
Abstract
Daratumumab in combination with bortezomib and dexamethasone (DVd) has demonstrated longer progression-free survival than combination of bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (RRMM). In this multicenter, open-label, phase-1 study, the safety, efficacy, and pharmacokinetics (PK) of DVd were evaluated in Japanese patients with RRMM. Eight patients with RRMM aged between 54 and 82 years were enrolled and treated with DVd regimen. Primary endpoints were tolerability and safety. Secondary endpoints were overall response rate (ORR), very good partial response (VGPR) or better, complete response (CR) or better, time to response (TTR), PK, and immunogenicity. All patients (n = 8) experienced Grade ≥ 3 treatment-emergent adverse events (TEAE), with thrombocytopenia (n = 6, 75%) being the most frequent. Mild Grade ≤ 2 infusion-related reactions were reported in five patients. Serious TEAEs were herpes zoster, nasopharyngitis, and prostate cancer (n = 1 each). Three dose-limiting toxicities were observed in two patients. No death or disease progression was reported as of the study cut-off date. ORR was 100% (2 CRs or better, 2 VGPRs, 4 PRs). The median TTR was 0.9 months. PK profiles were comparable to previous studies. The DVd regimen showed acceptable safety with favorable efficacy in Japanese patients with RRMM.
Clinical trial registration number
NCT02497378.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma (MM) is an incurable malignancy of plasma cells that is characterized by abnormal production of monoclonal immunoglobin and distinctive organ damages, mainly affecting bone tissues, hematopoietic system, and kidneys [1]. In Japanese population, the incidence of MM has increased from 0.92 to 5.2 per 100,000 during 1975 to 2010 [2]. Treatment outcomes in MM depend on the factors related to biologic profiles of disease, comorbidity of patients, and type of treatment such as the use of novel agents and autologous stem cell transplantation [3]. Although median overall survival of patients with MM has significantly improved subsequent to introduction of newer therapies such as proteasome inhibitors (PIs) [4] and immunomodulatory drugs (IMiDs), long-term prognosis is still unsatisfactory [2, 5,6,7]. Also, elderly and frail patients have poorer responses to the initial therapy and frequently develop resistance to these agents [8, 9].
CD38, a transmembrane glycoprotein possessing multifunctions as an ectoenzyme, is overexpressed in MM cells and suggested to support their growth and survival in vitro [1]. Daratumumab is an immunoglobulin (Ig) G1 kappa monoclonal antibody that specifically binds to CD38-expressing tumor cells. It facilitates tumor cell death through multiple immune-mediated mechanisms, including the induction of apoptosis directly through Fc-mediated cross-linking, antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) [1, 10]. Additionally, recent studies have revealed previously unknown immunomodulatory effects of daratumumab [10].
In an open-label, phase 2 trial, daratumumab monotherapy was found to be tolerable and efficacious with an overall response rate (ORR) of 29% (95% CI 21–39%) in patients with RRMM [11]. Based on these findings, US FDA granted an accelerated approval to daratumumab as a monotherapy for treatment of patients with MM refractory to a minimum of three prior lines of therapies, including a PI and an IMiD [12, 13]. Daratumumab in combination with a PI (bortezomib) or an IMiD (lenalidomide) has also been investigated separately in two randomized, open-label, phase 3 trials, CASTOR and POLLUX, respectively. In both these studies, the addition of daratumumab to a bortezomib- or lenalidomide-based regimen demonstrated a significant reduction in the risk of disease progression or death in patients with MM who have received at least one prior therapy [12, 14].
Daratumumab monotherapy has been found to be tolerable and safe in a phase 1 study in Japanese patients with RRMM [15]. However, daratumumab-containing combination therapy has not been investigated so far in Japanese patients. Therefore, this phase 1 study was conducted in Japanese patients with RRMM to evaluate the safety, tolerability, efficacy, immunogenicity, and PK of daratumumab combined with bortezomib and dexamethasone (DVd). In this manuscript, we present the results from data collected as of the clinical data cut-off date, 03 June 2016.
