
Abstract
A number of missense mutations in the two related small heat shock proteins HspB8 (Hsp22) and HspB1 (Hsp27) have been associated with the inherited motor neuron diseases (MND) distal hereditary motor neuropathy and Charcot-Marie-Tooth disease. HspB8 and HspB1 interact with each other, suggesting that these two etiologic factors may act through a common biochemical mechanism. However, their role in neuron biology and in MND is not understood. In a yeast two-hybrid screen, we identified the DEAD box protein Ddx20 (gemin3, DP103) as interacting partner of HspB8. Using co-immunoprecipitation, chemical cross-linking, and in vivo quantitative fluorescence resonance energy transfer, we confirmed this interaction. We also show that the two disease-associated mutant HspB8 forms have abnormally increased binding to Ddx20. Ddx20 itself binds to the survival-of-motor-neurons protein (SMN protein), and mutations in the SMN1 gene cause spinal muscular atrophy, another MND and one of the most prevalent genetic causes of infant mortality. Thus, these protein interaction data have linked the three etiologic factors HspB8, HspB1, and SMN protein, and mutations in any of their genes cause the various forms of MND. Ddx20 and SMN protein are involved in spliceosome assembly and pre-mRNA processing. RNase treatment affected the interaction of the mutant HspB8 with Ddx20 suggesting RNA involvement in this interaction and a potential role of HspB8 in ribonucleoprotein processing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Distal hereditary motor neuropathy (dHMN) is one type of motor neuron disease (MND) that is characterized by progressive weakness and wasting of the extensor muscles of the feet, frequently resulting in foot deformity (Harding and Thomas 1980; Irobi et al. 2004a). In due course, distal upper limb muscles often become involved, and additional features including paralysis of the vocal cords and diaphragm may occur. Charcot-Marie-Tooth disease (CMT) has similar symptoms, with additional involvement of the sensory nerves of the peripheral nervous system, although the clinical differentiation of both neuropathies sometimes can be difficult (Harding and Thomas 1980; Shy 2004). Both diseases are genetically heterogeneous. Among the proteins affected by mutations are the two small heat shock proteins (sHSP) of the HSPB family, HspB8 (synonyms: Hsp22, H11, E2IG1; gene: HSPB8) and HspB1 (synonyms: Hsp27, Hsp25; gene: HSPB1) (reviewed in Benndorf and Welsh 2004; Dierick et al. 2005; Benndorf 2010). To date, three missense mutations in HspB8 in five families with dHMN type II or CMT type 2L (OMIM 608014, #608673) have been reported (Irobi et al. 2004b; Tang et al. 2005). These mutations affect the "hot spot" amino acid residue Lys141 (cf. Fig. 1a) in the wild-type HspB8 (wtHspB8) protein changing it to either Glu (K141EHspB8) or Asn (K141NHspB8).

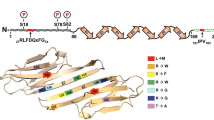
Sequences and domains of human HspB8 and Ddx20 and isoelectric points of wtHspB8 and muHspB8 forms. a HspB8 (GenBank accession: AF250138.1). The domains were designated as described previously (Fontaine et al. 2003). The known disease-associated mutations affect Lys141 (highlighted in black) in the conserved α-crystallin domain. b Ddx20 (GenBank accession: NM_007204.4). The N-terminal half contains the conserved DEAD box RNA helicase domain with the seven helicase motifs I, Ia, II, III, IV, V, and VI. The non-conserved C-terminal half contains the HspB8 binding region (positions 475–727; bold, single underline), the SMN protein-binding region (positions 456–547; dashed underline; cf. Charroux et al. 1999), and the SF-1 binding region (positions 719–824; dotted underline; corresponds to positions 721–825 in the mouse sequence BC137721.1; cf. Yan et al. 2003). In (a) and (b), cysteine residues reactive to the BMH cross-linking reagent are highlighted in gray (cf. Fig. 3). c Isoelectric focusing of wtHspB8 and muHspB8 forms. cDNAs of untagged wtHspB8 (construct 2), K141EHspB8 (construct 3), or K141NHspB8 (construct 4) were used to transfect HEK293T cells, and 48 h later the cell proteins were separated by IEF-PAGE/Western blotting using a specific anti-HspB8 antibody. K141EHspB8 and K141NHspB8 showed more acidic pIs (∼4.2 and ∼4.3, respectively) as compared to wtHspB8 (∼4.7). The approximate positions of the marker proteins β-lactoglobulin B (∼5.1), phycocyanin (∼4.6), and glucose oxidase (∼4.2) are indicated
Mammals contain ten sHSPs (Fontaine et al. 2003; Kappé et al. 2003; Kappé et al. 2010). These proteins are grouped together by the presence of a conserved stretch of ∼85 amino acid residues, the so-called α-crystallin domain, in their C-terminal halves (cf. Fig. 1a). Most of the studied sHSPs have chaperone-like properties that are thought to be the basis for their ability to protect cells from adverse conditions by inhibiting apoptosis (Rogalla et al. 1999; Bruey et al. 2000; Kim et al 2004; Carra et al. 2005), although HspB8 may act also through a non-canonical mechanism which is unrelated to the chaperone model, and may even promote apoptosis (Hase et al. 2005; Carra et al. 2009). sHSPs have been implicated in various additional functions, including consolidation of redox homeostasis, proteasome-mediated protein degradation, RNA processing, and cytoskeletal functions such as cell motility, and muscle contraction and relaxation (reviewed in Benndorf 2010).
Most of the sHSPs are expressed in many tissues, with differing expression profiles probably in each tissue (Verschuure et al. 2003). sHSPs have the ability to form homo- and hetero-dimers, and also homo- and hetero-oligomeric complexes (reviewed in Benndorf 2010). HspB8 was shown to form homo-dimers, and hetero-dimers with HspB1, HspB2, HspB5, HspB6, and HspB7 (Sun et al. 2004; Fontaine et al. 2005), although oligomeric forms could not be detected using recombinant HspB8 (Chowdary et al. 2004; Kim et al 2004). However, in extracts of cardiac and breast cancer cells, recruitment of HspB8 into high-molecular-mass material was reported, even though the nature of this material is not known (Fontaine et al. 2005; Sun et al. 2007). Association of HspB8 with other proteins may account for such high-molecular-mass species (see below).
sHSPs play distinguished roles in several tissues. Muscles, for example, contain extraordinary high amounts of the HspB5 (αB-crystallin), and a number of recently identified mutations in this sHSP are associated with myofibrillar myopathy or dilated cardiomyopathy (Inaguma et al. 1995; Vicart et al. 1998; Selcen and Engel 2003; Inagaki et al. 2006; Pilotto et al. 2006). Neuronal cells express several of the sHSPs, including HspB8, HspB1, and HspB5, and are efficiently protected from injury by sHSPs, notably HspB1 (Benn et al. 2002). Although involvement of HspB1 in growth and branching of neurons has been suggested, the precise role of sHSPs in the nervous system is not understood (Benn et al. 2002; Williams et al. 2006). Likewise, it is not known what events lead to the slow death of specifically motor neurons in dHMN and CMT resulting from the mutations in HspB8 or HspB1. Recent findings concerning the involvement of HspB8 in substrate sorting between renaturation and lysosomal degradation, or in proteasome activation, eventually may lead to a better understanding of its molecular and cellular functions (Carra et al. 2008; Hedhli et al. 2008).
The known mutant forms of HspB8 (muHspB8) have genetic dominant gain-of-function or dominant-negative characteristics suggesting that they exert their deleterious effects through abnormally altered properties, possibly in interaction with other proteins (Irobi et al. 2004b; Tang et al. 2005; Benndorf 2010). Previously, we have shown that both muHspB8 forms exhibited abnormally increased interactions with HspB1 and HspB5 (Fontaine et al. 2006). In addition to interactions among themselves, several sHSPs are known to interact also with other proteins, e.g., HspB1 interacts with PASS1 and Hic-5 (ARA55), and HspB8 with Sam68, TLR4, and Bag3 (Liu et al. 2000; Jia et al. 2001; Badri et al. 2006; Roelofs et al. 2006; Carra et al. 2009). This raises the question whether the muHspB8-associated pathology may also involve abnormal interactions with other, possibly unknown, proteins. To test this hypothesis, we performed a library-scale yeast two-hybrid (YTH) screen for proteins interacting with HspB8. We demonstrate herein that the RNA helicase Ddx20, a component of the survival-of-motor-neurons complex (SMN complex), interacts with HspB8, and that both muHspB8 forms have increased interaction stoichiometry with Ddx20. Ddx20 itself interacts with another component of the SMN complexes, the survival-of-motor-neurons protein (SMN protein; Charroux et al. 1999). Interestingly, mutations in the SMN1 gene are associated with spinal muscular atrophy (SMA), another MND (Lefebvre et al. 1995; Gubitz et al. 2004). Thus, these protein interaction data link seemingly unrelated etiologic factors (HspB8, HspB1, HspB5, and SMN protein) for several MNDs and myopathies, and we propose that they act through one common biochemical mechanism or signaling pathway.
