
Abstract
Safety, efficacy, and pharmacokinetics (PK) of daratumumab as a monotherapy were investigated in Japanese patients with relapsed/refractory multiple myeloma (MM). This multicenter, dose-escalation study included patients (age ≥20 years) with ≥2 prior therapies. Daratumumab was administered intravenously: 8 mg/kg (n = 4) and 16 mg/kg (n = 5). The primary endpoint was safety. Secondary endpoints included objective response, overall response rate (ORR), progression-free survival (PFS), PK, and immunogenicity. Daratumumab was well-tolerated. Eight patients experienced Grade ≥3 adverse event (AE). Four serious AEs were observed in three patients; no AEs leading to death. Infusion-related reactions occurred in four (44%) patients and were Grade 1 or 2. Mean (SD) cumulative dose of daratumumab was 132.3 (108.5) mg/kg. Median duration of follow-up was 10.5 months (range 2.3, 16.4) for 8 mg/kg cohort and 9.9 months (range 1.7, 13.2) for 16 mg/kg cohort. The ORR (44%) comprised 1 and 3 partial responses in 8 and 16 mg/kg cohorts, respectively. The median PFS was 6 months for 8 mg/kg cohort, 9.5 months for 16 mg/kg cohort. Daratumumab serum exposure was increased with increasing dose. Antibodies against daratumumab were not observed. Daratumumab was safe and well-tolerated in Japanese patients with relapsed /refractory MM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is less prevalent in Asia compared with Western countries; however, its occurrence has increased recently [1]. In Japan, the incidence of MM has increased 5–6 times during 1975–2010 [1]. Immunomodulatory agents (IMiDs) and proteasome inhibitors (PIs) are the novel agents approved for treatment of relapsed or refractory MM in Japan [1]. Introduction of these agents has led to a paradigm shift in the treatment of MM and significantly improved survival outcomes [1,2,3]. However, elderly and frail patients generally fail to achieve survival benefits [1]. Relapse and resistance are common after treatment with IMiDs and PIs and the median overall survival in patients double-refractory to these agents is approximately 9 months [4,5,6].
CD38 is a transmembrane glycoprotein that is overexpressed in hematological malignancies including MM [7]. It mediates adhesion, signaling mechanisms, and intracellular calcium metabolism in lymphoid and myeloid cells and is a potential target for MM treatments [7]. Daratumumab, a human IgG1κ monoclonal antibody binds to a CD38 epitope. The binding of daratumumab to CD38-expressing tumor cells results in complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), cell lysis by cross-linking and antibody-dependent cell phagocytosis (ADCP), and modulation of CD38 enzymatic activity [7, 8]. Daratumumab leads to the rapid and sustained elimination of highly immunosuppressive subsets of CD38+ Tregs, CD38+ MDSCs, and CD38+ regulatory B cells (Bregs) [9]. The elimination of these immunosuppressive cells and modulation of CD38 enzymatic activity leads to the increased clonal expansion of CD8+ and CD4+ T cells. Altogether, daratumumab’s converging mechanisms of actions are hypothesized to synergistically lead to the deep responses observed in patients.
In a previous phase 1/2 study, daratumumab exhibited an acceptable safety and efficacy profile in patients with relapsed or refractory MM [10]. In a phase 2, single-arm, monotherapy, open-label, multicenter study, an encouraging overall response rate (ORR 29%) and median duration of response (7.4 months) were observed in patients heavily pre-treated with other therapies, including new agents such as carfilzomib and pomalidomide [11]. Based on the findings of these studies, US FDA granted accelerated conditional approval and the European Commission granted a conditional approval to daratumumab as a monotherapy for treatment of patients with MM treated with at least three prior lines of therapy including a PI and IMiD or double-refractory to these agents [12]. More recently, daratumumab was approved by the US FDA in combination with bortezomib and dexamethasone or lenalidomide and dexamethasone in multiple myeloma patients that received at least one prior therapy based on the CASTOR and POLLUX studies [13, 14].
Based on these promising findings in the global population, this phase 1 study was conducted in Japanese patients with relapsed or refractory MM to evaluate the safety, tolerability, efficacy, immunogenicity, and PK of daratumumab.