Methods
Patients
Japanese patients aged ≥ 20 years with symptomatic MM (according to the International Myeloma Working Group [IMWG] diagnostic criteria) with measurable levels of M-component (M-protein ≥ 1 g/dL or urine M-protein ≥ 200 mg/24 h, serum free light chain ≥ 10 mg/dL with light chain MM), Eastern Cooperative Oncology Group (ECOG) performance status score of 0 or 1, and who had received at least 1 prior line of therapy were enrolled. Exclusion criteria included previous treatment with anti-CD38 therapy, refractoriness to PIs, meningeal involvement of MM, clinically significant cardiac or respiratory disease, absolute neutrophil count ≤ 1.0 × 109/L, hemoglobin level ≤ 8.0 g/dL, platelet count < 75 × 109/L, alanine aminotransferase or aspartate aminotransferase level ≥ 2.5 times of upper limit of normal (ULN), total bilirubin level ≥ 1.5 × ULN, creatinine clearance ≤ 20 mL/min/1.73 m2, and corrected serum calcium > 14.0 mg/dL.
The study protocol and amendments were approved by an Institutional Review Board of all participating centers, and the study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the protocol. All patients provided written informed consent to participate in the study.
Study design
This was a phase 1b, open-label, non-randomized, multicenter study conducted at 5 centers in Japan. The study included a screening phase (day −21 to day −1), a treatment phase (21 day cycle from cycle 1–8 and 28-day cycle from cycle 9 till end of treatment phase), and a follow-up phase (8 weeks). During the treatment phase, patients received daratumumab (16 mg/kg intravenous) every week for the first 3 cycles, on day 1 of every 3 weeks for the cycles 4–8, and then on day 1 of every 4 weeks from cycle 9. Patients were also treated with bortezomib (1.3 mg/m2, subcutaneous) on days 1, 4, 8, and 11 of each 21-day cycle (cycle 1–8) and dexamethasone (20 mg, oral) on days 1, 2, 4, 5, 8, 9, 11, and 12 of each 21-day cycle. Dexamethasone was also administered as a premedication on day 15 in cycles 1–3. To prevent infusion-related reactions (IRRs), all patients were pre-treated with dexamethasone 20 mg intravenously before the administration of daratumumab. The end of treatment (EoT) visit occurred 4 weeks (± 3 days) after the last dose of study treatment. The assessment of dose-limiting toxicities (DLTs) began with the first infusion of daratumumab and ended at the end of cycle 1. Dose-limiting toxicities were defined as one or more of the following during the evaluation period: Grade 4 or unmanageable Grade 3 IRRs occurring within 48 h of the daratumumab infusion; Grade 4 thrombocytopenia lasting > 7 days; Grade ≥ 3 thrombocytopenia with bleeding; Grade 4 neutropenia lasting > 7 days; Grade 3 febrile neutropenia or sepsis; Grade 4 anemia; any Grade ≥ 3 non-hematologic toxicity except Grade ≥ 3 fatigue/asthenia lasting < 7 days, and manageable Grade 3 nausea/vomiting or diarrhea, tumor lysis syndrome or hyperuricemia. Dose modifications of daratumumab for individual patients were not permitted, however, dose delay was permitted to manage daratumumab-related adverse events (AEs). Treatment with daratumumab was continued until disease progression, unacceptable toxicity, or other treatment discontinuation criteria.
Bortezomib dose modification was permitted based on the highest grade of toxicity. Treatment with bortezomib was withheld upon onset of any Grade 3 non-hematologic or Grade 4 hematologic toxicities, except neuropathy. The treatment could be reinitiated at a 25% reduced dose per approved labeling once the symptoms of toxicities were resolved (Supplementary file 1). Dose modification of dexamethasone was based on the toxicity. The dose could be initially reduced to 40 mg/week, and then 20 mg weekly if additional problems persisted, at the discretion of investigators.
Concomitant and prohibited medications
Diphenhydramine, paracetamol, dexamethasone, antihistamine, short-acting β2 adrenergic receptor agonist, and control medications for lung disease such as chronic obstructive pulmonary disease and asthma as pre- or postinfusion medications were all permitted during the study. However, therapy intended to treat MM, therapies targeting CD38, other investigational agents, and systemic corticosteroids (other than dexamethasone) were prohibited during the study. Strong CYP3A4 inducer was prohibited during the bortezomib treatment.
Assessments
Tolerability and safety
The primary endpoint was tolerability and safety of DVd. Patients were hospitalized from day 1 to day 8 of cycle 1 to allow close clinical monitoring for AEs, laboratory abnormalities, and clinical response in the treatment phase. A study evaluation team (SET), consisting of participating investigators and sponsor’s medical monitor, was formed to evaluate tolerability and safety in support of the Bayesian logistic regression model (BLRM) using escalation with overdose control (EWOC) scheme. Safety assessments included monitoring of treatment-emergent adverse events (TEAEs), deaths, serious AEs (SAEs), other significant AEs (TEAEs leading to treatment discontinuation, dose delay, dose reduction or infusion interruption), AEs of special interest (IRRs and infections), clinical laboratory tests, electrocardiograms, physical examination, ECOG performance status, and vital signs. Adverse events were graded for severity according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), Version 4.03.
