
Abstract
Leukemic stem cells (LSCs) in acute myeloid leukemia (AML) represent a low-frequency subpopulation of leukemia cells that possess stem cell properties distinct from the bulk leukemia cells, including self-renewal capacity and drug resistance. Due to these properties, LSCs are supposed to facilitate the development of relapse. The existence of LSCs is demonstrated by the ability to engraft and initiate human AML in immune-compromised mouse models. Although several lines of evidence suggest the complex heterogeneity of phenotypes displayed by LSC, many studies consider the CD34+/CD38− compartment as the most relevant. To increase the understanding of the true LSC, techniques such as multicolor flow cytometry, side-population assay and ALDH assay are utilized in many laboratories and could aid in this. A better understanding of different LSC phenotypes is necessary to enhance risk group classification, guide clinical decision-making and to identify new therapeutic targets. These efforts to eliminate LSC should ultimately improve the dismal AML outcome by preventing relapse development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
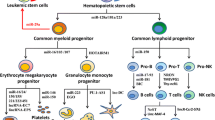
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by a multitude of molecular abnormalities. Despite increasing understanding of the pathogenesis no significant changes in treatment have been achieved as yet. With current treatment strategies, the percentage of adult patients achieving complete remission has increased, mainly due to better risk classifications and improvements in supportive care. However, with still roughly half of these patients relapsing, survival rates remain low. A rare population of therapy resistant cells is believed to be at the origin of the relapse. Since these cells have the self-renewal capacity to repopulate a leukemia despite their low frequency, they are considered leukemic stem cells (LSC), also referred to as the leukemia-initiating cells (LIC). Whether these cells originate from normal hematopoietic stem cells (HSC) or from more mature progenitors that gained stemness features remains elusive and may differ among patients. Currently, many studies reveal the importance of estimating LSC burden for prognostic purposes and strategies to eradicate these cells in order to completely eliminate the leukemia. In this review, we will focus mainly on the identification of these LSC using flow cytometry and summarize novel opportunities for elimination of these LSC.
Identification of leukemic stem cells
In order to identify LSC, knowledge of their specific characteristics is essential. The recognition of stemness features (e.g., drug resistance, self-renewal and undifferentiated state) [1] alone is not sufficient since those features are also characteristics for HSC coexisting in the bone marrow (BM) [2]. The existence of leukemic cells that meet these criteria was first demonstrated over 20 years ago. In the 1990s, Dick and colleagues demonstrated that a rare fraction of AML cells (i.e., cells with the CD34+/CD38− immunophenotype, similar to HSC) was capable of generating leukemia in immunodeficient mice [3, 4]. Since this discovery, these putative LSC have been the focus of extensive research. Golden standard property of LSC populations is the ability to engraft and initiate leukemia in a recipient mouse (initiation), to grow out after re-transplantation into secondary recipients (self-renewal) and preferable in tertiary recipients. Since normal CD34+/CD38− cells possess similar features as LSC and the design of new therapies require the specific eradication and monitoring of CD34+/CD38− LSC it is crucial to specifically discriminate LSC-containing fractions from HSC using cell-surface markers. Table 1 presents a summary of markers that are commonly used to distinguish LSC from HSC, thereby allowing to define the contribution of both the LSC and the HSC to the total CD34+/CD38− compartment. However, each marker allowed identification of LSC only in part of the AML patient population and often identified only part of the total LSC population in a particular patient [5, 6]. Combining all markers and other properties that distinguish LSC from HSC [5] allows to robustly identify the LSC and to estimate its frequency. Since the use of all markers to identify CD34+/CD38− LSC fraction of the total CD34+/CD38− compartment in each patient would require a dramatic amount of work and money, we developed a simplified comprehensive panel of markers that included only non-redundant LSC markers [6].
The existence of LSC outside the CD34+/CD38− fraction has been proposed as early as 1996, when CD34− cells were shown initiating leukemia in immune-deficient mice [6, 7]. Indeed, in less immunodeficient mice strains, both CD34+ and CD34− populations engraft [8–10]. Besides the influence of distinct properties of the mouse models used on engraftment, important studies of Bonnet et al. showed that there is another phenomenon that determines engraftment: they showed that the anti-CD38 antibody (used to purify CD34+/CD38−, CD34+/CD38+, CD34−/CD38− and CD34−/CD38+ cells prior to transplantation) inhibited subsequent engraftment [11, 12]. This is suggesting that earlier results could be based on technical interference rather than true biological differences of the CD38− and CD38+ populations.