Methods
Patients
The study included Japanese men and women aged ≥20 years with symptomatic MM according to International Myeloma Working Group (IMWG) diagnostic criteria, measurable levels of M-component (M-protein ≥1 g/dL or urine M-protein ≥200 mg/24 h), received at least two prior lines of therapy, an Eastern Cooperative Oncology Group (ECOG) performance status score of ≤2 and a life expectancy of >3 months. Exclusion criteria included meningeal involvement of MM, clinically significant cardiac disease, and respiratory conditions, absolute neutrophil count ≤1.0 × 109/L, hemoglobin level ≤7.5 g/dL, platelet count <75 × 109/L, alanine aminotransferase or aspartate aminotransferase level ≥2.5 times of upper limit of normal (ULN), total bilirubin level ≥2 × ULN, creatinine clearance ≤20 mL/min/1.73 m2, potassium level <3.0 mEq/L, and corrected serum calcium >14.0 mg/dL.
The study protocol was approved by the local Independent Ethics Committees of participating institutions, and the study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the protocol. All patients provided written informed consent to participate in the study.
Study design
This was a phase 1, open-label, single-arm, non-randomized, multicenter, dose-escalation monotherapy study conducted between 28 April 2014 and 25 September 2015. The study included a screening phase; an open-label, two-period treatment phase; and a follow-up phase. Screening phase included 21 days (day −21 to day −1) prior to first administration of daratumumab. The open-label treatment phase started on day 1 (week 0) with administration of daratumumab and extended until end of treatment (EoT). In the first period of treatment phase, patients received the first infusion of daratumumab at day 1 (week 0); after a 3-week resting period, 6 infusions were administered weekly until week 8 (a total of seven infusions). In the second period, patients received daratumumab in cycles of 28 days. From week 10 to 24 (cycles 1–4) patients received infusions every other week, and monthly infusions were given from week 26 (cycle 5) to EoT. The follow-up phase started after EoT and continued until 8 weeks after the last administration of daratumumab, death, loss to follow-up, withdrawal of consent for study participation, or study end, whichever occurred first. Dose limiting toxicities (DLTs) were defined as one or more of the following toxicities during the DLT evaluation period: (1) Grade 4 or unmanageable Grade 3 infusion-related reactions (IRRs) occurred within 48 h of the daratumumab infusion; (2) Grade 4 thrombocytopenia lasting >7 days; (3) Grade ≥3 thrombocytopenia with bleeding; (4) Grade 4 neutropenia lasting >7 days; (5) Grade 3 febrile neutropenia or sepsis; (6) Grade 4 anemia; and (7) any Grade ≥3 non-hematologic toxicity except isolated Grade 3 γ-glutamyltransferase elevation, Grade ≥3 fatigue/asthenia lasting <7 days, and manageable Grade 3 nausea/vomiting or diarrhea, tumor lysis syndrome or hyperuricemia. The DLT assessment began with the first infusion of daratumumab and ended immediately before initiation of the fourth infusion. DLT evaluation period was set to evaluate tolerability of a single dose of daratumumab for 3 weeks; thereafter, at least two consecutive doses in the most intensive dose period (first period of treatment phase).
Study drug and treatment
Daratumumab was administered as an intravenous infusion diluted in a sterile, pyrogen-free physiologic saline solution (0.9% NaCl). The patients were treated in two cohorts with different dose levels (8 and 16 mg/kg) of daratumumab. Treatment in the second cohort (16 mg/kg) was initiated only after no DLT reported with 8 mg/kg dose. Dose modification for individual patients was not permitted during the first period of treatment phase. Dose delay was permitted in the second period of treatment phase to manage daratumumab-related adverse events (AEs). Treatment with daratumumab was to be continued until the disease progresses, AEs were deemed unmanageable in the treatment phase or the study treatment was discontinued.
Concomitant medications
Throughout the study, any medications deemed necessary to provide adequate supportive care were prescribed by investigators. All patients received diphenhydramine, methylprednisolone, and paracetamol as preinfusion medications. For the prevention of delayed IRRs, all patients received corticosteroid (20 mg of methylprednisolone or equivalent) on the first and second day after all daratumumab infusions. For patients with a higher risk of respiratory complications antihistamines, short-acting β2 adrenergic receptor agonists, and inhaled corticosteroids were given as postinfusion medications. Any therapy intended to treat MM, therapies targeting CD38, antireceptor activator of nuclear factor-kappa B ligand (RANKL), systemic corticosteroids, and nonsteroidal anti-inflammatory drugs were prohibited during the study course.
Assessments
Tolerability and safety
The primary endpoint was tolerability and safety of daratumumab. Safety assessments included treatment emergent adverse events (TEAEs), clinical laboratory tests, vital signs, electrocardiograms (ECGs), physical examinations, and ECOG performance status. TEAEs were graded for severity according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), Version 4.03.