Efficacy
Efficacy assessment included overall response rate [defined as the proportion of response evaluable population who achieved a stringent complete response (sCR), complete response (CR), very good partial response (VGPR), and partial response (PR)] and time to response. Clinical benefit rate was evaluated as the proportion of response evaluable population who achieved overall response and minimal response (MR). Other efficacy endpoints included duration of response (DOR), time to disease progression, progression-free survival (PFS), overall survival (OS), and changes in serum and urine M-protein from baseline over time. The modified IMWG consensus recommendations for MM treatment response criteria were used for assessment of objective responses (only added MR criteria from European Group for Blood and Marrow Transplantation criteria) [16, 17]. Blood samples for β2-microglobulin and albumin were collected at screening to determine the International Staging System (ISS) stage.
Pharmacokinetics, immunogenicity, and biomarkers
Sampling
Venous blood samples (5 mL per sample) were collected at day 1 of cycles 1, 3, 6, 9, and 12, EoT, and follow-up for PK and immunogenicity assessments. Participants who discontinued the treatment were also asked to return for immunogenicity evaluation during the follow-up phase. All samples collected for immunogenicity analysis were also evaluated for serum daratumumab concentration to ensure appropriate interpretation of immunogenicity data. Blood samples for biomarker evaluations were collected at cycles 1, 2, 3, 8, and at EoT. Whole blood samples were used for immunophenotyping analysis.
Parameters assessed
The mean daratumumab serum concentration–time profiles following the first infusion were assessed and minimum observed serum concentration (C min) and maximum observed serum concentration (C max) were estimated. Biomarker assessment included analysis of natural killer (NK) cells, T cells, and B cells. CD3+CD4+, CD3+CD8+, CD3−CD16+CD56+, and CD19+ cells were analyzed by the central laboratory.
Statistical methods
Sample size
At least 6 patients were planned to be enrolled initially. If a dose reduction was needed, at least 3 patients could be enrolled in subsequent cohorts. The next dose level could be selected based on the BLRM using the EWOC scheme. A patient could be replaced after receiving study drug when the patient failed to satisfy the criteria for DLT evaluation.
Analysis sets
All patients who received at least 1 dose of daratumumab were considered for the safety and efficacy analysis. Patients who received at least one dose of daratumumab and had a disease assessment during treatment phase were included in response evaluation while, those who received at least 1 dose of daratumumab and had at least 1 daratumumab concentration data or immunogenicity data were considered for PK or immunogenicity assessments, respectively. All patients who received at least 1 dose of daratumumab and who did not meet the next patient replacement rule for dose escalation decision were considered for DLT evaluation.
Time to event parameters were analyzed using Kaplan–Meier method, median with 95% confidence intervals (CIs) were estimated. Adverse events were reported using frequency tables. Data related to all other parameters were summarized using descriptive statistics.
Results
Patient population
The study was initiated on 05 August 2015 and was ongoing at the time of reporting. Eight patients were enrolled in the study and all of them received at least 1 dose of daratumumab. As of the clinical cut-off date, 7 patients were continuing with the treatment while, 1 patient discontinued due to prostate cancer. The median age of the patients was 74.5 years (range 54–82). The majority of patients (n = 7, 87.5%) had stage I MM; median duration of MM since initial diagnosis was 3.7 years (range 2.3–11.5). Six patients (n = 6, 75%) were previously treated with 2 prior therapies including PIs (n = 4, 50.0%) and IMiDs (n = 5, 62.5%) (Table 1). Half of the patients (n = 4, 50.0%) had 1–3 lytic bone lesions. However, none of the patients had extramedullary plasmacytomas.
Treatment duration and exposure
The median number of treatment cycles received was 8.0 (range 6–12). The median duration of study treatment was 5.3 months (range 4.3–8.9). The median relative dose intensities of daratumumab, bortezomib, and dexamethasone were 97.1% (range 83.4–105.4), 72.2% (range 55.9–95.2), and 48.2% (range 34.2–100.0), respectively. The median duration of follow-up was 5.5 months (range 4.3–8.9).