Taken together, these studies advocate that LSC might co-exist in all CD34/CD38 defined subpopulations. In a recent study of Ng et al., the four CD34/CD38 defined cell populations of AML patients were sorted and were subsequently injected into mice and screened for their leukemia-initiating ability [13]. This exquisite approach confirmed that LSC activity was detected in all fractions; however, with a majority of CD34+ fractions, especially CD34+/CD38−, and minority of CD34− fractions containing LSC. The fact that there were hardly cases in which leukemia-initiating cells originated from CD34− and/or CD34+/CD38+ without concomitant activity in CD34+/CD38− suggests that the CD34+/CD38− fraction contains the most important leukemia-initiating cells when the other fractions are concomitantly present. This hypothesis is confirmed by other observations: in transplantation experiments using NOD/SCID mice of unfractionated AML, engraftment correlated only with the CD34+/CD38− frequency in the original sample, but not with the CD34+/CD38+ or CD34 frequency [14]. In addition, in line with the finding of in vitro and in vivo therapy resistance [15, 16], it was found that it is only the CD34+/CD38− LSC frequency that correlates with therapy outcome and minimal residual disease (MRD) levels, i.e., number of leukemic blasts detected after therapy [5]. This suggests that it is this fraction, and not the CD34+/CD38+ and CD34− fractions, that preferentially survives therapy and recapitulates leukemia.
The frequency of LSC within all mononuclear cells is shown to vary widely between patients (1 in 1.6 × 103 in 1.1 × 106) [10]. Since the CD34+/CD38− population frequency is 1 in 5 × 103 [17], we need, at least in part of the patients, assays to identify the smaller subpopulation of LSC within this population. Exploiting other (non-immunophenotypical) features of the LSC allows this.
About 20% [18] of AML cases are characterized by absence of neoplastic CD34+ cells [18, 19]. In these cases the commonly small CD34+ (<1%) blast population does not contain leukemic cells [18, 20, 21]. By definition, these CD34− patients lack CD34+/CD38− or CD34+/CD38+ leukemic populations; however, a potential LSC population should be found within the remaining CD34− fraction. Apparently, there are small and yet unidentified subpopulations to consider as most leukemogenic and therapy resistant in these AML cells.
Since LSC are supposed to be relatively chemotherapy resistant, the finding of a very small cellular compartment that is defined by high ABC drug transporter activity is of particular interest. Indeed a specific cell population [i.e., side population (SP)] could be identified using flow cytometry analysis in which the specific Hoechst dye 33342 [22, 23] is extruded efficiently by these drug pumps. These SP cells are resistant to AML therapies that include drugs that are used for treatment of AML patients like anthracyclines [24]. Purified SP cells were shown to have leukemic initiating capacity in NOD/SCID mouse models [24, 25] and contained both CD34+ cells and CD34− cells [26, 27] which are indeed in part neoplastic [28]. Although this suggest that a small part of the CD34− cells are therapy resistant, it remains to be established whether the SP cells are candidates for the leukemia-initiating cells in so-called CD34− leukemia.
Since the SP population can contain both HSC and LSC, inclusion of LSC-specific surface markers should aid in distinction between the LSC and HSC within the SP.
Next, stem cells are known to protect themselves by high expression of aldehyde dehydrogenase (ALDH), which is a cytosolic enzyme involved in retinoic acid metabolism maintaining cellular homeostasis. ALDH is shown to protect against DNA damage induced by reactive oxygen species and reactive aldehydes. In normal BM CD34+/CD38− HSC display high levels ALDH activity (ALDHhigh) [29]. In both normal BM and in the majority of AML BM cells, the CD34+/CD38−/ALDHhigh population is considered to contain only HSC [21, 29, 30]. In contrast to normal BM, in AML a second population can be discriminated with cells having intermediate ALDH expression [29]. When purified, this population was most potent in AML engraftment in immunodeficient mice and was generally found positive for leukemic cytogenetic markers [29]. Furthermore, presence of this population after therapy was highly predictive for relapse [29]. In conclusion, ALDH activity can be used as a functional stem cell marker, identifying HSC population and LSC population in AML. Validity to therapeutically target ALDH in AML treatment is controversial; a recent paper showed that in vitro and in vivo inhibition of ALDH selectively eradicates CD34+/CD38−ALDH+ cells [31]. In this study, the authors used the CD34+/CD38−/ALDH+ phenotype to describe LSC, which is distinct from most other studies that define CD34+/CD38−/ALDH+ to reflect HSC. To reveal whether these CD34+/CD38−/ALDH+ cells that are targeted, are indeed neoplastic cells, additional genetic characterization might be insightful.