Efficacy
Clinical efficacy of daratumumab was evaluated as a secondary endpoint including overall response rate (ORR), progression-free survival (PFS), and duration of response. The ORR was evaluated as the proportion of patients in the response evaluable population who achieved a stringent complete response (sCR), complete response (CR), very good partial response (VGPR), and partial response (PR). Clinical benefit rate was estimated as the number of patients in the response evaluable population who achieved a sCR, CR, VGPR, PR, and minimal response (MR). The modified IMWG consensus recommendations for MM treatment response criteria (only added MR criteria from European Group for Blood and Marrow Transplantation criteria) was used for response evaluation [15,16,17].
Pharmacokinetics
Pharmacokinetics of daratumumab were evaluated as secondary endpoints.
Sampling
Venous blood samples (5 mL/sample) for measuring serum daratumumab concentrations and immunogenicity (antibodies to daratumumab) were collected. Each serum sample was divided into three aliquots (one each for PK, immunogenicity, and a backup). Patients who discontinued treatment were also asked to return for PK and immunogenicity evaluation during the follow-up phase. Whole blood samples were used for immunophenotyping analysis for the assessment of natural killer (NK) and T cells.
Pharmacokinetic parameters
The mean daratumumab serum concentration–time profiles following the first infusion (from day 1 pre-dose to day 22 pre-dose in period 1) and the repeated infusions (from day 29 pre-dose in period 1 to cycle 9 day 1 pre-dose in period 2) were assessed. Pharmacokinetic parameters were assessed following the first infusion (day 1) and the 7th infusion (day 57). The following pharmacokinetic parameters were derived using the non-compartmental analysis methods for all patients who received a dose of daratumumab: area under the concentration–time curve (AUC) from time 0 to 7 days (AUC0–7day); AUC from time 0 to the last measurable concentration (AUClast); AUC from time 0 to infinity (AUC∞); total systemic clearance of drug after IV administration (CL); maximum observed concentration (C max); pre-dose concentration at steady state (C trough); elimination half-life (t 1/2); volume of distribution of drug after IV administration (V); time of maximum concentration (t max).
Immunogenicity and biomarkers
Immunogenicity assessments were secondary endpoints. Anti-daratumumab antibodies in human serum samples were evaluated using in-house developed ‘Bridging ECLIA’ (electrochemiluminescent immunoassay). Biomarkers to daratumumab response were also explored. Measured items for immunophenotyping included CD3+ (%), CD3+ CD4+ (%), CD3+ CD8+ (%), CD3−CD16+ CD56+ (%) T cell and NK cell populations.
Statistical methods
Sample size
At least three patients were planned to be enrolled in a cohort. The next dose level was selected based on the Bayesian logistic model using EWOC scheme [18]. The sample size was dependent on the number of patients enrolled in each cohort and the number of traveled dose levels, which was dependent on the number of DLT incidences in each cohort. The size of each cohort was three patients. The projected maximum sample size was 12 patients.
Statistical analysis
All TEAEs, Grade 3 or higher TEAEs, serious adverse events (SAEs), TEAEs leading to discontinuation of treatment, dose delay, dose reduction, and infusion interruption, or death were summarized with descriptive statistics. Serum daratumumab concentrations and the incidence of antibodies to daratumumab were summarized descriptively. The counts of NK cells (CD3−CD16+CD56+), ORR and objective responses were summarized. Progression-free survival was evaluated descriptively with the Kaplan–Meier method.
Analysis sets
All patients who received at least one dose of daratumumab were included in safety and efficacy analyses. All the patients who received at least one dose of daratumumab and who did not meet the next patient replacement rule for dose-escalation decision were considered for DLT evaluation. Patients who received at least one dose of daratumumab and had a disease assessment were included in response evaluation, and those who had at least one during-treatment-sample collected were considered for pharmacokinetic and immunogenicity assessments.
