cancer. However, the mechanism behind the CS-induced apoptosis and related proteins remains unclear. To elucidate the association between apoptosis and CS-induced lung pathogenesis, an animal exposure system was designed to investigate apoptosis and to evaluate the expression of related proteins using immunohistochemical detection method in the terminal bronchiole areas of the lung tissue of rats exposed to CS.
Materials and methods
Chemicals
Tris citrate buffer, xylene, hematoxylin, H2O2, and diaminobenzidine (DAB) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). In-situ apoptosis detection kit, TACS Blue Labe and Kit (TA300) was purchased from R and D Systems Inc. (MN, USA). Polyclonal antibody against phospho-p53 (Ser392) was obtained from Transduction Lab. (KY, USA). Antibodies against FasL, Bcl-2, Mcl-1, Bax, t-Bid, and cleaved caspase-3 were from Santa Cruz Biotechnology (CA, USA), and phospho-JNK (Thr183/Tyr185) was from Cell Signaling Tech. (MA, USA).
Animal treatment
Male Wistar rats (120±10 g body weight at 6-week old) were purchased from the National Science Council Animal Center, Taiwan. These animals were housed six per cage in an environmentally controlled animal room, and water was provided ad libitum. All animals were handled according to the guidelines of the Taiwan Society for Laboratory Animals Sciences for the care and use of laboratory animals. A total of 24 rats were divided into four exposure groups. For the exposure test, rats were placed in whole-body exposure chambers and exposed to smoke from 5, 10, or 15 cigarettes (New Paradise, Taiwan) simultaneously or filtered room air as control. The filtered breathing air was introduced into the chamber at a flow rate of 200 l/min. The exposure was carried out twice a day, 6 days per week for over 1 month, with each exposure lasting for 30 min. During the exposures, the temperature was maintained at 22–25°C, and the relative humidity was approximately 40%. One month after cessation of exposure, the rats were sacrificed by decapitation, and the lungs were immediately excised for immunohistochemistry determination of apoptosis-related proteins.
Autopsy and histology
Immediately after death, a complete necropsy was performed and the lung was examined. The lung was washed with physiologic saline and inspected for gross lesions. Tissues were fixed in 10% buffered formalin, processed for histological examination according to the lung tissue and stained with hematoxylin and eosin. The morphology, number, and location of any lesion observed were registered.
In-situ apoptosis detection
After excision, the lung tissues were fixed in 1,040 buffered-formalin solution for 18–24 h, dehydrated, embedded in paraffin, and cut into sections of 5 μm thickness. Prior to the labeling reaction, the samples were deparaffinized by xylenes and ethanol. The labeling reaction mixtures were prepared just before use according to the method of apoptosis detection kits (R and O Systems Inc., MN, USA). Small biopsy samples were covered with 50 μl labeling reaction mixtures and incubated at 37°C for 1 h. Samples were immersed in stop buffer for 5 min at 18–24°C and washed twice with dH2O. The samples were then mixed using 50 μl detection buffer and incubated at 18–24°C for 60 min. After washing, the samples were incubated with 50 μl of TACS Blue Label (TBL) for 2–5 min. Samples stained with TBL were counterstained with Nuclear Fast Red. Cells containing fragmented nuclear chromatin characteristic of apoptosis exhibit a blue nuclear staining that may become very dark after labeling, and this intense blue staining is typically associated with cell condensation. The preparation of solution and buffer was performed following the description for the commercial kit (see the procedure of Apoptosis Detection Kit TA300, R and D Systems Inc. for details). Six randomly selected fields (0.2 mm2) from six lung tissues of every group at a final magnification of 400 folds were counted in each section and the results were expressed as the total TBL-positive cells per mm2.