LSC heterogeneity of LSC within a patient
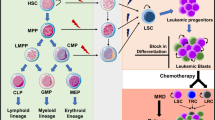
Recently it was shown that the constitution of AML at relapse may differ from diagnosis due to clonal changes including clonal evolution, clonal regression and clonal selection, with possible changes on immunophenotypic [32, 33], cytogenetic [34], genetic [34, 35] and epigenetic [35] level. Detailed whole genome sequencing studies, analyzing paired diagnosis–relapse samples, showed that at time of diagnosis, patients could present with a wide array of small subclones of which some remained in relapse [36]: indicative for clonal selection under therapy pressure. In 2012, we showed that immunophenotypically defined subpopulations of cells prominent at relapse could be traced back as very minor immature (CD34+/CD38−/dim) subpopulations of cells at diagnosis [32], again suggesting the importance of the CD34+/CD38− leukemic stem cell fraction. Since LSC are currently followed during therapy as biomarker of treatment efficacy and as prognostic factor for relapse, it is of great relevance to identify all (possibly minor) LSC populations that are potentially capable of causing relapse [37].
Clinical relevance of LSC load for prognosis
The CD34+/CD38− burden of AML patients is of strong prognostic value. In adult AML, patients with CD34+/CD38− frequencies higher than 3.5% at diagnosis had a median relapse-free survival of 5.6 months, compared to 16 months in those with lower CD34+/CD38− frequencies [14]. These results were later confirmed in other studies in adult AML [14] and in pediatric AML [38]. As knowledge on the makeup of the CD34+/CD38− fraction increased, other markers and properties were included anticipating better selectivity in defining LSC as previously summarized [5, 29]. In studies on the prognostic impact of CD34+/CD38− LSC on disease outcome, the prognostic influence of complete absence of this fraction was also discovered: CD34− status, characterized by the complete absence of neoplastic CD34+ cells [18], turned out to be an independent prognostic factor identifying patients with better prognosis in adult [17] and pediatric AML [39] compared to patient with high or low CD34+/CD38− LSC frequencies.
Despite the accumulating evidence of the prognostic relevance of LSC load at diagnosis, this feature is currently not included in risk group stratification. It is our assumption that implementation of flow cytometric quantification of LSC could be implemented without great effort using our one-tube assay [6]. Since prediction of outcome also greatly depends on many different factors during therapy, including LSC measurements during therapy (for instance at MRD time points) is warranted [40].
Impact of LSC frequency during therapy
Assessment of the frequency of remaining leukemic cells present during and after therapy (measurable/minimal residual disease, MRD) is increasingly used as an early read-out of therapy efficacy [6, 41]. MRD frequency measurement has been shown to have independent prognostic impact across different cytogenetic and molecular subgroups [42–45], and is currently used to refine risk group classifications after induction therapy. In particular, MRD is implemented in the HOVON/SAKK H132 study to guide decisions for transplantation type in intermediate risk patients. In this study, immunophenotypic MRD measurements are complemented with mutation analysis in NPM1-mutated patients, in which NPM1 status at MRD is leading for the clinical decision. In fact, many MRD studies are currently being performed, which use (or include) molecular assays [57].
It is remarkable that in all immunophenotype and/or molecular MRD studies still a proportion of MRD-negative patients develop a relapse. There are multiple possible reasons for this, e.g., low assay sensitivity, occurrence of mutational/immunophenotypic shifts or different kinetics of MRD disappearance. There may, however, also be a biological explanation: it may not only be the number of leukemic blast cells, reflecting MRD, that defines the risk of relapse, but also the number of LSC present within this blast cell population. As we argued earlier, stem cells have been demonstrated to be more therapy resistant than leukemic blast cells. The MRD population is thus likely enriched with LSC, but these are too low in frequency to contribute significantly to the total frequency of MRD cells. Indeed, when the number of CD34+/CD38− LSC after therapy was determined, LSC load was an independent predictive factor for patient survival [5]. Such was found by others too, be it with different assays and different immunophenotypical and functional definitions of stem cells [29, 46]. Remarkably, assessment of both LSC and MRD led to better separation of patients risk group classification than either MRD or LSC alone [5, 17].