Results
Patient population
Of 13 patients screened, 9 patients were treated with daratumumab. Four patients were first enrolled to the 8 mg/kg cohort. Following the confirmation of tolerability, five patients were enrolled to the 16 mg/kg cohort. The median age of the study population was 65 years (range 47, 73) with baseline ECOG performance status score 0 or 1. The majority of patients had MM stage I or II [n = 4 each (44.4%)]. The median duration of MM since initial diagnosis was 28 months (range 8, 88) in the 8 mg/kg level and 70 months (range 48, 139) in the 16 mg/kg level. The most common prior systemic MM therapies were PIs (bortezomib), IMiDs (lenalidomide, thalidomide), and conventional therapies (dexamethasone, cyclophosphamide, and vincristine) (Table 1). All patients discontinued treatment. All four patients in the 8 mg/kg cohort discontinued due to disease progression. In the 16 mg/kg cohort, one patient (20%) discontinued due to non-compliance, one patient (20%) due to protocol violation, and three patients (60%) due to disease progression.
Treatment duration and exposure
The median duration of treatment was 6.3 months (range 0.7, 10.6) for the 8 mg/kg level and 2.4 months (range 0, 9.5) for the 16 mg/kg level. The median cumulative dose of daratumumab administered was 127.10 mg/kg (range 16.3, 182.1) in the 8 mg/kg level and 122.68 mg/kg (range 16.2, 309.7) in the 16 mg/kg level. The median relative dose intensity was 100.62% (range 99.2, 113.8) for the 8 mg/kg dose and 96.78% (range 94.7, 104.7) for the 16 mg/kg level (Table 2).
Safety
All patients experienced at least one TEAE. Overall, treatment emergent SAEs were reported in 3 (33.3%) patients (8 mg/kg cohort: n = 1; 16 mg/kg: n = 2). IRRs were observed in 4 patients (44.4%; 8 mg/kg cohort: n = 1; 16 mg/kg cohort: n = 3). There were no deaths or TEAEs leading to study drug discontinuation. No DLTs were observed and no Grade ≥3 IRRs were reported in the study.
The most frequently reported TEAEs were lymphopenia (100% in both cohorts), neutropenia [8 mg/kg cohort: n = 2 (50%); 16 mg/kg cohort: n = 5 (100%)], leukopenia [16 mg/kg cohort: n = 3 (60%)], and pyrexia [16 mg/kg cohort: n = 3 (60%)].
Three patients (75%) in 8 mg/kg cohort and five patients (100%) in 16 mg/kg cohort had Grade ≥3 TEAEs. The most frequently reported Grade ≥3 hematological TEAEs were lymphopenia [8 mg/kg cohort: n = 2 (50%); 16 mg/kg cohort: n = 5 (100%)], neutropenia (16 mg/kg cohort: n = 4, 80%), leukopenia (16 mg/kg cohort: n = 3, 60%) (Table 3).
One patient (25%) in the 8 mg/kg cohort experienced an SAE of Grade 4 thrombocytopenia that was not resolved and considered not related to daratumumab by the investigator. Two patients (40%) in the 16 mg/kg cohort experienced 3 SAEs considered possibly related to the study drug: Grade 3 pneumonia (one case), Grade 2 headache (one case) and Grade 1 pyrexia (one case). No SAEs led to death or discontinuation of daratumumab.
Efficacy
The median duration of follow-up was 10.5 months (range 2.3, 16.4) for the 8 mg/kg cohort and 9.9 months (range 1.7, 13.2) for the 16 mg/kg cohort. The ORR was 25.0% (1 of 4 patients) in 8 mg/kg cohort and 60.0% (3 of 5 patients) in 16 mg/kg cohort; all 4 patients with ORR achieved a PR. Progressive disease was reported for one patient in 8 mg/kg cohort and stable disease was reported in two patients in each cohort. The median PFS was 6 months (95% CI 0.72, 10.64) in 8 mg/kg cohort and 9.5 months in 16 mg/kg cohort (95% CI 1.84, NE). Median time-to initial-response was 2.33 months (range 2.3, 2.3) in 8 mg/kg cohort and 1.84 months (range 1.2, 1.9) in 16 mg/kg cohort. The median duration of response was 2.8 months (95% CI NE, NE) in 8 mg/kg cohort and 7.7 months (95% CI 6.11, NE) in 16 mg/kg cohort. Median time to disease progression was 6.0 months (95% CI 0.72, 10.64) in 8 mg/kg cohort and 9.5 months (95% CI 1.84, NE) in 16 mg/kg cohort (Table 4). Overall, majority of patients [n = 5 (55.6%)] had reductions in serum M-protein from baseline. Three patients (33.3%) had >50% reduction in serum M-protein from baseline (8 mg/kg cohort: n = 1; 16 mg/kg cohort: n = 2) (Fig. 1).