Immunohistochemical analysis
After being excised from the animals, the lungs were fixed in 10% buffer-formalin solution for 18–24 h, dehydrated, embedded in paraffin and cut into sections of 5 μm thickness. To perform the immunohistochemistry, the sections were deparaffinized in xylene, rehydrated in 0.05 M Tris buffer, pH 7.6, for 10 min and boiled in 0.01 M citrate buffer, pH 6.0, for 5 min. The sections were then removed and allowed to cool at room temperature for 20 min and rinsed two times with TBS for a total of 30 min. Endogenous peroxidase activity was blocked by 15 min incubation in 3% hydrogen peroxide. To increase antigenic exposure, tissue sections were incubated in 0.1% Triton X-100 for 45 min at room temperature.
Following this, the samples were incubated with diluted primary antibodies, including anti-FasL, Bcl-2, t-Bid, Mcl-1, phospho-p53, phospho-JNK, Bax, cleaved caspase-3 polyclonal antibodies, for 45 min at room temperature. After rinsing two times with TBS for a total of 20 min the bound primary antibodies were detected by sequential incubation with biotinylated secondary antibody (Biotinylated anti-rabbit or anti-mouse immunoglobins, LSAB kit from DAKO) for 30 min, streptavidin peroxidase (LSAB kit from DAKO) for 15 min, and DAB for 5–10 min, at room temperature with two rinses of TBS in between. The sections were then washed with distilled water and counterstained with Mayer’s hematoxylin. After dehydration and mounting, the expressions of these detection proteins in rat lung tissue were assessed by microscopic examination of the immunoperoxidase staining. The negative control for each experiment was a DAKO antibody diluent with background reductional compound, which is intended for use as a diluent in the preparation of primary antibodies and negative control reagents. The positively staining area, brown in color, was determined by Leica Q500 MC Image processing and analysis system against the negatively staining region shown relatively as blue in color.
Statistical analysis
Data reported are means ± standard deviation of six different animals and evaluated by one-way analysis of variance (ANOVA). Significant differences were established at p<0.05.
Results
Lung injury caused by CS
To study the role of CS in lung pathogenesis in vivo, rats were exposed to various concentrations of CS. The rats were exposed to gas-phase CS in a chamber twice a day, 30 min for each time and 6 days per week for over 1 month. The experimental conditions were designed to mimic passive smoke exposure of humans in a confined room. One month after cessation of exposure, injury induced by CS included lung inflammation, emphysema dilatation and peribronchiole fibrosis in the terminal bronchioles and alveolar duct area. The injury caused by CS was directly related to the number of cigarettes (data not shown).
In-situ apoptosis induced by CS
Following 1 month of CS treatment, the rat lung tissue showed typical apoptotic features: membrane blebbing, chromatin condensation, nuclear DNA fragmentation, and apoptotic bodies. A histogram of TBL-positive cells per mm2 with representative photographs, as shown in Fig. 1, reveals that exposure of rats to the smoke from 5 to 15 cigarettes produced a concentration-dependent increase in the TBL-positive cells. Table 1 showed a significant increase of about 1.26-, 1.47-, and 1.85-folds (p<0.001 for all). These results indicated that expression of apoptosis was significantly increased in rat lungs exposed to the smoke from 5 to 15 cigarettes.

Immunohistochemical examination of the expression of apoptosis in rat exposed smoking lung tissue .The rats were exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, twice a day, 6 days a week for 1 month. The arrow indicates myoepithelial cells undergoing apoptosis. The level of positive stain was quantified and shown in Table 1. Mean ± SD of six different animals.* p<0.001 compared with the control
Effect of CS on bcl-2 family
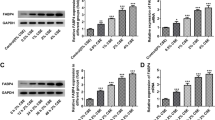
Investigations of the bcl-2 gene family have shown a complex network regulating apoptosis. The Bcl-2 protein family is an integral membrane protein that regulates apoptosis in several numerous biological systems (Danial and Korsmeyer 2004). We examined the cellular level of Bcl-2, Mcl-1, Bax, and t-Bid in lung tissues from CS-exposed rats. The expression of Bcl-2 and Mcl-1 (anti-apoptotic proteins) were significantly decreased in rat lungs exposed to the smoke from 5 to 15 cigarettes (Fig. 2) with maximal reduction folds of control about 0.75- and 0.49-folds, respectively (Table 2); in addition, the expressions of Bax and t-Bid (proapoptotic proteins) were elevated after exposure to CS (Fig. 3A, B) and reached the maximum levels at about 3.29- and 1.75-folds in rat lungs exposed to the smoke from 15 cigarettes (p<0.001 for all, Table 3).