For newly tested therapies, survival end point is the most important determinant of the therapeutic effectiveness. However, large clinical trials are needed with high numbers of included patients. At best, it than takes approximately 2–3 years to predict survival [47]. With increasing numbers of tested therapies, specifically targeting LSC, usage of LSC frequencies as surrogate intermediate endpoint for survival would be highly beneficial [48].
Therapeutic opportunities eliminating LSC
General principles and challenges faced by targeting LSC
With the poor prognosis of AML and only little improvements in therapeutic options, there is a pressing need for novel therapies. Therapies targeting LSC offer hope for such improvement. Fundamental to LSC therapy is the selection of the target and the timing of the therapy. Ideally, the target is highly expressed by LSC, highly selective, i.e., absence of expression on other cells in particular HSC and no circulating antigens, and preferentially expressed by high numbers of patients. Acknowledging the many similarities that LSC and HSC share, it is not surprising that current treatment approaches are limited. As the specific identification and, with that, the characterization of LSC has become more detailed, therapies directed to LSC, while sparing normal HSC, are becoming reality and are currently investigated as delineated below.
Distinct cell-surface markers have been proposed as potential LSC-specific targets (Table 1) and several approaches targeting some of these LSC surface markers are currently in clinical trials (see Table 2). Of these markers, therapies targeting CD33 are possibly the most studied in AML patients. Although targeting CD33 was originally not meant as an anti-LSC therapy, it turned out that CD33 was overexpressed in LSC compared to HSC [6]. Treatment with Gemtuzumab ozogamicin (GO) treatment was associated with reduced relapse risk and improved overall survival in patient subgroups [49, 50]. Whether GO targets CD33+ LSC, causing the reduction in relapse risk, remains unclear [50] as higher numbers of CD34+/CD38−/CD33+ cells and high CD33 expression levels decreased GO sensitivity in vitro [51]. One lesson that can be learned from GO treatment is clear: high specificity of the therapy is important. CD33 is, next to leukemic cells, also present on most HSC, on mature and immature myeloid cell and on various progenitors [52], possibly underlying toxicities as found in earlier studies [53]. Anti-CD123 therapy may have similar disadvantages [6], while results of clinical trials targeting newer discovered surface markers more specific for LSC (including CCL-1 [54, 55], TIM3 [55–57], CD96 [58]), will provide important insights in validity of therapies targeting immunophenotypic markers.
Next to specificity, the design of the antibody in terms of conjugates is of importance for effectiveness. Novel engineering of antibodies has potential to improve efficacy and reducing immunogenicity (mechanisms and constructs are reviewed by Scott et al. [59]. and Tiller and Tessier [60]).
One alternative way of direct LSC targeting is with the use of small-molecule inhibitors interfering in key signaling pathways altered in LSC (see Table 2). Using this strategy the leukemic progenitor cells are also targeted since mutations found in signaling pathways in AML are not limited to the LSC, but are inherited by their progeny. Recent studies have also indicated the relevance of splicing on signaling pathways [61]; therefore, small molecules that affect the spliceosome are also investigated as novel therapeutics to eradicate LSC [62].
Future perspectives
LSC maintenance and functioning is related, at least in part, to signals from the BM microenvironment [63–65]. Therapeutic targeting is therefore not only directed to LSC. Initial studies inhibiting factors necessary for LSC homing (e.g., CXCR4, CXCL12) have shown to abrogate chemoresistance [66], suggesting combination therapies with LSC-specific targets. Clinical trials targeting the LSC niche are in progress [67] (see Table 2).