2 Material and methods
2.1 Origin of cDNAs and cloning information
HspB8 cDNA was obtained from a human heart cDNA library (Benndorf et al. 2001). Full-length Ddx20 cDNA was bought from OriGene (Rockville, MD, USA). More information on the used cloning procedures, plasmid constructs and PCR primers is given in Table 1. The vectors TOPO TA, pcDNA4/HisMAx TOPO TA, pcDNA3.1(+), pcDNA3.1myc (all from Invitrogen, Carlsbad, CA, USA), and pFlag-CMV2 (Sigma, St. Louis, MO, USA) were used for cloning purposes and for expression in mammalian cells. pACT2 and pGBKT7 (Clontech, Mountain View, CA, USA) were used for YTH experiments, and peCFP-N1 and peCIT-C1 (Invitrogen) for quantitative fluorescence resonance energy transfer (qFRET) experiments. For site-directed mutagenesis the PfuTurbo Hotstart PCR Master Mix (Stratagene, La Jolla, CA, USA) was used.
2.2 Yeast two-hybrid library screen
The human heart cDNA library (Clontech) in the "prey" vector pACT2 with the Gal4-transcription activation domain was propagated as described in the manufacturer’s instruction. The YTH screening was performed by transforming the yeast strain AH109 first with the “bait” plasmid (pGBKT7-wtHspB8, construct 1) followed by a second transformation with the library plasmids. More than 106 independent clones were screened. Primary positive clones were obtained with nutrition deficiency (-Trp, -Leu, -His) selection (growth) and the X-α-gal reporter gene expression assay (blue colonies) according to the manufacturer’s instruction. These primary positive colonies were depleted of the “bait” vectors and, in order to identify false-positive colonies, mated with yeast strain Y187 carrying human lamin C cDNA in the vector pGBKT7 as unrelated control construct. From the remaining “true” positive colonies, the plasmids were recovered and sequenced. Out of ∼20 “true” positive clones, one clone contained a fragment of the Ddx20 sequence as shown in Fig. 1.
2.3 Cell culture and transfection
HEK293T cells were maintained at 37°C in a 5% CO2 humidified atmosphere in Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal calf serum. To prevent cell detachment, culture dishes, coverslips, and glass bottom dishes (MatTek, Ashland, MA) were treated with poly-d-lysine before seeding the cells. Cells grown in six-well plates were transiently transfected with 1 or 2 μg vector cDNA for single or double transfections, respectively. For expression of cyan fluorescent protein (CFP) or citrin (CIT) fusion proteins, cells were transfected with FuGENE 6 (Roche, Indianapolis, IN, USA) at ∼40% confluency, and analyzed or harvested 10 h later. For expression of all other constructs, cells were transfected with Lipofectamine 2000 (Invitrogen) at ∼90% or ∼40% confluency and analyzed 24 or 48 h later as specified.
2.4 Co-immunoprecipitation
Forty-eight hours after transfection, HEK293T cells were washed three times with PBS and lysed on ice for 30 min in 500 μl buffer A (low stringency buffer, 50 mM Tris–HCl, pH 8.0; 100 mM NaCl; 1% Triton X-100; 5 mM EDTA; 1 mM EGTA; 1× protease inhibitor mix from Roche). Cell lysates were centrifuged at 4°C for 10 min at 14,000×g and the supernatants were recovered.
For immunoprecipitation with the anti-Xpress monoclonal antibody (Invitrogen) as shown in Fig. 2, the supernatants were pre-cleared by incubation with 50 μl protein G-Sepharose slurry (Sigma) for 2 h at 4°C. Two micrograms of the anti-Xpress antibody was bound to 50 µl of protein G-Sepharose by incubating for 2 h at 4°C in 500 µl of buffer A. These antibody-coated beads then were added to the cell lysates and incubated over night at 4°C on a rotating platform. The beads were collected by centrifugation and washed three times with either buffer A or buffer B (high stringency buffer: buffer A plus 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulfate (SDS)).

Abnormally increased interaction of muHspB8 with Ddx20 in cell extracts. HEK293T cells were singly or doubly transfected with vector cDNA of Xpress-tagged Ddx20 (construct 5) and myc-tagged wtHspB8 (construct 6), K141EHspB8 (construct 7), or K141NHspB8 (construct 8), as indicated. Forty-eight hours later, the presence of the expressed proteins in cell extracts was confirmed by SDS-PAGE/Western blotting using Xpress- and myc-specific antibodies (I, II). wtHspB8 and muHspB8 occur in similar amounts in the cell extracts. Subsequently, Ddx20 was immunoprecipitated from the cell extracts using both low and high stringency conditions (III and V, respectively), and its presence in the precipitates was confirmed in cells transfected with Ddx20 indicating proper functioning of the method. The Western blots of the precipitates were then developed for co-precipitated HspB8. The various HspB8 forms were detected only in the precipitates of those cells which expressed both proteins, both under low and high stringency conditions (IV and VI, respectively). This co-IP indicates interaction between Ddx20 and the various forms of HspB8. At low stringency conditions (IV), all HspB8 forms were co-precipitated to a similar extent, while at high stringency conditions (VI), the amounts of co-precipitated K141NHspB8, and to a lesser extent also of K141EHspB8, were greater than that of wtHspB8. The blot is representative of three independent experiments
For treatment of cell extracts with RNase as shown in Fig. 5, the supernatants were divided into two aliquots. One aliquot was incubated at 37°C for 30 min with 1 μl of RNase (500 U/ml RNase A; 20,000 U/ml RNase T1) cocktail (Ambion, Austin, TX, USA), the other aliquot (control) was incubated without RNase. Thereafter, the supernatants were pre-cleared by incubation with 25 µl of protein G-Sepharose for 1 h at 4°C. One microgram of the anti-myc monoclonal antibody 9E10 (American Type Culture Collection, Manassas, VA, USA) was bound to 25 µl of protein G-Sepharose by incubating for 2 h at 4°C in 500 µl of buffer A. These antibody-coated beads then were added to the cell lysates and the samples were incubated for 2 h at 4°C on a rotating platform. The beads were collected by centrifugation and washed three times with buffer B.
After washing, the bound immunocomplexes were released from the beads by 3 min boiling in 80 µl of buffer C (62.5 mM Tris–HCl, pH 6.8; 2% SDS; 10% glycerol; 200 mM dithiothreitol; 0.002% bromophenol blue). Expressed and immunoprecipitated proteins were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting, using the primary anti-Xpress and anti-myc antibodies (Invitrogen/American Type Culture Collection), and the corresponding secondary goat Fc-specific anti-mouse antibody coupled to horseradish peroxidase (Jackson, West Grove, PA, USA). Protein bands were visualized using the ECL system (GE Healthcare, Uppsala, Sweden). In negative controls, mouse serum was used for the immunoprecipitation instead of the anti-Xpress and anti-myc antibodies.
2.5 Isoelectric focusing polyacrylamide gel electrophoresis analysis
Forty-eight hours after transfection, HEK293T cells were harvested in 100 μl of solution D (8 M urea; 2% ampholytes 3–10; 3% CHAPS; 2% β-mercaptoethanol). Cell proteins were separated by isoelectric focusing polyacrylamide gel electrophoresis (IEF-PAGE) followed by Western blotting as described previously (Benndorf et al. 2000). Goat anti-HspB8 antibody (Abcam, Cambridge, MA, USA) and donkey anti-goat antibody coupled to horseradish peroxidase (Jackson) were used as primary and secondary antibodies, respectively.
2.6 Cross-linking
Forty-eight hours after transfection, HEK293T cells grown in six-well plates were washed three times with ice-cold buffer E (20 mM Hepes–KOH, pH 7.5; 125 mM KCl; 5 mM NaCl; 11 mM glucose). Cells were permeabilized by adding 1 ml of digitonin (Sigma) solution (50 µg/ml in buffer E) per well. After incubation for 10 min at room temperature, cells were washed with 1 ml ice-cold buffer E. Thereafter, 1 ml of a freshly prepared 10 μM bismaleimido-hexane (BMH; Pierce, Rockford, IL, USA) solution in buffer E was added to the cells and incubation was continued on ice for 30 min. After washing with 1 ml ice-cold buffer E, cells were dissolved at room temperature in 100 μl of buffer C and boiled for 3 min. The samples were analyzed by SDS-PAGE/Western blotting using Xpress- and myc-specific antibody systems.