Immunohistochemical evaluation of the effect of gas-phase CS on the expression of (A) Bcl-2 and (B) Mcl-1. Lung sections obtained from the rats exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, twice a day, 6 days a week for 1 month. The expressions of Bcl-2 and Mcl-1 were detected by anti-Bcl-2 and Mcl-1 antibodies and biotin-avidin peroxidase system. Arrows indicated positive stain areas apparent in the terminal bronchioles of lung. The level of positive stain was quantified and shown in Table 2. Mean ± SD of six different animals. * p<0.001 compared with the control

Immunohistochemical evaluation of the effect of gas-phase CS on the expression of (A) Bax and (B) t-Bid. Lung sections obtained from the rats exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, twice a day, 6 days a week for 1 month. The expressions of Bax and t-Bid were detected by anti-Bax and t-Bid antibodies and biotin-avidin peroxidase system. Arrows indicated positive stain areas apparent in the terminal bronchioles of lung. The level of positive stain was quantified and shown in Table 3. Mean ± SD of six different animals. * p<0.001 compared with the control
Effect of CS on caspase cleavage
Caspases play a pivotal role during apoptosis after treatment with an inducing agent. Immunohistochemical analysis was used to understand whether caspases were activated by CS in rat lungs. We next examined the expression of cleaved caspase-3. Caspases-3 are cytosolic proteins that exist normally as inactive precursors with higher molecular weight. It is cleaved proteolytically into low molecular weight (20 kDa) when cell undergoes apoptosis. Data show that the cleavage caspase-3 was apparently increased after exposure to CS (Fig. 4 and Table 4). The result suggested that CS induced the activation of caspase-3 in rat lungs.

Immunohistochemical evaluation of the effect of gas-phase CS on the expression of cleaved caspase-3. Lung sections obtained from the rats exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, twice a day, 6 days a week for 1 month. The expression of cleaved caspase-3 was detected by anti-cleaved caspase-3 antibody and biotin-avidin peroxidase system. Arrows indicated positive stain areas apparent in the terminal bronchioles of lung. The level of positive stain was quantified and shown in Table 4. Mean ± SD of six different animals.* p<0.001 compared with the control
Effect of CS on p53 phosphorylation
The overexpression of Bax accelerates apoptotic death induced by different stress in many cell lines. It has been reported that in cells with stably expressed p53 a continuous increase in Bax expression might be responsible for the apoptosis (Oltvai et al. 1993). Our findings showed that rat lungs expressed relatively high level of Bax (Fig. 3A) and an increase in phosphorylation of p53 (Fig. 5) showed a significant and gradual increase of about 1.50-, 2.00-, and 2.43-folds (p<0.001 for all, Table 5) following CS treatment that, thereby, facilitated the induction of apoptosis.