In this review, we have conveyed the important role of LSC in AML with emphasis on the identification of LSC using flow cytometry. As the identification of CD34+/CD38− LSC allows for the identification of patients with a poor prognosis, we consider LSC measurements as valuable asset for clinical decision-making. This concerns both risk group classification at diagnosis or definition of risk groups after therapy (in an MRD situation). In view of the large heterogeneity of LSC within and among patients, the identification of all specific LSC would be too costly in terms of AML cells, time and money. For that reason, a broadly applicable simple one-tube approach has been developed, which can easily be implemented in routine diagnostics [6]. Additionally, screening for CD34+/CD38− LSC also enables identification of CD34− patients, who generally have a better prognosis [18]. Furthermore, as LSC-specific therapies—targeting LSC-specific surface markers—become available, individualized therapy may come in view. To select the most effective marker-directed therapy, the LSC phenotype of the individual patients needs to be determined. With increasing numbers of markers becoming available, innovations in flow cytometers will continue to support a growing number of channels/colors available in simultaneous measurements. The currently available multicolor flow cytometry approach used in AML does not exceed ten colors [68]. While this allows a universal screening, for precise characterization of the most pure (very minor) LSC population, multiple markers are needed. Current technological advances will come from high-number-multicolor flow cytometry or CyTOF approaches [69] in which an extensive panel of LSC markers will be available.
References
Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2.
Vormoor J, Lapidot T, Pflumio F, Risdon G, Patterson B, Broxmeyer HE, et al. Immature human cord blood progenitors engraft and proliferate to high levels in severe combined immunodeficient mice. Blood. 1994;83:2489–97.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8.
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7.
Terwijn M, Zeijlemaker W, Kelder A, Rutten AP, Snel AN, Scholten WJ, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9:e107587.
Zeijlemaker W, Kelder A, Oussoren-Brockhoff YJM, Scholten WJ, Snel AN, Veldhuizen D, et al. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia. 2016;30:439–46.
Terpstra W, Prins A, Ploemacher RE, Wognum BW, Wagemaker G, Löwenberg B, et al. Long-term leukemia-initiating capacity of a CD34− subpopulation of acute myeloid leukemia. Blood. 1996;87:2187–94.
Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, Keefer C, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2R γ c-deficient mice. J Clin Investig. 2011;121:384–95.
Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chainnull mice. Blood. 2005;106:1565–73.
Eppert K, Takenake K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–93.
Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, et al. Anti-CD38 antibody mediated clearance of human repopulating cells masks the heterogeneity of leukemia initiating cells anti-CD38 antibody mediated clearance of human repopulating cells masks the heterogeneity of leukemia initiating cells. Blood. 2008;112:568–76.
Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34—fraction leukemia initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34 fraction. Blood. 2010;115:1976–85.
Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7.
van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. 2005;11:6520–7.
Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA, et al. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–11.
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21.
Zeijlemaker W, Meijer R, Kelder A, Ham J, Oussoren-Brockhoff YJM, Snel AN, et al. Manuscript (submitted).
Zeijlemaker W, Kelder A, Wouters R, Valk PJM, Witte BI, Cloos J, et al. Absence of leukaemic CD34+ cells in acute myeloid leukaemia is of high prognostic value: a longstanding controversy deciphered. Br J Haematol. 2015;171:227–38.
Zhu H, Liu Y, Jiang H, Lu J, Qin Y, Jiang Q, et al. CD34 expression on bone marrow blasts is a novel predictor of poor prognosis independent of FlT3-ITD in acute myeloid leukemia with the NPM1-mutation. Leuk Res. 2013;37:624–30.
van der Pol MA, Feller N, Roseboom M, Moshaver B, Westra G, Broxterman HJ, et al. Assessment of the normal or leukemic nature of CD34+ cells in acute myeloid leukemia with low percentages of CD34 cells. Haematologica. 2003;88:983–93.
Schuurhuis GJ, Meel MH, Wouters F, Min LA, Terwijn M, de Jonge NA, et al. Normal hematopoietic stem cells within the AML bone marrow have a distinct and higher ALDH activity level than co-existing leukemic stem cells. PLoS One. 2013;8:e78897.
Boesch M, Reimer D, Rumpold H, Zeimet AG, Sopper S, Wolf F, et al. DyeCycle violet used for side population detection is a substrate of P-glycoprotein. Cytom Part A. 2012;81A:517–22.
Richard V, Nair MG, Santhosh Kumar TR, Pillai MR. Side population cells as prototype of chemoresistant, tumor-initiating cells. Int: Biomed Res; 2013.
Wang Y, Yin C, Feng L, Ma L, Wei Y, Sheng Q. Sorting, identification and enrichment of side population cells in THP-1 acute monocytic leukemia cells. Oncol Rep. 2013;29:1923–31.