2.7 Immunofluorescence
Twenty-four hours after transfection, HEK293T cells grown on glass coverslips were washed three times with PBS and fixed with 4% paraformaldehyde at room temperature for 30 min. After washing with PBS, the coverslips were incubated for 30 min in buffer F (5% BSA; 0.1% Triton X-100 in PBS) followed by sequential incubations with the primary antibodies (monoclonal anti-Xpress, rabbit anti-Flag; 1:200 dilutions in buffer F; antibodies from Invitrogen and Sigma, respectively) and the corresponding secondary antibodies (donkey anti-mouse-cy3, donkey anti-rabbit-FITC; 1:200 dilutions in buffer F; antibodies from Jackson). The coverslips were mounted on slides using Prolong Gold antifade reagent containing DAPI (Invitrogen), and sealed with nail polish. For fluorescence microscopy an inverted epifluorescence microscope Eclipse TE-2000 U (Nikon, Melleville, NY, USA) was used equipped with filter sets for FITC (exciter 480/40, emission 535/50), cy3 (exciter 560/55, emission S620/60), and DAPI (exciter 360/40, emission 460/50). Images were collected using a digital CoolSnap CCD camera (Photometrics, Tucson, AZ, USA) and Metamorph image processing software version 6.2r5 (Molecular Devices, Sunnyvale, CA, USA).
2.8 Live cell imaging and quantitative fluorescence resonance energy transfer analysis
Ddx20 and the HspB8 species were tagged with CIT and CFP, respectively (cf. Table 1). Ten hours after transfection, HEK293T cells grown in glass bottom six-well culture plates (MatTek) were washed two times with PBS and kept at 37°C in DMEM medium without phenol red for collecting the fluorescence images. The configuration of the microscope, image collection, and processing were as described above, with the exception that exciter filters 430/25 and 500/20, and a 505dcxr Dual View Micro Imager MSMI.DV.CC (Optical Insights, Tucson, AZ, USA) equipped with the emission filters 470/30 and 535/30 were used.
The qFRET method was applied to quantify apparent average fluorescence resonance energy transfer efficiencies (AAFE) as indicators of protein interactions (Hoppe et al. 2002). In this analysis, only cells or areas of cells were included that showed a relatively uniform cytoplasmic distribution of the expressed proteins and that did not contain any inclusion bodies or aggresomes. Acquired I A, I D, and I F images from at least 27 microscopic fields per sample group were used for the computation by the qFRET algorithm. The calculated output data were expressed as AAFE values ([E A + E D]/2). These AAFE values for the energy acceptor (E A) and donor (E D) are proportional to the fraction of the interaction partners in complex. As negative controls, the cells were transfected with the “empty” peCFP-N1/peCIT-C1 vectors (control 1), with wtHspB8-CFP/peCIT-C1 (control 2), or with Ddx20-CIT/peCFP-N1 (control 3) which defined the baseline signal. AAFE values which were significantly different from that baseline signal indicated interaction.
In order to verify expression of the correct Ddx20-CIT and HspB8-CFP fusion proteins, HEK293T cells were harvested 10 h after transfection, solubilized in buffer C, and processed for SDS-PAGE/Western blotting. Rabbit anti-GFP antibody (Cell Signaling, Beverly, MA, USA) and monoclonal anti-Ddx20 antibody (Sigma) were used as primary antibodies, and goat anti-rabbit and goat anti-mouse antibodies (both from Jackson) as secondary antibodies.
2.9 Statistics
The data collected by the qFRET method were expressed as mean AAFE ± SE. These data were analyzed using one-way ANOVA to compare the mean AAFE values between the different experimental groups and control 1 as shown in Fig. 4d (27 < n < 41). When overall significance was detected, a post-hoc multiple group comparison was conducted using Tukey HSD adjustment. The collected qFRET data were also checked for normality within each group. The histograms and Q–Q plots did not show severe violation of normality assumption. Additionally, unpaired student’s t test was applied to compare results between sample groups. Differences between groups were considered statistically significant if P < 0.05.
3 Results
3.1 Identification of Ddx20 as protein interacting with HspB8
Full-length human wtHspB8 (GenBank accession: AF250138.1, cf. Fig 1A) cDNA (construct 1; cf. Table 1) was used to screen a human cDNA library by the YTH method. Among the identified proteins was a C-terminal fragment (positions 475–727 in Fig. 1b) of the DEAD box protein Ddx20 (synonyms: gemin3, DP103; GenBank accession: NM_007204.4). Ddx20 itself interacts with SMN protein (Charroux et al. 1999), and loss-of-function mutations in the SMN1 gene are associated with SMA, another MND (Lefebvre et al. 1995; Gubitz et al. 2004). Thus, these protein interaction experiments apparently establish a linkage between HspB8, its binding partner HspB1, Ddx20, and the SMN protein, and mutations in any of these proteins, with the exception of Ddx20, cause various forms of MND. Because of the potential significance for the understanding of the pathomechanism of the associated MNDs, dHMN, CMT, and SMA, we then studied the interaction of wtHspB8 and both of its known mutant forms, K141EHspB8 and K141NHspB8, with Ddx20 in greater detail.
3.2 Acidic shift of the isoelectric points of mutant HspB8 species
As reported previously, wtHspB8 is an acidic protein with an isoelectric point (pI) of ∼4.7 (Benndorf et al. 2001). IEF-PAGE/Western blotting of extracts of HEK293T cells expressing untagged K141EHspB8 (construct 3) and K141NHspB8 (construct 4) revealed significant acidic shifts of their pIs to ∼4.2 and ∼4.3, respectively, as compared to wtHspB8 (construct 2; Fig. 1c). This difference of their pIs may well be the physical basis for the aberrant interaction properties of both muHspB8 forms as shown below, and also as reported previously (Fontaine et al. 2006).
3.3 Abnormal interaction of mutant HspB8 with Ddx20
Co-immunoprecipitation (co-IP) was used to verify the interaction of wtHspB8 with full-length Ddx20 (Fig. 2). HEK293T cells were singly (lanes 2 and 4) or doubly (lane 3) transfected with Xpress- and myc-tagged constructs of Ddx20 (construct 5) and wtHspB8 (construct 6) cDNAs. Forty-eight hours later, cells were harvested and cell extracts were processed for immunoprecipitation. Immunoprecipitated proteins were analyzed by SDS-PAGE followed by Western blotting. Appropriate controls verified the expression of Ddx20 and wtHspB8 in transfected cells (panels I and II) and the correct working of the immunoprecipitation method using both low and high stringency conditions (panels III and V, respectively). Immunoprecipitated Ddx20 was detected in cells transfected with Ddx20 (lanes 2 and 3) but not in non-transfected control cells (lane 1), and co-IP of wtHspB8 was detected only in cells transfected with both Ddx20 and wtHspB8, at both low and high stringency conditions (lane 3 in panels IV and VI, respectively). At high stringency conditions, the amount of co-immunoprecipitated HspB8 was less compared to low stringency conditions. This co-IP of wtHspB8 with Ddx20 suggested interaction between these proteins.
The ability of both disease-associated muHspB8 forms (K141EHspB8, construct 7; K141NHspB8, construct 8) to bind to Ddx20 was tested by the same method (lanes 5–8). The controls showed that both forms of muHspB8 were expressed to a similar level as wtHspB8 (panel II) and that the immunoprecipitation of Ddx20 occurred as expected, at both low and high stringency conditions (panels III and V, respectively). Co-IP of K141EHspB8 or K141NHspB8 was detected only in cells transfected with both Ddx20 and muHspB8 forms at both low and high stringency conditions (lanes 5 and 6 in panels IV and VI). At low stringency conditions, similar amounts of both forms of muHspB8 were detected as compared to wtHspB8 (panel IV). At high stringency conditions, however, slightly more K141EHspB8 and considerably more K141NHspB8 were found to co-immunoprecipitate with Ddx20, as compared to wtHspB8 (panel VI). Similar results were obtained by the reciprocal experiment, the co-IP of Ddx20 with wtHspB8 or muHspB8 forms (cf. Fig. 5).
Taken together, the co-IP data suggest interaction between the various HspB8 species and Ddx20. The strength of the interaction with Ddx20 appears to increase in the order wtHspB8 < K141EHspB8 < K141NHspB8.
3.4 Recruitment of HspB8 and Ddx20 into high-molecular-mass complexes
As both HspB8 and Ddx20 are components of larger supramolecular structures (Pellizzoni et al. 2002; Fontaine et al. 2005; Gubitz et al. 2004; Sun et al. 2004), other components of these structures may play a role in the interaction of both proteins. In order to learn about the possible involvement of additional components, the proteins of HEK293T cells expressing both Ddx20 and HspB8 species were chemically cross-linked with the homo-bifunctional reagent BMH that reacts with free sulfhydryls of cysteines. Human Ddx20 contains three cysteines in its C-terminal part which has been identified in the YTH screen as site interacting with HspB8 (Fig. 1b), and human HspB8 contains three cysteines, one each in the N terminus, the α-crystallin domain, and the C terminus (Fig. 1a). This distribution of reactive cysteines renders BMH a suitable reagent for cross-linking these two proteins.