Immunohistochemical evaluation of the effect of gas-phase CS on the expression of phospho-p53. Lung sections obtained from the rats exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, twice a day, 6 days a week for 1 month. The expression of phospho-p53 was detected by anti-phospho-p53 antibody and biotin-avidin peroxidase system. Arrows indicated positive stain areas apparent in the terminal bronchioles of lung. The level of positive stain was quantified and shown in Table 5. Mean ± SD of six different animals. * p<0.001 compared with the control
Effect of CS on JNK/ FasL signaling
Previous studies have also shown that an increase in the phosphorylation of JNK was observed in the cigarette-treated endothelial cells (Hoshino et al. 2005; Raveendran et al. 2005), and the persistence of this activation led to the activation of FasL, an AP-1 target gene. Release of FasL initiated apoptosis through Fas death receptor, which, subsequently, truncated Bid, implying an association between the cigarette-induced apoptosis and the activated JNK/FasL signaling (Lei et al. 1998; Raveendran et al. 2005). Whether CS-induced apoptosis is also modulated by JNK/FasL was examined. In rat lung tissues, the phosphorylation of JNK was significantly increased after exposure to CS (p<0.001). The expression of phospho-JNK showed dose-dependence in CS-exposed rats (Fig. 6A). Similar results were also observed in the study of FasL (Fig. 6B). There was a 1.78–2.89 folds expression of phospho-JNK and 1.53–2.47 folds expression of FasL (Table 6) in the CS-exposed rats compared with that of the control, respectively.

Immunohistochemical evaluation of the effect of gas-phase CS on the expression of (A) phospho-JNK and (B) FasL. Lung sections obtained from the rats exposed to the smoke from (a) 0; (b) 5; (c) 10; or (d) 15 cigarettes for 0.5 h per day, 6 days a week, twice a day, for 1 month. The expression of phospho-JNK was detected by anti-phospho-JNK and anti-FasL antibodies and biotin-avidin peroxidase system. Arrows indicated positive stain areas apparent in the terminal bronchioles of lung. The level of positive stain was quantified and shown in Table 6. Mean ± SD of six different animals. * p<0.001 compared with the control
Discussion
Cigarette smoking is known to be a risk factor in cancer development. However, the underlying mechanisms, in particular the contribution of gas-phase smoke constituents, have not yet been completely identified. The experimental conditions were designed to mimic passive smoke exposure of humans in a confined room. The rats were exposed to gas-phase CS in a chamber 30 min twice a day and 6 days per week for over 1 month. These rat lung lesions showed blue positive staining indicating the level of apoptosis in a dose-dependent manner by in-situ apoptosis detection (Fig. 1). Apoptosis was reported to contribute to the high rate of cell loss in malignant tumors and, moreover, promote tumor progression in previous studies (Lowe and Lin 2000; Thompson 1995).
What triggers apoptosis during lung tumor development? A variety of signals appear to be important. Increasing evidence suggests that free radicals are involved in many of the chronic diseases associated with smoking. Molecular toxicological investigations demonstrated that CS can produce DNA single strand breaks (Fielding et al. 1989; Leanderson and Tagesson 1992; Nakayama et al. 1984; Spencer et al. 1995; Stone et al. 1994, 1995), elevated expression of heme oxygenase (Muller and Gebel 1994) and altered regulation of the c-fos gene at the transcriptional level (Muller 1995). Data also showed that exposure to CS results in oxidant stress leading to the stimulation of iNOS and c-fos via regulation of protein tyrosine phosphorylation and MAP kinase, which in turn may promote lung pathogenesis (Chang et al. 2001).
During the process of apoptosis induced by stress from sources such as ROS, anticancer drugs, UV irradiation, Fas (Seimiya et al. 1997; Butterfield et al. 1997; Juo et al. 1997), were associated with or required for apoptosis. The goal of the following set of experiments was to determine which pathway is associated with the CS-induced apoptosis in rat lungs. Apoptosis-related molecules may have an opportunity to be the candidate. The Bcl-2 family of proteins constitutes a critical intracellular checkpoint in the intrinsic pathway of apoptosis, as the mitochondria pathway. Mammals possess an entire family of Bcl-2 proteins including proapoptotic as well as antiapoptotic members. Importantly, it has been demonstrated that the gene products of Bcl-2 and Bax play important roles in apoptotic cell death (Oltvai et al. 1993; Jacobson and Raff 1995). In the Bcl-2 family, the ratio of Bcl-2 and Bax proteins has been recognized as a key factor in regulating the apoptotic process (Adams and Cory 1998; Green and Reed 1998). In the present study, the increase in CS-induced apoptosis was associated with an increase in levels of Bax protein (Fig. 3A), which heterodimerizes with and thereby inhibits Bcl-2 (Fig. 2A). Our study has demonstrated that CS may alter the expression of Bcl-2 and Bax and, therefore, lead to the apoptosis of rat lung tissues.