Roshal M, Chien S, Othus M, Wood BL, Fang M, Appelbaum FR, et al. The proportion of CD34(+)CD38(low or neg) myeloblasts, but not side population frequency, predicts initial response to induction therapy in patients with newly diagnosed acute myeloid leukemia. Leukemia. 2013;27:728–31.
Feuring-Buske M, Hogge DE. Hoechst 33342 efflux identifies a subpopulation of cytogenetically normal CD34+ CD38− progenitor cells from patients with acute myeloid leukemia. Blood. 2001;97:3882–90.
Wulf GG, Wang R, Kuehnle I, Weidner D, Marini F, Brenner MK, et al. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood. 2001;98:1166–73.
Moshaver B, van Rhenen A, Kelder A, van der Pol M, Terwijn M, Bachas C, et al. Identification of a small subpopulation of candidate leukemia-initiating cells in the side population of patients with acute myeloid leukemia. Stem Cells. 2008;26:3059–67.
Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34+ CD38− cells in acute myeloid leukemia. Blood. 2012;119:3571–7.
Hoang VT, Buss EC, Wang W, Hoffmann I, Raffel S, Zepeda-Moreno A, et al. The rarity of ALDH+ cells is the key to separation of normal versus leukemia stem cells by ALDH activity in AML patients. Int J Cancer. 2015;137:525–36.
Venton G, Pérez-Alea M, Baier C, Fournet G, Quash G, Labiad Y, et al. Aldehyde dehydrogenases inhibition eradicates leukemia stem cells while sparing normal progenitors. Blood Cancer J. 2016;6:e469.
Bachas C, Schuurhuis GJ, Assaraf YG, Kwidama ZJ, Kelder A, Wouters F, et al. The role of minor subpopulations within the leukemic blast compartment of AML patients at initial diagnosis in the development of relapse. Leukemia. 2012;26:1313–20.
Ho T, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128:1671–9.
Parkin B, Ouillette P, Li Y, Keller J, Lam C, Roulston D, et al. Clonal evolution and devolution after chemotherapy in adult acute myelogenous leukemia. Blood. 2013;121:369–77.
Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F, et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22:792–9.
Ding L, Ley TJ, Larson DE, Miller CA, Kobolt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukemia revealed by whole genome sequencing. Nature. 2012;481:506–10.
Krӧnke J, Bullinger L, Teleanu V, Tschürtz F, Gaidzik VI, Kühn MWM, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122:100–9.
Witte KE, Ahlers J, Schäfer I, André M, Kerst G, Scheel-Walter HG, et al. High proportion of leukemic stem cells at diagnosis is correlated with unfavorable prognosis in childhood acute myeloid leukemia. Pediatr Hematol. 2011;28:91–9.
Hanekamp DW, Denys B, Kaspers GJL, te Marvelde JG, Schuurhuis GJ, de Haas V, et al. The relevance of stem cell load at diagnosis for the development of relapse in pediatric acute myeloid leukemia. Blood. 2015;126:2584.
Schuurhuis GJ. A gene-expression profile for leukaemia. Nature. 2016;540:346–8.
Buldini B, Rizzati F, Masetti R, Fagioli F, Menna G, Micalizzi C, et al. Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br J Haematol. 2017. doi:10.1111/bjh.14523.
Terwijn M, van Putten WLJ, Kelder A, van der Velden VHJ, Brooimans RA, Pabst T, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31:3889–97.
Klco JM, Miller CA, Griffith M, Petti A, Spencer DH, Ketkar-Kulkarni S, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314:811–22.
Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374:422–33.
Freeman SD, Virgo P, Couzens S, Grimwade D, Russell N, Hills RK, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol. 2013;31:4123–31.
Khan N, Freeman SD, Virgo P, Couzens S, Richardson P, Thomas I, et al. An immunophenotypic pre-treatment predictor for poor response to induction chemotherapy in older acute myeloid leukaemia patients: blood frequency of CD34+ CD38 low blasts. Br J Haematol. 2015;170:80–4.
Estey E, Othus M, Lee SJ, Appelbaum FR, Gale RP. New drug approvals in acute myeloid leukemia: what’s the best end point? Leukemia. 2015;30:521–5.
Han L, Jorgensen JL, Brooks C, Shi C, Zhang Q, Nogueras GM, et al. Anti-leukemia efficacy and mechanisms of action of SL-101, a novel anti-CD123 antibody-conjugate in acute myeloid leukemia. Clin Cancer Res. 2017. doi:10.1158/1078-0432.CCR-16-1904.