Safety
There was no death reported during the study as of the clinical data cut-off date. All patients experienced at least one TEAE. The most commonly reported (n ≥ 4, ≥ 50%) TEAEs of any grade were thrombocytopenia (n = 7, 87.5%) and lymphopenia (n = 5, 62.5%). All patients experienced at least 1 Grade 3 or 4 TEAE. The most commonly reported Grade 3 or 4 TEAEs were thrombocytopenia (n = 6, 75.0%) and lymphopenia (n = 5, 62.5%). Grade 3 or 4 treatment-emergent infections and infestations were reported in 2 patients (25.0%) comprising herpes zoster and nasopharyngitis, respectively. Of the 7 patients who received prophylactic antivirals, one developed abovementioned herpes zoster. IRRs were experienced by 5 patients (62.5%). Frequently reported IRRs were hypoxia, wheezing, and chills, reported in 2 patients (25.0%) each, all were non-severe (Grade ≤ 2). All IRRs occurred at the time of first infusion; no IRRs were observed at the subsequent infusions. One patient (12.5%) discontinued treatment due to the SAE of prostate cancer that occurred on study day 103.
Treatment cycle delays or dose modifications (i.e., dose delays, dose skipping, schedule change, or dose reduction) due to TEAEs were reported in all 8 patients, most frequently due to thrombocytopenia (n = 4, 50.0%). Bortezomib, dexamethasone, and daratumumab doses were modified in 8, 7, and 4 patients, respectively, due to TEAEs. One patient with prior exposure to bortezomib experienced treatment-emergent peripheral sensory neuropathy of toxicity Grade 2, which was considered to be related to bortezomib and the dose was reduced due to the event.
Thrombocytopenia (n = 7, 87.5%), lymphopenia (n = 5, 62.5%), anemia, neutropenia, and leukopenia (each n = 3, 37.5%) were commonly reported (n ≥ 3, > 30%) hematological toxicities. Dysesthesia, hyperglycemia, fatigue, blood lactate dehydrogenase increased, and weight loss were commonly reported non-hematological toxicities (each n = 3, 37.5%) (Table 2).
A total of 3 DLT events were observed in 2 patients (25.0%) during the DLT assessment period: Grade 3 thrombocytopenia with bleeding, Grade 3 gamma-glutamyl transferase increased and AST increased. None of the patients discontinued the study treatment due to the DLTs.
Efficacy
The ORR was 100.0% (sCR = 12.5%, CR = 12.5%, VGPR = 25.0%, PR = 50%). None of the patients were reported to have disease progression at the time of cut-off date. The median time to first response (PR or better) was 0.9 months (95% CI 0.8–1.5). The median duration of response was 4.1 months (range 3.5–8.1) (Table 3). Progression-free survival could not be estimated as there were no deaths or disease progressions. At the time of cut-off date, 5 out of 8 patients showed evaluable serum M-protein response. All 5 patients had ≥ 50.0% reduction from baseline, 3 patients (60.0%) had ≥ 90% reduction from baseline, and 1 patient (20.0%) had 100.0% reduction from baseline.
Pharmacokinetics, immunogenicity, and biomarkers
During the weekly dosing periods, the mean (SD) peak daratumumab concentrations were 354.10 (61.757) μg/mL at cycle 1, day 1 and 864.98 (167.806) μg/mL at cycle 3, day 1. The serum daratumumab concentration was maintained during the subsequent 3-weekly dosing periods (Fig. 1). The mean (SD) C max and C min of serum daratumumab from cycles 1–6 were 937.25 (160.965) µg/mL and 350.75 (63.539) µg/mL, respectively.

Peak and trough serum daratumumab concentration (linear scale) (pharmacokinetic evaluable analysis population)
Immunogenicity testing was performed on 5 patients after the first infusion. Antibodies to daratumumab were not detected in all 5 patients.
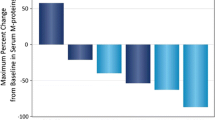
Regarding biomarkers, a marked decrease in the total NK-cell count was observed from baseline over time (Fig. 2). There was a decreasing trend in CD3+CD4+ and CD19+ lymphocyte subsets. No steady trend was observed with CD3+CD8+ lymphocytes (data not shown).