Because CS increased the amount of Bax, which is one of the p53 target genes, we next examined whether the CS-induced apoptosis may be mediated by the activation of p53. The phosphorylation level of p53 showed a significant increase of about 1.50-, 2.00-, and 2.43-folds (p<0.001 for all) following exposure of 5–15 cigarettes (Fig. 5). These results indicated that the CS-induced apoptosis was associated with an induction of p53 phosphorylation that subsequently modulated the expression of Bcl-2 family.
Of the death receptor pathway processes, FasL-mediated apoptosis followed a death-inducing signal complex (DISC) formation showing that the members presented have a death receptor Fas, FADD and casapse-8. The activated form of caspase-8 may activate in turn either caspase-3 or cleave Bid. Activated Bid triggers apoptosis by mitochondrial translocation, which stimulates cytochrome c release (Yin 2000). In Fas/TNF system, caspase-3 activation is mediated by caspase-8 or caspase-10 (Alnemri 1997). Caspase-3 has been shown to play a central role in the process of apoptosis induced by many stimuli (Tewari et al. 1995), as an effector caspase. Moreover, it has been demonstrated that cleavage of Bid mediates caspase-8-induced cytochrome c release from the mitochondria (Luo et al. 1998). It strongly suggested that Bid cleavage is necessary for CS-related caspase-3 activation and apoptosis release (Fig. 4). Some studies reported that caspases can modulate JNK activation in Fas- or TNFα-induced apoptosis (Juo et al. 1997; Roulston et al. 1998), indicating that the stress-activated kinases perhaps induced caspase activation. Thus, we observed that CS induced an increase in JNK activity related to FasL (Fig. 6 and Table 6).
The data obtained in the present study led to the following conclusions regarding the regulation of rat lung tissues destroyed by CS: (1) CS can induce apoptosis of rat lung tissues in a concentration-dependent manner, (2) CS-induced caspase activation leads to death of rat lung tissues, (3) CS induces apoptosis through a mechanism involving the release of apoptosis-related proteins from the mitochondria, and (4) CS-induced death in rat lung tissues requires JNK/FasL to activate an upstream of caspases.
In summary, this study is consistent with other studies demonstrating that CS induces lung injury including apoptosis via the two mechanisms involved, that is, (1) phosphorylation of JNK MAPK pathway followed by either activation of FasL or Bcl-2, and (2) an increase in Bax with the expression of phospho-p53 stimulated, thus leading to the activation of cleaved caspase-3. This effect may induce apoptosis and may be an important pathway in the lung pathogenesis of CS. These findings may help develop a novel strategy for prevention of lung cancer by understanding the signal cascade of CS-induced lung pathogenesis.