Jedema I, Barge RMY, van der Velden VHJ, Nijmeijer BA, van Dongen JJM, Willemze R, et al. Internalization and cell cycle-dependent killing of leukemic cells by Gemtuzumab Ozogamicin: rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia. 2003;18:316–25.
Khan N, Hills RK, Virgo P, Couzens S, Clark N, Gilkes A, et al. Expression of CD33 is a predictive factor for effect of Gemtuzumab Ozogamicin at different doses in adult acute myeloid leukemia. Leukemia. 2016. doi:10.1038/leu.2016.309.
Jawad M, Seedhouse C, Mony Y, Grundy M, Russell NH, Pallis M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia. 2010;24:74–80.
Giles F, Estey E, O’Brien S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer. 2003;98:2095–104.
de Witte T, Amadori S. The optimal dosing of gemtuzumab ozagamicin: where to go from here? Haematologica. 2016;101:653–4.
van Rhenen A, van Dongen GAMS, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–66.
Darwish NHE, Sudha T, Godugu K, Elbaz O, Abdelghaffar HA, Hassan EEA, et al. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget. 2016;7:57811–20.
Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708–17.
Kikushige Y, Miyamoto T. Identification of TIM-3 as a leukemic stem cell surface molecule in primary acute myeloid leukemia. Oncology. 2015;89(suppl 1):28–32.
Du W, Hu Y, Lu C, Li J, Liu W, He Y, et al. Cluster of differentiation 96 as a leukemia stem cell-specific marker and a factor for prognosis evaluation in leukemia. Mol Clin Oncol. 2015;3:833–8.
Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev. 2012;12:278–87.
Tiller KE, Tessier PM. Advances in antibody design. Annu Rev Biomed Eng. 2015;17:191–216.
Wojtuszkiewicz A, Assaraf YG, Maas MJP, Kaspers GJL, Jansen G, Cloos J. Pre-mRNA splicing in cancer: the relevance in oncogenesis, treatment and drug resistance. Expert Opin Drug Metab Toxicol. 2015;11:673–89.
Lee SCW, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–86.
Lee GY, Kim JA, Oh IH. Stem cell niche as a prognostic factor in leukemia. BMB Rep. 2015;48:427–8.
Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–9.
Sánchez-Aguilera A, Méndez-Ferrer S. The hematopoietic stem-cell niche in health and leukemia. Cell Mol Life Sci. 2017;74:579–90.
Rashidi A, DiPersio JF. Targeting the leukemia-stroma interaction in acute myeloid leukemia: rationale and latest evidence. Ther Adv Hematol. 2016;7:40–51.
Barbieri F, Bajetto A, Thellung S, Würth R, Florio T. Drug design strategies focusing on the CXCR4/CXCR7/CXCL12 pathway in leukemia and lymphoma. Expert Opin Drug Discov. 2016;11:1093–109.
Getta BM, Devlin S, Maloy MA, Mohanty A, Arcila M, Tallman MS, et al. Multicolor flow cytometry and multi-gene next generation sequencing are complimentary and highly predictive for relapse in acute myeloid leukemia following allogeneic hematopoietic stem cell transplant. Blood. 2016;128:834.
Han L, Qiu P, Zeng Z, Jorgensen JL, Mak DH, Burks JK, et al. Single-cell mass cytometry reveals intracellular survival/proliferative signaling in FLT3-ITD-mutated AML stem/progenitor cells. Cytometry A. 2015;87:346–56.
Askmyr M, Ågerstam H, Hansen N, Gordon S, Arvanitakis A, Rissler M, et al. Selective killing of candidate AML stem cells by antibody targeting of IL1RAP. Blood. 2013;121:3709–13.
Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, von Palffy S, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci. 2015;112:10786–91.
Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–8.
Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci. 2011;108:5009–14.
Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells yoriko. Sci Transl Med. 2010;2:179.
Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218.
Kersten B, Valkering M, Wouters R, van Amerongen R, Hanekamp D, Kwidama Z, et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br J Haematol. 2016;173:219–35.
Ponce LP, Fenn NC, Moritz N, Krupka C, Kozik JH, Lauber K, et al. SIRPα-antibody fusion proteins stimulate phagocytosis and promote elimination of acute myeloid leukemia cells. Oncotarget. 2017;8:11284–301.