HEK293T cells were doubly transfected to express both Xpress-tagged Ddx20 (construct 5), and myc-tagged wtHspB8 (construct 6), K141EHspB8 (construct 7), or K141NHspB8 (construct 8), or the cells were singly transfected for control purposes. Cross-linked proteins were analyzed by SDS-PAGE/Western blotting using anti-Xpress-and anti-myc-specific antibodies (Fig. 3). In control cells (not treated with BMH) expressing only Xpress-tagged Ddx20, one Xpress-positive band representing Ddx20 at ∼100 kDa was detected (Fig. 3a, upper panel, lane 1). While cross-linking of proteins in such cells resulted in the appearance of an additional poorly resolved band above 250 kDa, this did not result in any bands in the 120 to 250 kDa region (lane 2). Co-expression of Ddx20 with either wtHspB8, K141EHspB8, or K141NHspB8 had no visible effects on Ddx20 in non-cross-linked cells (lanes 3–5, respectively). Cross-linking in doubly transfected cells resulted in the appearance of three major Ddx20-containing bands in the 120 to 250 kDa region (bands 1–3) and in poorly resolved material above 250 kDa (band region 4; lanes 6–8). Thus, cross-linking of Ddx20 into several high-molecular-mass species depends on the presence of wtHspB8 or muHspB8. Equal loading of the samples was verified by visualization of the actin bands (lower panel).

Formation of heterogeneous high-molecular-mass complexes containing Ddx20 and HspB8 species. a HEK293T cells were singly or doubly transfected with vector cDNA of Xpress-tagged Ddx20 and myc-tagged HspB8 species as indicated using the same constructs as in Fig. 2. Forty-eight hours later, cells were treated for 30 min with the cross-linker BMH, and the cross-linked proteins were analyzed by SDS-PAGE/Western blotting using an Xpress-specific antibody to detect Ddx20. Only in the presence of either wtHspB8 or muHspB8, Ddx20 was cross-linked into high-molecular mass material (bands 1–3, band region 4 in lanes 6–8). On the same blots, actin was visualized to demonstrate equal loading of protein onto gels. b The experiment was performed as in (a) using a myc-specific antibody to detect the various HspB8 forms on the Western blots. Only in the presence of Ddx20, were wtHspB8 or muHspB8 cross-linked into high-molecular mass material (bands 2, 3 in lanes 10–12), in addition to the formation of HspB8 dimers (upper panel). Analysis of the same samples revealed that actin was not cross-linked into any high-molecular weight material suggesting that cross-linking of the HspB8 species with Ddx20 resulted from a specific interaction, and not from a general, non-specific cross-linking of cell proteins (lower panel). c The same sample as shown in lanes 6 and 10 in (a) and (b), respectively, was loaded onto two adjacent lanes of a SDS gel. One lane of the Western blot was developed for Ddx20, the other for wtHspB8. The identical position of bands 2 and 3 suggests that this cross-linked material contains both Ddx20 and wtHspB8. The positions of molecular mass markers (kDa), Ddx20 monomers, and of HspB8 monomers and dimers are indicated in a–c. The results are representative of three independent experiments
The samples of lanes 3–8 (Fig. 3a) and additional controls (HspB8 species alone, without and with BMH treatment), were analyzed for the shift of HspB8 species into the high-molecular-mass material (Fig. 3b, upper panel). Expression of either form of HspB8 alone or together with Ddx20 yielded only signals at the position of the HspB8 monomers (lanes 1–6). Cross-linking of either form of HspB8 alone yielded signals at the position corresponding to HspB8 dimers (lanes 7–9), similar to what we observed previously (Sun et al. 2004). Cross-linking of either HspB8 species in the presence of Ddx20 resulted in the appearance of additional high-molecular-mass bands in the 120 to 250 kDa region (bands 2 and 3), indicating cross-linking of HspB8 with Ddx20. Again, equal loading of the samples was verified by visualization of the actin bands (lower panel). After cross-linking, no actin-containing material was found in the high-molecular mass region thus suggesting that the cross-linked HspB8–Ddx20 species resulted from a specific interaction and not from non-specific cross-linking of cell proteins. In order to confirm that the high-molecular-mass bands contain material from both HspB8 and Ddx20, the same cross-linked sample shown in lanes 6 and 10 in Fig. 3a and b, respectively, was loaded onto two adjacent lanes of an SDS gel, and the blot was developed for Xpress (Ddx20) and myc (HspB8)(Fig. 3c). Bands 2 and 3 contained both wtHspB8 and Ddx20 thus suggesting interaction of both proteins. The apparent molecular masses of bands 2 and 3 are greater than the sum of HspB8 + Ddx20 (approximately 125 kDa) indicating the presence of additional components in these cross-linked complexes. This is not unexpected given the known supramolecular structure of both the sHSP complexes and the SMN complexes. Candidates are another subunit of HspB8, another sHSP, or components of the SMN complexes. Interestingly, a fraction of the Ddx20-containing high-molecular-mass material that formed in the presence of HspB8 did not contain HspB8 (band 1, band region 4). This suggests that HspB8 induced a rearrangement of the Ddx20-containing complexes, although their nature is not known. The cross-linking experiments did not reveal any differences between wtHspB8 and muHspB8.
Taken together, the cross-linking data confirmed that wild-type and mutant forms of HspB8 and Ddx20 are interacting proteins. Additionally, these data suggest that other, although unknown, factors are involved in the HspB8–Ddx20 interaction.
3.5 Interaction of wtHspB8 and muHspB8 with Ddx20 in live cells
Interaction of Ddx20 with HspB8 in live cells requires the presence of at least a fraction of both proteins in the same cell compartment. In order to determine the subcellular location, HEK293T cells were singly (not shown) and doubly transfected with Ddx20 and the various HspB8 species fused to the small tags Xpress and FLAG, which are not expected to affect the intracellular locations. Twenty-four hours after transfection, expressed Xpress-tagged Ddx20 (construct 5) and FLAG-tagged wtHspB8 (construct 9), K141EHspB8 (construct 10), and K141NHspB8 (construct 11) all showed largely uniform, cytoplasmic distribution in most of the cells (Fig. 4a). Thus, all expressed proteins occur in the cytoplasm and may interact.

In vivo interaction of wtHspB8 and muHspB8 species with Ddx20. a HEK293T cells grown on coverslips were doubly transfected with vector cDNA of Xpress-tagged Ddx20 (construct 5) and FLAG-tagged wtHspB8 (construct 9), K141EHspB8 (construct 10), or K141NHspB8 (construct 11), as indicated. Twenty-four hours later, cells were fixed and processed for immunostaining. In the selected cells, expressed Ddx20 (red) and all HspB8 species (green) showed a relatively even distribution in the cytoplasm (co-localization in yellow). For comparison, nuclei of the same cells stained with DAPI are shown. The bar indicates 50 μM. b HEK293T cells grown on glass bottom culture plates were doubly transfected with vector cDNA of CIT-tagged Ddx20 (construct 12) and CFP-tagged wtHspB8 (construct 13), K141EHspB8 (construct 14), or K141NHspB8 (construct 15), or with control vectors, as indicated. Ten hours later, in vivo images of selected cells with relatively even cytoplasmic distribution of the expressed proteins were collected. Such cells were used for the qFRET analysis as shown in (d). c SDS-PAGE/Western blotting of the cells as shown in (b). For visualization of the HspB8-CFP species, an anti-GFP antibody was used. For visualization of the Ddx20-CIT, an anti-Ddx20 antibody was used which detected also the endogenous Ddx20 (control lane). Note that the transfected cells contained similar amounts of wtHspB8-CFP, K141EHspB8-CFP, and K141NHspB8-CFP (left panel), and also similar amounts of Ddx20-CIT (right panel). The positions of molecular mass marker proteins are indicated. d AAFE values for the interactions of Ddx20 with wtHspB8, K141EHspB8, and K141NHspB8 as determined by the qFRET method. The used cells and constructs were as in (b). One-way ANOVA analysis revealed significant differences between the four groups, control 1, wtHspB8, K141EHspB8, and K141NHspB8 [F(5, 234) = 269.25; P < 0.001]. Post-hoc pairwise group comparisons showed significant differences between the sample values and the control (P < 0.001), between K141NHspB8 and wtHspB8 (P < 0.001), and between K141NHspB8 and K141EHspB8 (P < 0.001). By this method, no significant difference between K141EHspB8 and wtHspB8 (P = 0.099) was revealed, while the direct comparison between both sample groups by the Student’s t test indicated a significant difference (P = 0.033)
In order to confirm the Ddx20–HspB8 interaction in live cells, and also to detect possible differences between wtHspB8 and muHspB8 binding to Ddx20, the in vivo qFRET method was applied (Hoppe et al. 2002). This is a stoichiometric method to determine the relative fractions of energy donor and acceptor molecules in complex in live cells using CIT (energy acceptor) and CFP (energy donor) fusion proteins. HEK293T cells were co-transfected with vector cDNAs to co-express Ddx20-CIT (construct 12) and wtHspB8-CFP (construct 13), K141EHspB8-CFP (construct 14), or K141NHspB8-CFP (construct 15). Control cells were transfected with the “empty” CIT and CFP vectors (control 1), with wtHspB8-CFP (construct 13) and the "empty" CIT vector (control 2), or with Ddx20-CIT (construct 12) and the "empty" CFP vector (control 3).