Waterfall plot of individual maximum percent change in serum M-proteins from baseline (serum M-protein evaluable patients). Six of nine cases were serum M-protein evaluable patients. Free light chain (FLC) only: n = 3 patients
Pharmacokinetics, immunogenicity, and biomarkers
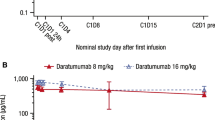
In patients treated with the 16 mg/kg dose regimen, accumulation of daratumumab continued throughout the weekly dosing, and decreased slightly as patients entered the every 2 weeks and subsequently every 4-week dosing periods (Fig. 2). Mean C max (~2.3-fold) and AUClast (~4.2-fold) were higher and t 1/2 (~2-fold) was prolonged in 16 mg/kg cohort compared to 8 mg/kg cohort. Mean value of clearance decreased with increasing dose (0.678 to 0.247 mL/h/kg), while V d was comparable in both cohorts (Table 5).

Mean daratumumab serum concentration (μg/mL) following the first (a) and repeated infusion (b)
Immunogenicity testing for daratumumab was performed on day 1 pre-dose and at follow-up (week 4 and 8). Antibodies to daratumumab were absent in all patients.
In both 8 and 16 mg/kg cohorts, a marked decrease in total NK cells was observed until day 8, and the decrease was sustained throughout the treatment phase (Fig. 3). Both CD3+ and CD3+CD8+ cells increased, whereas CD3+CD4+ and CD3−CD16+CD56+ decreased over time.

Total natural killer cells over time in blood (all treated analysis population)
Discussion
This was the first study conducted among Japanese patients with relapsed or refractory MM to evaluate safety, tolerability, efficacy, and PK of daratumumab monotherapy. The target doses of daratumumab used in the present study were selected based on the results of the phase 1/2 study [10] in which a maximum dose of 24 mg/kg was administered without limiting toxicity. A dose level of 8 mg/kg was considered as starting dose. A second dose level of 16 mg/kg was selected because at this dose daratumumab was previously shown to saturate the CD38 tumor target. This dose level also results in deeper responses than that achieved with 8 mg/kg dose [10].
Both dose levels of daratumumab were found well-tolerated in this study. As observed in the previous global phase 1/2 study [10], neutropenia, lymphopenia, leukopenia were commonly reported as hematological TEAEs in the present study [10]. Similar as previous global phase 1/2 study, infusion-related reactions were reported in 4 of 9 patients (1 patient in the 8 mg/kg level and 3 patients in the 16 mg/kg level). All IRRs were Grade 1 or 2 in severity and could be managed by pre- and postinfusion medications. The frequency of Grade ≥3 toxicities was 100% (in 5 patients) in 16 mg/kg cohort and 75% (in 4) in 8 mg/kg cohort. In the phase 1/2 study, the frequency of Grade ≥3 toxicities was 26% (in 42) in 16 mg/kg cohort and 53% (in 30) in 8 mg/kg cohort [10]. Although the frequency of Grade ≥3 toxicities was relatively higher in the Japanese patients, all but two (Grade 3 pneumonia and Grade 3 osteitis) were hematological TEAEs, and all were clinically manageable. Nevertheless, the smaller sample size is a limitation of current study that may contribute to higher reported rates of AEs.
The ORR in the 16 mg/kg cohort was 60.0% (n = 5). However, the best response was PR only, none of the patients achieved deep responses as the duration of treatment was shorter. The ORR observed in phase 2 randomized study in non-Japanese patients was 29.2% (n = 106); 3% patients achieved sCR, 9% achieved VGPR, and 17% had PR in 16 mg/kg dose cohort [11]. The higher response seen in Japanese patients was likely due to the small sample size tested, and perhaps a more refractory population in previous studies [11]. Nevertheless, the ORR obtained with daratumumab in present study was encouraging considering the heavily pre-treated patients.
Median time-to-initial-response in our study (1.8 months) was comparable to the findings of phase 2 monotherapy SIRIUS study (1.0 month) [11] a phase 1/2 study GEN501 study (0.9 month) [10] in 16 mg/kg cohort, and several phase 1/2 and phase 3 combination studies [13, 14, 19].
In the present study, four patients (44.4%) had >50% reduction in serum M-protein or free light chains level from baseline [8 mg/kg cohort: n = 1 (25%); 16 mg/kg cohort: n = 3 (60%)]. Similar results were observed in the phase 1/2 study where 19 patients (46%) in 16 mg/kg cohort and 4 patients (15%) in 8 mg/kg cohort had >50% reduction in serum M-protein or free light chains level from baseline [10].