Abbreviations
- CS:
-
Cigarette smoke
- Bax:
-
Bcl-2 associated X protein
- t-Bid:
-
Truncated Bid
- Bcl-2:
-
B cell lymphoma-2
- MAPK:
-
Mitogen-activated protein kinases family
- JNK:
-
c-Jun N-terminal kinase
- Fas-L:
-
Fas ligand
- DAB:
-
Diaminobenzidine
- iNOS:
-
Inducible NO synthase
- NOx:
-
Nitrogen oxides
- HPF:
-
High-power-field
References
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Alnemri ES (1997) Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem 64:33–42
Asami S, Hirano T, Yamaguchi R, Tomioka Y, Itoh H, Kasai H (1996) Increase of a type of oxidative DNA damage, 8-hydroxyguanine and its repair activity in human leukocytes by cigarette smoking. Cancer Res 56:2546–2549
Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, Yamaguchi R, Itoh H, Kasai H (1997) Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis 18:1763–1766
Butterfield L, Storey B, Maas L, Heasley LE (1997) c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J Biol Chem 272:10110–10116
Chang WC, Lee YC, Liu CL, Hsu JD, Wang HC, Chen CC, Wang CJ (2001) Increased expression of iNOS and c-fos via regulation of protein tyrosine phosphorylation and MEK1/ERK2 proteins in terminal bronchiole lesions in the lungs of rats exposed to cigarette smoke. Arch Toxicol 75:28–35
Chan-Hui PY, Weaver R (1998) Human mitogen-activated protein kinase kinase kinase mediates the stress-induced activation of mitogen-activated protein kinase cascades. Biochem J 336:599–609
Cobb MH, Goldsmith EJ (1995) How MAP kinases are regulated. J Biol Chem 270:14843–14846
Czekaj P, Palasz A, Lebda-Wyborny T, Nowaczyk-Dura G, Karczewska W, Florek E, Kaminski M (2002) Morphological changes in lungs, placenta, liver and kidneys of pregnant rats exposed to cigarette smoke. Int Arch Occup Environ Health 75(Suppl):S27–S35
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219
De Flora S, D’Agostini F, Balansky R, Camoirano A, Bennicelli C, Bagnasco M, Cartiglia C, Tampa E, Longobardi MG, Lubet RA, Izzotti A (2003) Modulation of cigarette smoke-related end-points in mutagenesis and carcinogenesis. Mutat Res 523–524:237–252
Fielding S, Short C, Davies K, Wald N, Bridges BA, Waters R (1989) Studies on the ability of smoke from different types of cigarettes to induce DNA single-strand breaks in cultured human cells. Mutat Res 214:147–151
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Greenlee RT, Hill-Harmon MB, Murray T, Thun M (2001) Cancer statistics, CA. Cancer J Clin 51:15–36
Herr I, Debatin KM (2001) Cellular stress response and apoptosis in cancer therapy. Blood 98:2603–2614
Hoshino S, Yoshida M, Inoue K, Yano Y, Yanagita M, Mawatari H, Yamane H, Kijima T, Kumagai T, Osaki T, Tachiba I, Kawase I (2005) Cigarette smoke extract induces endothelial cell injury via JNK pathway. Biochem Biophys Res Commun 329:58–63
Hsieh YS, Wang HC, Tseng TW, Chang WC, Wang CJ (2001) Gaseous nitric oxide-induced 8-nitroguanine formation in human lung fibroblast cells and cell-free DNA. Toxicol Appl Pharmacol 172:210–216
Hsieh YS, Chen BC, Shiow SJ, Wang HC, Hsu JD, Wang CJ (2002) Formation of 8-nitroguanine in tobacco cigarette smokers and in tobacco smoke-exposed Wistar rats. Chem Biol Interact 140:67–80
Ichijo H (1999) From receptors to stress-activated MAP kinases. Oncogene 18:6087–6093
Jacobson MD, Raff MC (1995) Programmed cell death and Bcl-2 protection in very low oxygen. Nature 374:814–816
Juo P, Kuo CJ, Reynolds SE, Konz RF, Raingeaud J, Davis RJ, Biemann HP, Blenis J (1997) Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol 17:24–35
Ko YC, Lee CH, Chen MJ, Huang CC, Chang WY, Lin HJ, Wang HZ, Chang PY (1997) Risk factors for primary lung cancer among non-smoking women in Taiwan. Int J Epidemiol 26:24–31
Leanderson P, Tagesson C (1992) Cigarette smoke-induced DNA damage in cultured human lung cells: role of hydroxy radicals and endonuclease activation. Chem Biol Interact 81:197–208