Previously, we had determined that the used K141EHspB8-CFP and K141NHspB8-CFP constructs showed an increased (∼4% and ∼37%, respectively) tendency to form inclusion bodies or aggresomes, as compared to wtHspB8 (Fontaine et al. 2006). Increased tendency of aggresome formation was also reported for endogenous muHspB8 in fibroblasts derived from affected patients (Irobi-Devolder et al. 2008). The expressed Ddx20-CIT construct as used in this study also tended to form aggresomes. Co-expression of HspB8-CFP species and Ddx20-CIT resulted in the formation of aggresomes affecting virtually all cells 24 h after transfection (not shown). Since the aggresomes may interfere with the applied interaction assay, we made efforts to reduce their formation. Aggresome formation of ectopically expressed proteins is a time-dependent process starting across the cytoplasm with the formation of multiple foci that subsequently coalesce into large amorphous perinuclear aggregates (Chávez-Zobel et al. 2003). Approximately 10 h after transfection we obtained a sufficient number of cells with apparently even distribution of both fusion proteins. Selected cell images as they were used for the qFRET measurements are shown in Fig. 4b. SDS-PAGE/Western blotting revealed the presence of similar amounts of the different HspB8-CFP species and of Ddx20-CIT in the cells used for the qFRET measurements (Fig. 4c). The AAFE values obtained for all tested interactions were significantly different from the negative controls thus indicating interaction of Ddx20 with wtHspB8 and muHspB8 (Fig. 4d). Results from one-way ANOVA revealed that there was significant difference in AAFE values between the groups (control 1, wtHspB8, K141EHspB8, and K141NHspB8). Post-hoc pairwise group comparisons showed that the fraction of K141NHspB8 in complex with Ddx20 is significantly higher than that of wtHspB8 and K141EHspB8, although this method did not reveal a significant difference between the fractions of K141EHspB8 and wtHspB8 with Ddx20 in complex (P = 0.099). Using the Student’s t test, direct comparison of the AAFE values of K141EHspB8 and wtHspB8 indicated that this difference is significant (P = 0.033). In spite of this uncertainty, in conjunction with the co-IP data (Fig. 2), our results suggest that both mutant forms of HspB8 show aberrantly increased interaction with Ddx20.
Taken together, the presented data in live cells support the hypothesis that HspB8 interacts with Ddx20. They also support an increased interaction stoichiometry of muHspB8 species with Ddx20, as compared to wtHspB8. Even if formation of pre-aggresome material contributes to these data, they identify differences in the association behavior of wtHspB8 and the muHspB8 forms. Similar to the results of the co-IP experiments (Fig. 2), the strength of the interaction with Ddx20 appears to increase in the order wtHspB8 < K141EHspB8 < K141NHspB8.
3.6 RNA involvement in the HspB8–Ddx20 interaction
DEAD box proteins are a subgroup of the DExH/D protein family which is characterized by evolutionary conserved sequence motifs (Jankowsky and Jankowsky 2000). Most DEAD box proteins were implicated in various RNA-related processes (see below), and Ddx20 binds, like other DEAD box proteins, RNA and has an RNA helicase activity (Grundhoff et al. 1999; Yan et al. 2003). We therefore reasoned that RNA may be involved in the HspB8–Ddx20 interaction. In order to test this, cell extracts containing Ddx20–HspB8 complexes were treated with RNase followed by immunoprecipitation experiments. HEK293T cells were doubly transfected to express Xpress-tagged Ddx20 (construct 5) and myc-tagged wtHspB8 (construct 6), K141EHspB8 (construct 7), or K141NHspB8 (construct 8). The expression of the transfected fusion proteins (Fig. 5, panels I and II) and the immunoprecipitation of HspB8 species in the presence vs. absence of RNase was verified by SDS-PAGE/Western blotting (panel III). Immunoprecipitation of wtHspB8 resulted in a similar co-IP of Ddx20 with and without RNase treatment of the cell extracts (panel IV), suggesting that RNA either is not involved in this interaction, or that both proteins, HspB8 and Ddx20 together, protect bound RNA from the RNase. In contrast, using K141EHspB8 and K141NHspB8 under otherwise identical conditions revealed that co-IP of Ddx20 is largely sensitive to the treatment of the cell extracts with RNase.

Involvement of RNA in the Ddx20–HspB8 interaction. HEK293T cells were singly or doubly transfected with vector cDNA of Xpress-tagged Ddx20 (construct 5) and myc-tagged wtHspB8 (construct 6), K141EHspB8 (construct 7), or K141NHspB8 (construct 8), as indicated. Forty-eight hours later, the presence of the expressed proteins in cell extracts was confirmed by SDS-PAGE/Western blotting using Xpress- and myc-specific antibodies (I, II). Immunoprecipitation of the various HspB8 species after RNase treatment vs. the controls without treatment resulted in similar amounts of precipitated proteins (III). The amounts of Ddx20 co-immunoprecipitated with wtHspB8 with and without RNase treatment were similar (IV, lanes 3 and 4), while the amounts of Ddx20 co-immunoprecipitated with K141EHspB8 and K141NHspB8 were decreased after RNase treatment as compared to the untreated samples (IV, lanes 5–8). The blot is representative of three independent experiments
Collectively, these data tentatively suggest that RNA is involved in the interaction of HspB8 with Ddx20, and that muHspB8 species no longer protect bound RNA from the RNase.
4 Discussion
The known missense mutations in both HspB8 and HspB1 result in clinically indistinguishable forms of MND, dHMN, and CMT (Benndorf and Welsh 2004; Irobi et al. 2004b; Tang et al. 2005). Three facts suggest that both proteins are part of the same biochemical mechanism leading to disease: (1) HspB8 and HspB1 interact with one another (Sun et al. 2004); (2) as far as studied, mutations in either protein result in abnormally increased interaction between both proteins (Irobi et al. 2004b; Fontaine et al. 2006); and (3) among all proteins in the databases, HspB8 and HspB1 are the most similar to each other (Sun et al. 2004). Thus, abnormally altered interaction between HspB8 and HspB1 may be at the base of these MNDs. This concept is supported by studies on the intersubunit contacts of sHSPs. A number of disease-associated mutations in HspB8 (K141E and K141N) and HspB1 (S135F, R136W, R140G, and K141Q) are positioned in the β6/β7 strands of their α-crystallin domains (Benndorf 2010). This region was shown to be critical for the homo-dimer formation of several sHSPs, including HspB8 and HspB1, and possibly also for the hetero-dimer formation (Mymrikov et al. 2010). The mutation K141E in HspB8 resulted in significant changes in the protein structure and properties that affected also the intersubunit contacts (Kim et al. 2006; Kasakov et al. 2007). A similar situation was found for the R120G mutation in HspB5 (references in Mymrikov et al. 2010). This concept of abnormal sHSP interactions, however, does not explain the molecular mechanisms by which mutations in these sHSPs lead to the slow and selective loss of motor neurons. To identify potential factors in this process, we searched for proteins that interact with HspB8 by using a YTH screen. Among the co-transformants was a yeast clone containing the C-terminal part of the DEAD box protein Ddx20 (gemin3).
By independent methods, we have verified the HspB8–Ddx20 interaction in both cell extracts and live cells. The co-IP and qFRET data also indicate increased interaction of disease-associated muHspB8 forms with Ddx20 as compared to wtHspB8. Additionally, the cross-linking experiments suggest that both HspB8 and Ddx20 were recruited into the same high-molecular-mass structures implying that the HspB8–Ddx20 interaction occurs, at least in part, in larger structures or complexes. This outcome is not entirely unexpected, since Ddx20 and sHSPs in general are known to be components of supramolecular structures. While the Ddx20-containing structure, the SMN complexes, is relatively well defined (see below), the precise nature of the HspB8-containing structure(s) remains elusive. Associations with the identified interacting sHSPs, including HspB1, HspB2, HspB5, HspB6, and HspB7, or with the other identified interacting proteins, Sam68, TLR4, or Bag3, may play a role (Sun et al. 2004; Fontaine et al. 2005; Badri et al. 2006; Roelofs et al. 2006; Carra et al. 2008). Whether Ddx20 competes with these proteins on a limited number of common binding sites on HspB8 or whether some or all of these proteins form a common complex, is not known.
The DEAD box proteins form a subgroup of the DExD/H box family of helicases and have an ATP-dependent RNA unwinding (helicase) activity (Grundhoff et al. 1999; Jankowsky and Jankowsky 2000; Yan et al. 2003). DExD/H box proteins may act as RNA chaperones and support optimal RNA structures. Processes that involve DExD/H box proteins include pre-mRNA processing, ribosome biogenesis, RNA turnover, RNA export and translation, and the organization of complex RNA structures. The helicase core sequences of DExD/H box proteins are conserved across all kingdoms, as opposed to the flanking domains which are highly divergent binding sites for a number of other proteins. Through such interactions DExD/H box proteins have the ability to become involved in additional processes, e.g., in regulation of transcription. These "secondary" functions appear to be independent of the RNA helicase activity.