The pharmacokinetic profile of daratumumab in this study was consistent with the phase 1/2 study [10]. A decrease in total NK cells following daratumumab treatment in both dose levels supports the observation that NK cells express CD38. Though the NK cell subtype responsible for ADCC induced by daratumumab is not known [20], increased peripheral level of CD3+ and CD3+CD8+ cells with daratumumab exposure implies their role as effector in daratumumab induced cytotoxicity against MM cells.
A small sample size is characteristic to a phase 1 study. However, findings from this small sample size need to be interpreted carefully and further investigations in larger samples are warranted.
Overall, daratumumab as monotherapy was found to be well-tolerated and safe in Japanese patients with relapsed or refractory MM, previously treated with therapies including PIs, IMiDs, or chemotherapy. Clinical efficacy was observed in these heavily pre-treated patients. The exposure of serum daratumumab was increased with increasing dose and no human antibodies to daratumumab were apparent. Results observed in this study were consistent with similar studies in non-Japanese populations.
References
Ozaki S, Handa H, Saitoh T, Murakami H, Itagaki M, Asaoku H, et al. Trends of survival in patients with multiple myeloma in Japan: a multicenter retrospective collaborative study of the Japanese Society of Myeloma. Blood Cancer J. 2015;5(9):e349.
San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906–17.
Sonneveld P, Goldschmidt H, Rosiñol L, Bladé J, Lahuerta JJ, Cavo M, et al. Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: a meta-analysis of phase III randomized controlled trials. J Clin Oncol. 2013;31(26):3279–87.
Laubach JP, Voorhees PM, Hassoun H, Jakubowiak A, Lonial S, Richardson PG. Current strategies for treatment of relapsed/refractory multiple myeloma. Expert Rev Hematol. 2014;7(1):97–111.
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26(1):149–57.
Iida S. Mechanisms of action and resistance for multiple myeloma novel drug treatments. Int J Hematol. 2016;104(3):271–2.
de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol Baltim Md 1950. 2011;186(3):1840–8.
Nijhof IS, Groen RWJ, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. 2015;29(10):2039–49.
Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384–94.
Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207–19.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet Lond Engl. 2016;387(10027):1551–60.
Research C for DE. Approved drugs—daratumumab injection [Internet]. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm472904.htm. Accessed 17 Jun 2016.
Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754–66.
Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for mulitple myeloma. N Engl J Med. 2016;375:1319–31.
Bladé J, Samson D, Reece D, Apperley J, Björkstrand B, Gahrton G, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102(5):1115–23.
Durie BGM, Harousseau J-L, Miguel JS, Bladé J, Barlogie B, Anderson K, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467–73.
Rajkumar SV, Harousseau J-L, Durie B, Anderson KC, Dimopoulos M, Kyle R, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117(18):4691–5.
Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27(13):2420–39.
Plesner T, Arkenau HT, Gimsing P, Krejcik J, Lemech C, Minnema MC, et al. Phase 1/2 study of daratumumab, lenalidomide, and dexamethasone for relapsed multiple myeloma. Blood. 2016;128:1821–28.
Dosani T, Carlsten M, Maric I, Landgren O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J. 2015;5(4):e306.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M.A. and M.I. are employees of Janssen Pharmaceutical. K.S. received honoraria from Celgene and Janssen; S.I. from Celgene, Janssen, Ono Pharmaceutical, Takeda Pharmaceutical and Bristol-Myers; S.I., S.K., and M.R. received research funding from Ono Pharmaceutical, Lilly, Celgene, Janssen, Kyowa Hakko Kirin, Sanofi, Takeda Pharmaceutical, Novartis; S.I., S.K., and M.R. received subsidies or donations from Chugai Pharmaceutical, Kyowa Hakko Kirin, Teijin Pharma, Astellas, Toyama Chemical. The study was funded by Janssen Pharmaceutical. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. All authors met the International Council of Medical Journal Editors’ criteria for authorship, and anyone who met those criteria is listed as an author. All authors have read and approved the final manuscript for submission.
About this article

Cite this article
Iida, S., Suzuki, K., Kusumoto, S. et al. Safety and efficacy of daratumumab in Japanese patients with relapsed or refractory multiple myeloma: a multicenter, phase 1, dose-escalation study. Int J Hematol 106, 541–551 (2017). https://doi.org/10.1007/s12185-017-2281-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2281-6