Total natural killer cells over time in blood (all-treated analysis set)
Discussion
This was the first study conducted in Japanese patients with RRMM to evaluate safety, tolerability, efficacy, PK, and immunogenicity of DVd therapy. It is hypothesized that targeting both the tumor cells and the surrounding stromal compartment should lead to higher efficacy in MM. Daratumumab directly targets tumor cells by selectively binding to CD38 molecules, which are overexpressed by malignant plasma cells in MM [18]. Combination of bortezomib and dexamethasone is the standard-of-care for patients with relapsed MM [19,20,21]. Daratumumab in combination with bortezomib and other MM backbone therapies has shown encouraging results in newly diagnosed MM patients [22]. The efficacy and safety of this daratumumab and bortezomib combination has been confirmed in a phase 3 study (CASTOR) conducted in non-Japanese patients with RRMM [14]. Also, daratumumab monotherapy at the dose of 16 mg/kg was found to be safe and tolerable in Japanese patients with RRMM in a phase 1 study [15]. So far, no drug interaction between daratumumab and other drugs has been reported. Therefore, DVd combination was also expected to be safe and efficacious in Japanese patients with RRMM. Our findings in this study show that DVd is a promising therapy for Japanese patients with RRMM.
Although the dose of bortezomib and/or dexamethasone was reduced in all the patients due to TEAEs, treatment could be continued by their appropriate management. The median relative dose intensity of dexamethasone was lower (48.2%) compared with that of daratumumab (97.1%) and bortezomib (72.2%). This may be due to a small sample size that was likely to be affected by dose modifications, as dexamethasone doses were skipped in 7 patients (87.5%), and reduced in 6 patients (75.0%) due to TEAEs such as hyperglycemia. Another possible reason could be the impaired glucose tolerance; 2 patients (25%) had history of diabetes mellitus and 4 patients (50%) were older than 75 years. No death was reported at the clinical cut-off date. Moreover, incidence rates of hematological toxicities reported in this study appeared to be slightly higher compared with CASTOR study [thrombocytopenia (87.5 vs 58.8%), anemia (37.5 vs 26.3%), and neutropenia (37.5 vs 17.7%)] [14]. The sample size in our study was very low and these estimates should be confirmed in further studies with a larger Japanese population.
IRRs were reported in 5 patients (62.5%), all were Grade 1 or 2 and no patient discontinued due to IRRs. The median time to onset of IRRs at the time of first infusion was 85.0 min (range 20.0–184.0) and all IRRs resolved within 24 h of onset. These findings were comparable with the results of CASTOR study [14].
One patient discontinued the study treatment due to Grade 3 serious TEAE of prostate cancer, which was considered not related to the study treatment because the patient had indicated the abnormal finding of right prostate by FDG-PET at baseline screening. Three DLT events were observed in 2 patients (25%), all of which were resolved and no patient discontinued due to the DLTs. The SET evaluated these DLTs and concluded that 16 mg/kg dose of daratumumab in combination with Vd was safe and tolerable.
The combination therapy showed promising efficacy results. The ORR was 100%, which was similar to the efficacy reported in CASTOR study (ORR, 82.9%) [14]. The better efficacy may be attributed to the enhanced direct cytotoxicity of combination as reported in preclinical studies, [23, 24] and lesser exposure of patients to prior treatments. PK results were as expected and consistent with prior studies [15, 25]. The immunogenicity profile and biomarker analysis of the combination therapy was also consistent with the phase 1 daratumumab monotherapy study conducted in Japanese patients. Both the studies observed a decrease in CD3+CD4+ and CD19+ lymphocytes count, and in total NK-cell count from baseline at all time points [15].
In conclusion, daratumumab 16 mg/kg in combination with bortezomib and dexamethasone was found to be tolerable in Japanese patients with RRMM who had received at least one prior therapy. Clinical efficacy results were also favorable for this combination therapy.
Further studies are required to confirm long-term efficacy and safety of this promising regimen in Japanese patients with RRMM, particularly in those who are ineligible for high-dose melphalan supported with autologous hematopoietic stem cell transplantation.
References
Raedler LA. Darzalex (Daratumumab): first anti-CD38 monoclonal antibody approved for patients with relapsed multiple myeloma. Am Health Drug Benefit. 2016;9(Spec Feature):70–3.
Ozaki S, Handa H, Saitoh T, Murakami H, Itagaki M, Asaoku H, et al. Trends of survival in patients with multiple myeloma in Japan: a multicenter retrospective collaborative study of the Japanese Society of Myeloma. Blood Cancer J. 2015;5:e349.
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–60.