Ddx20 is obligatory for early embryonic development (Mouillet et al. 2008). It associates with the SMN protein, a component of the SMN complexes of which it is a core component (Charroux et al. 1999; Fuller-Pace 2006). SMN complexes are involved in assembly and processing of diverse RNPs, including snRNPs (spliceosomes), snoRNPs, hnRNPs, transcriptosomes, and miRNPs (Pellizzoni et al. 2002; Gubitz et al. 2004; Nelson et al. 2004; Shpargel and Matera 2005; Wan et al. 2005). Through its interaction with Ago2, Ddx20 may be involved in gene silencing by RNA interference (Donker et al. 2007). In addition to its role in RNA processing, Ddx20 may play diverse roles in cell biology as indicated by its interactions with a number of other proteins. Ddx20 can interact with several nuclear proteins and transcription factors, including Epstein–Barr virus nuclear proteins EBNA2 and EBNA3C (Grundhoff et al. 1999), the orphan nuclear receptor steroidogenic factor SF-1 (Ou et al. 2001), early growth response transcription factors (Egr1-4; Gillian and Svaren 2004), transcription factor FOXL2 (Lee et al. 2005b), and the mitogenic Ets repressor METS and several related components of a co-repressor complex including N-coR, Sin3A, and the histone-deacetylases (HDAC) 2 and 5 (Klappacher et al. 2002). As far as has been determined, interactions with these factors are mediated by the C-terminal flanking domain of Ddx20 (cf. Fig. 1) that has an intrinsic transcriptional repression activity and may involve sumoylation via an E3 SUMO ligase (Lee et al. 2005a; Fuller-Pace et al. 2007). An overall picture emerges that Ddx20 is a dual function molecule, with its RNA helicase activity being located in the N-terminal part and its transcriptional and translational regulation activities being located in the C-terminal part. The interaction of wtHspB8 with Ddx20 in its C-terminal part theoretically can modulate all functions executed by Ddx20. Accordingly, abnormally increased interaction of muHspB8 with Ddx20 may contribute to the disease phenotype. A scheme of Ddx20-interacting proteins and cellular processes that may be affected by binding of HspB8 species is shown in Fig. 6a.

Schematic of protein interactions involving Ddx20 and HspB8. a Identified proteins that interact with Ddx20 and the cellular processes in which they are involved. Upon binding to Ddx20, HspB8 theoretically may modulate these cellular processes. Gemin4 and SMN protein are components of the SMN complexes that are involved in spliceosome assembly and RNP processing. Upon binding to gemin4 and eIF2C2, Ddx20 forms complexes with microRNAs that are involved in gene silencing and translational repression, and upon binding to Ago2, Ddx20 may modulate siRNA biogenesis. Ddx20 can also interact with the nuclear proteins and transcription factors that are involved in transcriptional repression, including Epstein–Barr virus nuclear proteins EBNA2 and 3C, nuclear receptor steroidogenic factor SF-1, transcription factor FOXL2, early growth response transcription factors Egr1-4, and the mitogenic Ets repressor METS with its co-repressor components N-coR and Sin3A, and with the histone-deacetylases HDAC 2 and 5. b Through protein interactions, four etiologic factors HspB1, HspB8, HspB5, and SMN protein have been linked that are associated with the various forms of motor neuropathy (dHMN, CMT, SMA) or myopathy. Based on the identified interactions, Ddx20 is a candidate gene for mutations in neuromuscular or muscular diseases of unknown cause. While sHSP complexes may contain various family members, the recruitment of HspB8 into true hetero-oligomeric complexes, together with HspB1 and HspB5, remains to be determined. SMN complexes contain a number of additional core components and associated proteins (not shown). The HspB8–Ddx20 interaction apparently involves RNA. c Changed conformation of muHspB8 leads to increased interactions with HspB1 and HspB5 (Fontaine et al. 2006), and with Ddx20. At the same time, the associated RNA becomes exposed to RNase
Homozygous deletion or loss-of-function mutations in the SMN1 gene cause SMA, another MND and one of the most prevalent genetic causes of infant mortality (Lefebvre et al. 1995; Gubitz et al. 2004). Thus, the interaction data presented in this study link the etiologic factors HspB8 and HspB1 with SMN protein suggesting that these mutations affect a common biochemical mechanism, be it through a common protein complex or signaling pathway. Because HspB5 interacts with both HspB8 and HspB1, it may also be a component of this complex or pathway (Simon et al. 2007). Figure 6b shows a scheme of the linked etiologic factors HspB8, HspB1, HspB5, and SMN protein. However, if HspB8 forms true hetero-oligomeric complexes with HspB1 and HspB5 remains to be determined. Based on the identified interactions, DDX20 is a candidate gene for mutations in neuromuscular or muscular diseases of unknown cause.
With SMN protein, HspB8 and HspB1 being expressed in most if not all cells in mammals, it is not clear why reduced expression of SMN protein and expression of the mutant sHSPs affect specifically the function of motor neurons. The prominent and specific role of both SMN protein and sHSPs (in particular HspB1) in growth, branching, axogenesis, and survival of neurons was demonstrated previously, tentatively suggesting a specialized function of these proteins in motor neurons (Benn et al. 2002; Pagliardini et al. 2000; McWhorter et al. 2003; Williams et al. 2006). The transport of SMN protein- and Ddx20-containing granules into neuronal processes and growth cones could constitute such a specialized function (Zhang et al. 2003). Similarly, the involvement of SMN complexes in the transport of actin mRNPs along the axon and of snRNPs into the nucleus seems to constitute such a specialized function (Sharma et al. 2005). In mouse models of SMA, motor neuron degeneration was attributed to defects associated with the specific axonal transport, translational control, and/or regulation of stability of mRNAs rather than with defects in spliceosome assembly function (Jablonka et al. 2004). Neuropathy-associated mutant HspB1 lead to the formation of insoluble intracellular aggregates in primary neuronal cells with negative consequences for axonal structure and transport (Ackerley et al. 2006). Thus, the available data tentatively suggest an impaired axonal transport as a common motif of neuron damage in mutant SMN protein- and mutant sHSP-associated MND.
HspB5 is also expressed in most mammalian cells including neurons and muscle cells. The fact that mutations in HspB5 affect primarily muscle rather than neuronal tissues may simply result from the extraordinary high abundance of this protein in muscles (in which it may exceed 2% of total protein) as compared to neurons. For example, the ratios of HspB5: HspB1 in the rat soleus muscle (∼6.9) and heart (∼5.3) are approximately an order of magnitude higher than in the neuronal tissues of the cerebral cortex (∼0.16) and hippocampus (∼0.80) (Inaguma et al. 1995).
A startling observation is that most known MND-associated mutations in HspB8 and HspB1 are missense mutations with dominant gain-of-function characteristics, whereas MND-associated mutations in the SMN1 gene have recessive loss-of-function characteristics (Gubitz et al. 2004; Irobi et al. 2004b; Tang et al. 2005; Benndorf 2010). In most SMA patients, both copies of exon 7 of the SMN1 gene are absent, while in the remaining SMA patients nonsense, missense, and frameshift mutations are distributed over the entire length of the protein (Prior 2007). This discrepancy is currently not understood, the more so if HspB8, HspB1, and SMN protein would act through the same mechanism or signaling pathway as proposed in this study. The formation of hetero-oligomeric sHSP complexes may explain the genetic dominance seen with most of the mutant sHSPs. Recruitment of one mutant sHSP subunit may compromise both structure and function of the whole sHSP complex (Fig. 6c). The result may be the formation of cytoplasmic inclusion bodies or aggresomes as has been shown for mutant forms of HspB8, HspB1, and HspB5 (Vicart et al. 1998; Irobi et al. 2004b; Fontaine et al. 2006; Ackerley et al. 2006; Irobi-Devolder et al. 2008). These aggresomes, a hallmark of many neurodegenerative disorders, can recruit also other proteins which may include RNPs or their components, and thus prevent their proper transport along the axons of the motor neurons. Although speculative at this time, SMN complexes may become trapped in these aggresomes resulting in a depletion of functionally active SMN complexes. Such secondary depletion of the SMN complexes may have similar consequences as the loss of functional SMN complexes that result from mutations in the SMN1 gene in SMA.
The finding of RNA involvement in the interaction of muHspB8 with Ddx20 is remarkable on two accounts: it potentially implies a role for HspB8 in RNA processing, and it identifies a difference between muHspB8 (interaction with Ddx20 is RNase-sensitive) and wtHspB8 (interaction with Ddx20 is RNase-insensitive). The fact that the muHspB8–Ddx20 interaction is sensitive to RNase (Fig. 5) does not necessarily contradict the findings shown in Figs. 2 and 4 according to which muHspB8 has an increased interaction with Ddx20, as compared to wtHspB8. wtHspB8 may bind to RNA which itself binds to Ddx20, and muHspB8 might bind abnormally stronger to this RNA. The formation of these ternary complexes involving Ddx20, RNA, and wtHspB8 or muHspB8 would result in co-IP and qFRET data as shown in Figs. 2 and 4, respectively. While the conformation of wtHspB8 would protect the RNA in the ternary complex, muHspB8 would adopt an abnormal conformation that no longer can protect the RNA from RNase, in spite of the increased binding, thus resulting in the disassembly of the ternary complexes in the presence of RNase. These findings have been included in the schematic in Fig. 6b and c.