Chung SS, Tavakkoli M, Klimek VM, Park CY. CD99 is a therapeutic target on disease stem cells in acute myeloid leukemia and the myelodysplastic syndromes. Blood. 2014;124:3760.
van Rhenen A, Moshaver B, Kelder A, Feller N, Nieuwint AWM, Zweegman S, et al. Aberrant marker expression patterns on the CD34+ CD38− stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia. 2007;21:1700–7.
Harrington KH, Gudgeon CJ, Laszlo GS, Newhall KJ, Sinclair AM, Frankel SR, et al. The broad anti-AML activity of the CD33/CD3 BiTE antibody construct, AMG 330, is impacted by disease stage and risk. PLoS One. 2015;10:1–13.
Sutherland MSK, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122:1455–63.
Noordhuis P, Terwijn M, Rutten AP, Smit L, Ossenkoppele GJ, Schuurhuis GJ. Targeting of CLEC12A in acute myeloid leukemia by antibody-drug-conjugates and bispecific CLL-1×CD3 BiTE Antibody. Blood. 2010;116:2890.
Sutherland MSK, Yu C, Walter RB, Westendorf L, Valliere-Douglass J, Pan L, et al. SGN-CD123A, a pyrrolobenzodiazepine dimer linked anti-CD123 antibody drug conjugate, demonstrates effective anti-leukemic activity in multiple preclinical models of AML. Blood. 2015;126:330.
Gadhoum SZ, Madhoun NY, Abuelela AF, Merzaban JS. Anti-CD44 antibodies inhibit both mTORC1 and mTORC2: a new rationale supporting CD44-induced AML differentiation therapy. Leukemia. 2016;30:2379–401.
Blackburn JS, Liu S, Wilder JL, Dobrinski KP, Lobbardi R, Moore FE, et al. Clonal evolution enhances leukemia propagating cell frequency in T-cell acute lymphoblastic leukemia through akt/mtorc1 pathway activation. Cancer Cell. 2014;25:366–78.
Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia. 2008;22:147–60.
Zeng Z, Shi YX, Tsao T, Qiu Y, Kornblau SM, Baggerly KA, et al. apoptosis in AML cells under conditions mimicking the bone marrow microenvironment targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood. 2012;120:2679–89.
Zeng Z, Wang Y, Qiu YH, Mak DH, Coombes K, You SY, et al. MLN0128, a novel mTOR kinase inhibitor, disrupts survival signaling and triggers apoptosis in AML and AML stem/progenitor cells. Oncotarget. 2016;7:55083–97.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41.
Etchin J, Berezovskaya A, Conway AS, Galinsky IA, Stone RM, Baloglu E, et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia. 2017;31:143–50.
Etchin J, Montero J, Berezovskaya A, Le BT, Kentsis A, Christie AL, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia. 2016;30:190–9.
Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–9.
Fukushima N, Minami Y, Kakiuchi S, Kuwatsuka Y, Hayakawa F, Jamieson C, et al. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016;107:1422–9.
van der Helm LH, Bosman MCJ, Schuringa JJ, Vellenga E. Effective targeting of primitive AML CD34+ cells by the second-generation proteasome inhibitor carfilzomib. Br J Haematol. 2015;171:652–5.
Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. J Clin Investig. 2014;124:528–42.
Li Y, Chen K, Zhou Y, Xiao Y, Deng M, Jiang Z, et al. A new strategy to target acute myeloid leukemia stem and progenitor cells using chidamide, a histone deacetylase inhibitor. Curr Cancer Drug Targets. 2015;15:493–503.
Rau RE, Rodriguez BA, Luo M, Jeong M, Rosen A, Rogers JH, et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood. 2016;128:971–81.
Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19:357–66.
Hsieh YT, Jiang E, Pham J, Kim HN, Abdel-Azim H, Khazal S, et al. VLA4 blockade in acute myeloid leukemia. Blood. 2013;122:3944.
Acknowledgements
D. Hanekamp receives a grant from the Dutch Cancer Society (KWF)/Alpe d’HuZes; Contract Grant Number: ALPE 2013-6371.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Hanekamp, D., Cloos, J. & Schuurhuis, G.J. Leukemic stem cells: identification and clinical application. Int J Hematol 105, 549–557 (2017). https://doi.org/10.1007/s12185-017-2221-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2221-5