Lei W, Yu R, Mandlekar S, Kong AN (1998) Induction of apoptosis and activation of interleukin 1beta-converting enzyme/Ced-3 protease (caspase-3) and c-Jun NH2-terminal kinase 1 by benzo(a)pyrene. Cancer Res 58:2102–2106
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of Bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501
Lowe SW, Lin AW (2000) Apoptosis in cancer. Carcinogenesis 21:485–495
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94:481–490
Muller T, Gebel S (1994) Heme oxygenase expression in Swiss 3T3 cells following exposure to aqueous cigarette smoke fractions. Carcinogenesis 15:67–72
Muller T (1995)Expression of c-fos in quiescent Swiss 3T3 cells exposed to aqueous cigarette smoke fractions. Cancer Res 55:1927–1932
Muller T, Haussmann HJ, Schepers G (1997) Evidence for peroxynitrite as an oxidative stress-inducing compound of aqueous cigarette smoke fractions. Carcinogenesis 18:295–301
Nakayama T, Kaneko M, Kodama M, Nagata C (1984) Cigarette smoke induced DNA single-strand breaks in human cells. Nature 314:462–464
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74:609–619
Raveendran M, Wang J, Senthil D, Wang J, Utama B, Shen Y, Dudley D, Zhang Y, Wang XL (2005) Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS Lett 579:733–740
Roulston A, Reinhard C, Amiri P, Williams LT (1998) Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor alpha. J Biol Chem 273:10232–10239
Seimiya H, Mashima T, Toho M, Tsuruo T (1997) c-Jun NH2-terminal kinase-mediated activation of interleukin-1beta converting enzyme/CED-3-like protease during anticancer drug-induced apoptosis. J Biol Chem 272:4631–4636
Shishodia S, Aggarwal BB (2004) Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-kappaB by suppressing activation of IkappaBalpha kinase in human non-small cell lung carcinoma: correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res 64:5004–5012
Spencer JP, Jenner A, Chimel K, Ardoma OL, Cross CE, Wu R, Halliwell B (1995) DNA damage in human respiratory tract epithelial cells: damage by gas phase cigarette smoke apparently involves attack by reactive nitrogen species in addition to oxygen radicals. FEBS Lett 375:179–182
Stone KK, Bermudez E, Pryor WA (1994) Aqueous extracts of cigarette tar containing the tar free radical cause DNA nicks in mammalian cells. Environ Health Perspect (Suppl)102:173–178
Stone K, Bermudez E, Zang LY, Carter KM, Queenan KE, Pryor WA (1995) The ESR properties, DNA nicking, and DNA association of aged solutions of catechol versus aqueous extracts of tar from cigarette smoke. Arch Biochem Biophys 319:196–203
Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog for CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81:801–809
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 226:1097–1099
Villeneuve PJ, Morrison HI (1995) Trends in mortality from smoking related cancers, 1950 to 1991. Can Soc Trends 39:8–11
Vineis P, Caporaso N (1995) Tobacco and cancer epidemiology and the laboratory. Environ Health Perspect 103:156–160
Yermilov V, Rubio J, Becchi M, Friesen MD, Pignatelli B, Ohshima H (1995a) Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis 16:2045–2050
Yermilov V, Rubio J, Ohshima H (1995b) Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett 376:207–210
Yin XM (2000) Signal transduction mediated by Bid, a pro-death Bcl-2 family protein, connects the death receptor and mitochondria apoptosis pathways. Cell Res 10:161–167
Acknowledgment
This work was supported by a grant from the National Science Council (NSC 94-2320-B040-039), Taiwan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, CH., Lin, HH., Yan, FP. et al. Immunohistochemical Detection of Apoptotic Proteins, p53/Bax and JNK/FasL Cascade, in the Lung of Rats Exposed to Cigarette Smoke. Arch Toxicol 80, 328–336 (2006). https://doi.org/10.1007/s00204-005-0050-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-005-0050-4