While not in the mainstream of research on sHSPs, there are several other reports concerning their role in RNA functions and processing. HspB5 was found to interact with the SMN protein, and both proteins co-localized in nuclear speckles which are sites of RNA processing (den Engelsman et al. 2005). This finding is an independent confirmation of a linkage between a sHSP and components of the SMN complexes. Similarly, HspB1 and HspB7 were found to be recruited to the nuclear speckles (Bryantsev et al. 2007; Vos et al. 2009). In activated monocytes, HspB1 was identified as protein-binding AU-rich elements in mRNAs in AUF1 protein complexes which function in mRNA degradation (Sinsimer et al. 2008). Similarly, in association with AUF1, HspB1 bound to the 3’-untranslated regions (UTR) of the cell-death-inhibiting RNA CDIR and of the c-YES mRNA in HeLa cells and breast cancer cells, respectively (Shchores et al. 2002; Sommer et al. 2005). In the latter case, this resulted in degradation of the c-YES mRNA. In contrast, a mimic of phosphorylated HspB1 stabilized a chimeric mRNA which contained a 3’-UTR element of the cyclooxygenase-2 mRNA (Lasa et al. 2000). HspB1 was also shown to enhance recovery of splicing activity after heat shock by regulating the splicing factor SRp38 (Marin-Vinader et al. 2006). HspB1 inhibited translation during heat shock by specifically binding the eukaryotic initiation factor 4G and facilitating dissociation of cap-initiation complexes (Cuesta et al. 2000). Finally, HspB8 was shown to interact with Sam68, another RNA-binding protein that has been implicated in cell proliferation and tumorigenesis (Badri et al. 2006).
In summary, results presented in this study link the three etiologic factors, HspB8, HspB1, and SMN protein. Mutations in each of these genes are associated with related disease phenotypes and are proposed to affect a common biochemical mechanism or signaling pathway involving both sHSP complexes and SMN complexes, although the underlying biochemical processes remain to be determined.
References
Ackerley S, James PA, Kalli A, French S, Davies KE, Talbot KA (2006) A mutation in the small heat-shock protein HSPB1 leading to distal hereditary motor neuronopathy disrupts neurofilament assembly and the axonal transport of specific cellular cargoes. Hum Mol Genet 15:347–354
Badri KR, Modem S, Gerard HC, Khan I, Bagchi M, Hudson AP, Reddy TR (2006) Regulation of Sam68 activity by small heat shock protein 22. J Cell Biochem 99:1353–1362
Benn SC, Perrelet D, Kato AC, Scholz J, Decosterd I, Mannion RJ, Bakowska JC, Woolf CJ (2002) Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 36:45–56
Benndorf R (2010) HspB1 and HspB8 Mutations in Neuropathies. In: Simon S, Arrigo A-P (eds) Small stress proteins in human diseases. Nova Publishers (NY) (in press)
Benndorf R, Welsh MJ (2004) Shocking degeneration. Nat Genet 36:547–548
Benndorf R, Engel K, Gaestel M (2000) Analysis of small Hsp phosphorylation. Methods Mol Biol 99:431–434
Benndorf R, Sun X, Gilmont RR, Biederman KJ, Molloy MP, Goodmurphy CW, Cheng H, Andrews PC, Welsh MJ (2001) HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 (3DHSP27). J Biol Chem 276:26753–26761
Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C (2000) Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2:645–652
Bryantsev AL, Chechenova MB, Shelden EA (2007) Recruitment of phosphorylated small heat shock protein Hsp27 to nuclear speckles without stress. Exp Cell Res 313:195–209
Carra S, Sivilotti M, Chavez-Zobel AT, Lambert H, Landry J (2005) HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet 14:1659–1669
Carra S, Seguin SJ, Lambert H, Landry J (2008) HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem 283:1437–1444
Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2α phosphorylation. J Biol Chem 284:5523–5532
Charroux B, Pellizzoni L, Perkinson RA, Shevchenko A, Mann M, Dreyfuss G (1999) Gemin3: A novel DEAD box protein that interacts with SMN, the spinal muscular atrophy gene product, and is a component of gems. J Cell Biol 147:1181–1193
Chávez-Zobel AT, Loranger A, Marceau N, Thériault JR, Lambert H, Landry J (2003) Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G αB-crystallin mutant. Hum Hum Mol Genet 12:1609–1620
Chowdary TK, Raman B, Ramakrishna T, Rao CM (2004) Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem J 381(Pt2):379–387
Cuesta R, Laroia G, Schneider RJ (2000) Chaperone hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev 14:1460–1470
den Engelsman J, Gerrits D, de Jong WW, Robbins J, Kato K, Boelens WC (2005) Nuclear import of αB-crystallin is phosphorylation-dependent and hampered by hyperphosphorylation of the myopathy-related mutant R120G. J Biol Chem 280:37139–37148
Dierick I, Irobi J, De Jonghe P, Timmerman V (2005) Small heat shock proteins in inherited peripheral neuropathies. Ann Med 37:413–422
Donker RB, Mouillet JF, Nelson DM, Sadovsky Y (2007) The expression of Argonaute2 and related microRNA biogenesis proteins in normal and hypoxic trophoblasts. Mol Hum Reprod 13:273–279
Fontaine J-M, Rest JS, Welsh MJ, Benndorf R (2003) The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones 8:62–69
Fontaine J-M, Sun X, Benndorf R, Welsh MJ (2005) Interactions of HSP22 with HSP20, αB-crystallin, and HSPB3. Biochem Biophys Res Commun 337:1006–1011
Fontaine J-M, Sun X, Hoppe AD, Simon S, Vicart P, Welsh MJ, Benndorf R (2006) Abnormal small heat shock protein interactions involving neuropathy-associated HSP22 (HSPB8) mutants. FASEB J 20:2168–2170
Fuller-Pace FV (2006) DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res 34:4206–4215
Fuller-Pace FV, Jacobs AM, Nicol SM (2007) Modulation of transcriptional activity of the DEAD-box family of RNA helicases, p68 (Ddx5) and DP103 (Ddx20), by SUMO modification. Biochem Soc Trans 35(Pt6):1427–1429
Gillian AL, Svaren J (2004) The Ddx20/DP103 dead box protein represses transcriptional activation by Egr2/Krox-20. J Biol Chem 279:9056–9063
Grundhoff AT, Kremmer E, Türeci O, Glieden A, Gindorf C, Atz J, Mueller-Lantzsch N, Schubach WH, Grässer FA (1999) Characterization of DP103, a novel DEAD box protein that binds to the Epstein-Barr virus nuclear proteins EBNA2 and EBNA3C. J Biol Chem 274:19136–19144
Gubitz AK, Feng W, Dreyfuss G (2004) The SMN complex. Exp Cell Res 296:51–56
Harding AE, Thomas PK (1980) The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 103:259–280
Hase M, Depre C, Vatner SF, Sadoshima J (2005) H11 has dose-dependent and dual hypertrophic and proapoptotic functions in cardiac myocytes. Biochem J 388:475–483
Hedhli N, Wang L, Wang Q, Rashed E, Tian Y, Sui X, Madura K, Depre C (2008) Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc Res 77:497–505
Hoppe A, Christensen K, Swanson JA (2002) Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys J 83:3652–3664
Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, Teraoka K, Chikamori T, Yamashina A, Kimura A (2006) αB-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun 342:379–386
Inaguma Y, Hasegawa K, Goto S, Ito H, Kato K (1995) Induction of the synthesis of hsp27 and αB- crystallin in tissues of heat-stressed rats and its suppression by ethanol or an alpha 1-adrenergic antagonist. J Biochem 117:1238–1243
Irobi J, De Jonghe P, Timmerman V (2004a) Molecular genetics of distal hereditary motor neuropathies. Hum Mol Genet 13(Suppl 2):R195–R202
Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, Vennekens K, Mazanec R, Tournev I, Hilton-Jones D, Talbot K, Kremensky I, Van Den Bosch L, Robberecht W, Van Vandekerckhove J, Broeckhoven C, Gettemans J, De Jonghe P, Timmerman V (2004b) Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet 36:597–601
Irobi-Devolder J, Krishnan J, Almeida-Souza L-SC, Dierick I, Ceuterick-de Groote C, van den Bosch L, Timmermans J, Robberecht W, de Jonghe P, Janssens S, Timmerman VL. Mutant heat shock protein HSPB8 induces aggregation and a pro-apoptotic phenotype in distal motor neuropathy. 38th Annual Meeting of the Society for Neuroscience, Washington, DC, Nov. 15–19, 2008.