Yoshizawa K, Mukai HY, Miyazawa M, Miyao M, Ogawa Y, Ohyashiki K, et al. Bortezomib therapy-related lung disease in Japanese patients with multiple myeloma: incidence, mortality and clinical characterization. Cancer Sci. 2014;105:195–201.
San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–17.
Sonneveld P, Goldschmidt H, Rosiñol L, Bladé J, Lahuerta JJ, Cavo M, et al. Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: a meta-analysis of phase III randomized, controlled trials. J Clin Oncol. 2013;31:3279–87.
Kumar SK, Dimopoulos MA, Kastritis E, Terpos E, Nahi H, Goldschmidt H, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31:2443–8.
Kumar N, Zaw AS, Reyes MR, Malhotra R, Wu PH, Makandura MC, et al. Versatility of percutaneous pedicular screw fixation in metastatic spine tumor surgery: a prospective analysis. Ann Surg Oncol. 2015;22:1604–11.
Laubach JP, Voorhees PM, Hassoun H, Jakubowiak A, Lonial S, Richardson PG. Current strategies for treatment of relapsed/refractory multiple myeloma. Expert Rev Hematol. 2014;7:97–111.
Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128:384–94.
Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387:1551–60.
Approved drugs—daratumumab (DARZALEX). https://www.fda.gov/Drugs/informationOnDrugs/ApprovedDrugs/ucm530249.htm. Accessed 17 May 2017.
Janssen Biotech Inc. DarzalexTM (daratumumab): prescribing information. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761036s004lbl.pdf. Accessed 17 May 2017.
Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754–66.
Iida S, Suzuki K, Kusumoto S, Ri M, Tsukada N, Abe Y, et al. Safety and efficacy of daratumumab in japanese patients with relapsed or refractory multiple myeloma: a multicenter, phase 1, dose-escalation study. Int J Hematol. 2017;106:541–51.
Durie BGM, Harousseau J-L, Miguel JS, Bladé J, Barlogie B, Anderson K, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–73.
Rajkumar SV, Harousseau J-L, Durie B, Anderson KC, Dimopoulos M, Kyle R, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117:4691–5.
de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–8.
Kropff MH, Bisping G, Wenning D, Volpert S, Tchinda J, Berdel WE, et al. Bortezomib in combination with dexamethasone for relapsed multiple myeloma. Leuk Res. 2005;29:587–90.
Dimopoulos MA, Orlowski RZ, Facon T, Sonneveld P, Anderson KC, Beksac M, et al. Retrospective matched-pair analysis of bortezomib plus dexamethasone versus bortezomib monotherapy in relapsed multiple myeloma. Haematologica. 2015;100:100–6.
Moreau P, San Miguel J, Ludwig H, Schouten H, Mohty M, Dimopoulos M, et al. Multiple myeloma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl_6):vi133–7.
Mateos M-V, Moreau P, Comenzo R, Bladé J, Benboubker L, de La Rubia J, et al. An open-label, multicenter, phase 1B study of daratumumab in combination with pomalidomide-dexamethasone and with backbone regimens in patients with multiple myeloma. European Hematology Association; 2015. Abstract p. 275.
Endell J, Samuelsson C, Boxhammer R, Strauss S, Steidl S. Effect of MOR202, a human CD38 antibody, in combination with lenalidomide and bortezomib, on bone lysis and tumor load in a physiologic model of myeloma. J Clin Oncol. 2011;29: 2011 (suppl; abstr 8078) suppl; abstr 8078. http://meetinglibrary.asco.org/content/80499-102. Accessed 17 May 2017.
van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PWHI, et al. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood Cancer J. 2011;1:e41.
Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207–19.
Funding
This study was sponsored by Janssen Research & Development, LLC.
Author information
Authors and Affiliations
Contributions
Dr. Aoki had primary role in the study design, analysis and data interpretation. Drs Iida, Ichinohe, Shinagawa, Suzuki and Takezako were involved in study design, data interpretation and also involved as investigators. All authors contributed to the development of the manuscript and approved the final manuscript for submission. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Corresponding author
Additional information
Previous publication: The abstract submitted to the JSH, 20–22th October, 2017 in Yokohama, Japan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Iida, S., Ichinohe, T., Shinagawa, A. et al. Safety and efficacy of daratumumab in combination with bortezomib and dexamethasone in Japanese patients with relapsed or refractory multiple myeloma. Int J Hematol 107, 460–467 (2018). https://doi.org/10.1007/s12185-017-2390-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2390-2