Jablonka S, Wiese S, Sendtner M (2004) Axonal defects in mouse models of motoneuron disease. J Neurobiol 58:272–286
Jankowsky E, Jankowsky A (2000) The DExH/D protein family data base. Nucleic Acid Res 28:333–334
Jia Y, Ransom RF, Shibanuma M, Liu C, Welsh MJ, Smoyer WE (2001) Identification and characterization of hic-5/ARA55 as an hsp27 binding protein. J Biol Chem 276:39911–39918
Kappé G, Franck E, Verschuure P, Boelens WC, Leunissen JA, de Jong WW (2003) The human genome encodes 10 α-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 8:53–61
Kappé G, Boelens WC, de Jong, WW (2010) Why proteins without an a-crystallin domain should not be included in the human small heat shock protein family HSPB. Cell Stress Chaperones, (in press)
Kasakov AS, Bukach OV, Seit-Nebi AS, Marston SB, Gusev NB (2007) Effect of mutations in the beta5-beta7 loop on the structure and properties of human small heat shock protein HSP22 (HspB8, H11). FEBS J 274:5628–5642
Kim MV, Seit-Nebi AS, Marston SB, Gusev NB (2004) Some properties of human small heat shock protein Hsp22 (H11 or HspB8). Biochem Biophys Res Commun 315:796–801
Kim MV, Kasakov AS, Seit-Nebi AS, Marston SB, Gusev NB (2006) Structure and properties of K141E mutant of small heat shock protein HSP22 (HspB8, H11) that is expressed in human neuromuscular disorders. Arch Biochem Biophys 454:32–41
Klappacher GW, Lunyak VV, Sykes DB, Sawka-Verhelle D, Sage J, Brard G, Ngo SD, Gangadharan D, Jacks T, Kamps MP, Rose DW, Rosenfeld MG, Glass CK (2002) An induced Ets repressor complex regulates growth arrest during terminal macrophage differentiation. Cell 109:169–180
Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR (2000) Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol Cell Biol 20:4265–4274
Lee MB, Lebedeva LA, Suzawa M, Wadekar SA, Desclozeaux M, Ingraham HA (2005a) The DEAD-box protein DP103 (Ddx20 or Gemin-3) represses orphan nuclear receptor activity via SUMO modification. Mol Cell Biol 25:1879–1890
Lee K, Pisarska MD, Ko JJ, Kang Y, Yoon S, Ryou SM, Cha KY, Bae J (2005b) Transcriptional factor FOXL2 interacts with DP103 and induces apoptosis. Biochem Biophys Res Commun 336:876–881
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, Le Paslier D, Frezal J, Cohen D, Weissenbach J, Munnich A, Melki J (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–165
Liu C, Gilmont RR, Benndorf R, Welsh MJ (2000) Identification and characterization of a novel protein from Sertoli cells, PASS1, that associates with mammalian small stress protein hsp27. J Biol Chem 275:18724–18731
Marin-Vinader L, Shin C, Onnekink C, Manley JL, Lubsen NH (2006) Hsp27 enhances recovery of splicing as well as rephosphorylation of SRp38 after heat shock. Mol Biol Cell 17:886–894
McWhorter ML, Monani UR, Burghes AH, Beattie CE (2003) Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162:919–931
Mouillet JF, Yan X, Ou Q, Jin L, Muglia LJ, Crawford PA, Sadovsky Y (2008) DEAD-box protein-103 (DP103, Ddx20) is essential for early embryonic development and modulates ovarian morphology and function. Endocrinology 149:2168–21675
Mymrikov EV, Bukach OV, Seit-Nebi AS, Gusev NB (2010)The pivotal role of the beta7 strand in the intersubunit contacts of different human small heat shock proteins. Cell Stress Chaperones, (in press)
Nelson PT, Hatzigeorgiou AG, Mourelatos Z (2004) miRNP:mRNA association in polyribosomes in a human neuronal cell line. RNA 10:387–394
Ou Q, Mouillet JF, Yan X, Dorn C, Crawford PA, Sadovsky Y (2001) The DEAD box protein DP103 is a regulator of steroidogenic factor-1. Mol Endocrinol 15:69–79
Pagliardini S, Giavazzi A, Setola V, Lizier C, Di Luca M, DeBiasi S, Battaglia G (2000) Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum Mol Genet 9:47–56
Pellizzoni L, Baccon J, Rappsilber J, Mann M, Dreyfuss G (2002) Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J Biol Chem 277:7540–7545
Pilotto A, Marziliano N, Pasotti M, Grasso M, Costante AM, Arbustini E (2006) αB-crystallin mutation in dilated cardiomyopathies: low prevalence in a consecutive series of 200 unrelated probands. Biochem Biophys Res Commun 346:1115–1157
Prior TW (2007) Spinal muscular atrophy diagnostics. J Child Neurol 22:952–956
Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, van den Berg WB, Radstake TR (2006) Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol 176:7021–7027
Rogalla T, Ehrnsperger M, Préville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo A-P, Buchner J, Gaestel M (1999) Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J Biol Chem 274:18947–18956
Selcen D, Engel AG (2003) Myofibrillar myopathy caused by novel dominant negative αB-crystallin mutations. Ann Neurol 54:804–810
Sharma A, Lambrechts A, le Hao T, Le TT, Sewry CA, Ampe C, Burghes AH, Morris GE (2005) A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp Cell Res 309:185–197
Shchores K, Yehiely F, Kular RK, Kotlo KU, Brewer G, Deiss LP (2002) Cell death inhibiting RNA (CDIR) derived from a 3′-untranslated region binds AUF1 and heat shock protein 27. J Biol Chem 277:47061–47072
Shpargel KB, Matera AG (2005) Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci USA 102:17372–17377
Shy ME (2004) Charcot-Marie-Tooth disease: an update. Curr Opin Neurol 17:579–585
Simon S, Fontaine JM, Martin JL, Sun X, Hoppe AD, Welsh MJ, Benndorf R, Vicart P (2007) Myopathy-associated alphaB-crystallin mutants: abnormal phosphorylation, intracellular location, and interactions with other small heat shock proteins. J Biol Chem 282:34276–34287
Sinsimer KS, Gratacós FM, Knapinska AM, Lu J, Krause CD, Wierzbowski AV, Maher LR, Scrudato S, Rivera YM, Gupta S, Turrin DK, De La Cruz MP, Pestka S, Brewer G (2008) Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol Cell Biol 28:5223–5237
Sommer S, Cui Y, Brewer G, Fuqua SA (2005) The c-Yes 3′-UTR contains adenine/uridine-rich elements that bind AUF1 and HuR involved in mRNA decay in breast cancer cells. J Steroid Biochem Mol Biol 97:219–229
Sun X, Fontaine J-M, Rest JS, Shelden EA, Welsh MJ, Benndorf R (2004) Interaction of human HSP22 (HSPB8) with other small heat shock proteins. J Biol Chem 279:2394–2402
Sun X, Fontaine J-M, Bartl I, Behnam B, Welsh MJ, Benndorf R (2007) Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 12:307–319
Tang BS, Zhao GH, Luo W, Xia K, Cai F, Pan Q, Zhang RX, Zhang FF, Liu XM, Chen B, Zhang C, Shen L, Jiang H, Long ZG, Dai HP (2005) Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2 L. Hum Genet 116:222–224
Verschuure P, Tatard C, Boelens WC, Grongnet JF, David JC (2003) Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur J Cell Biol 82:523–530
Vicart P, Caron A, Guicheney P, Li Z, Prévost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M (1998) A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20:92–95
Vos MJ, Kanon B, Kampinga HH (2009) HSPB7 is a SC35 speckle resident small heat shock protein. Biochim Biophys Acta 1793:1343–1353
Wan L, Battle DJ, Yong J, Gubitz AK, Kolb SJ, Wang J, Dreyfuss G (2005) The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol 25:5543–5551
Williams KL, Rahimtula M, Mearow KM (2006) Heat shock protein 27 is involved in neurite extension and branching of dorsal root ganglion neurons in vitro. J Neurosci Res 84:716–723
Yan X, Mouillet JF, Ou Q, Sadovsky Y (2003) A novel domain within the DEAD-box protein DP103 is essential for transcriptional repression and helicase activity. Mol Cell Biol 23:414–423
Zhang HL, Pan F, Hong D, Shenoy SM, Singer RH, Bassell GJ (2003) Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J Neurosci 23:6627–6637
Acknowledgments
We thank L. Zhang (UM Center for Statistical Consultation and Research) for assistance with the statistical analysis of the qFRET data. The critical reading of the manuscript by Dr. D. Chandler (Columbus, OH) is gratefully acknowledged. The work was supported by Public Health Service grant P01ES11188 from the National Institute of Environmental Health Sciences to M.J.W. (PI) and R.B.; by a Munn Idea grant from the University of Michigan Comprehensive Cancer Center to R.B.; by Canadian Institutes of Health Research grant MOP-7088 to J.L.; by the French Ministry of Research and by the Association française contre les myopathies (AFM) to S.S.; and by the Centre national de la recherche scientifique (CNRS) and by the AFM grant No. 11764 to P.V.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, X., Fontaine, JM., Hoppe, A.D. et al. Abnormal interaction of motor neuropathy-associated mutant HspB8 (Hsp22) forms with the RNA helicase Ddx20 (gemin3). Cell Stress and Chaperones 15, 567–582 (2010). https://doi.org/10.1007/s12192-010-0169-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-010-0169-y