
Abstract
Allogeneic hematopoietic cell transplantation (allo-HCT) can be a curative strategy for hematological diseases, and the indications for allo-HCT have broadened widely due to recent progress in supportive strategies. However, patients must overcome various complications and chronic graft-versus-host disease (cGVHD) remains the most common allo-HCT cause of long-term morbidity and mortality. cGVHD is difficult to biologically assess due to the heterogeneity of cGVHD symptoms, and the pathogenesis of cGVHD has yet to be established. Recent experimental model progress has suggested that B-cells play a critical role in cGVHD development. Consistent with these experimental results, some clinical studies investigating B-cell depletion and modulation of B-cell signaling pathways have decreased cGVHD incidence and provided some therapeutic benefit. However, randomized control studies are necessary to confirm the efficacy of B-cell targeting drugs for cGVHD. Here, we review the pathophysiology of cGVHD, especially focusing on the role of B-cell immunity, and discuss the efficacy of both B-cell depletion and modulation of B-cell signaling pathways in human cGVHD prevention, initial treatment, and salvage treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 20,000 allogeneic hematopoietic cell transplantations (allo-HCT) are performed annually in the world, and the indications for allo-HCT have broadened. Progress within the past half century regarding alternative donor sources, reduced-intensity conditioning (RIC), immunosuppressive agents, and other variable supportive cares has led to the expanded therapeutic use of allo-HCT [1, 2]. While allo-HCT has curative potential and has become one of standard treatments for hematological diseases, patients must overcome various complications including relapse, infection, and graft-versus-host disease (GVHD). Approximately, half of patients post-allo-HCT suffer from chronic graft-versus-host disease (cGVHD) [3–7], and cGVHD still remains associated with a high degree of long-term morbidity and mortality [8]. Patients who suffer from cGVHD often require a prolonged period of corticosteroid therapy, which may have adverse effects on diabetes, infections, osteoporosis, and mental status, all of which may lead to an impaired quality of life (QOL) [9, 10]. However, cGVHD pathogenesis and treatment remain poorly understood. Therefore, more efficient approaches to predict, prevent, and treat cGVHD should be explored. The role of B-cells in cGVHD has been suggested in both preclinical and clinical studies [11–17]. Here, we will review the recent findings of the pathophysiology of cGVHD and progress in use of B-cell modulating drugs to manage cGVHD.
Chronic graft-versus-host disease
Incidences and features of cGVHD
In general, cGVHD develops 100 days or later following allo-HCT and remains a major concern among long-term survivors. The incidence ranges between 30 and 70 % [18, 19], and the established risk factors for cGVHD development include age of patients and donors, disease types, cytomegalovirus serostatus, HLA-mismatch, male patients with female donors (F→M HCT), donor sources (use of mobilized peripheral blood or cord blood), use of total body irradiation (TBI), use of anti-thymocyte globulin (ATG), conditioning intensity, and grade of aGVHD [6, 18, 20, 21].
The National Institute of Health (NIH) consensus development project proposed diagnostic criteria in for cGVHD in 2005, and these criteria have recently been revised [19, 22]. According to the NIH consensus, the development of cGVHD may involve inflammation, fibrosis, cell-mediated immunity, and humoral immunity [19, 22]. Chronic GVHD affects various organs throughout body. The symptoms, which include sicca syndrome, skin sclerosis, and pulmonary involvement, bear resemblance to those of autoimmune diseases. The NIH consensus criteria include distinctive diagnostic organ-specific manifestations for cGVHD, including poikiloderma, lichen planus, sclerosis, and depigmentation for skin involvement, bronchiolitis obliterans syndrome for lung involvement, esophageal web for gastrointestinal tract involvement, fasciitis, joint stiffness, and myositis for muscle/joint involvement [19, 22]. Erythema, maculopapular rash, gingivitis, anorexia, weight loss syndrome, and hepatic dysfunction are also observed commonly but not included in the NIH diagnostic criteria. On the other hand, cryptogenic organizing pneumonia (previously known as bronchiolitis obliterans organizing pneumonia) [23], pancreatic atrophy [24, 25], and thrombocytopenia are defined as other features or unclassified entities of cGVHD.
The NIH scoring system estimates global severity of cGVHD by considering eight organs (skin, mouth, eyes, gastrointestinal tract, liver, lungs, joint and fascia, and genital tract) and defines mild as ≤2 organs with ≤score 1, moderate as ≥3 organs with ≤score 1 or at least 1 organ with a score 2 or lung score 1 and severe as at least 1 organ with score 3 or lung score ≥2 [19]. It should be noted that any pulmonary involvements increase the global severity score. Bronchiolitis obliterans syndrome is thought to drastically impair recipients’ QOL and is associated with inferior survival [26–28].
Pathophysiology
While the pathophysiology of aGVHD has been established as an allo-reactive T-cell inflammatory cascade, the pathophysiology of cGVHD remains to be elucidated [8]. Acute GVHD is thought to involve immunological inflammatory interactions between donor immune cells, recipient’s antigen-presenting cells (APCs), and host tissues [29]. In accordance with this principle, many animal studies for aGVHD have so far shown the critical role of (1) host APC activation by tissue damage due to conditioning, (2) donor T-cell activation through interactions with host APC, and (3) excessive production of cellular and inflammatory mediators [29–34]. In fact, it has been established that TBI and T-cell depletion using ATG and alemtuzumab clinically affect the incidence of aGVHD [35–37]. Thus far, both clinical and experimental studies of aGVHD development have been consistent with an allo-reactive T-cell inflammatory cascade. On the other hand, there remains much debate regarding the pathophysiology of cGVHD [29, 38]. The difficulty in understanding cGVHD pathogenesis may be due to the focused nature of reductionist animal models providing restricted insights while human cGVHD is more heterogeneous and temporally evolving [39]. Human cGVHD symptoms vary by organ involvements, timepost-HCT, and inflammatory vs. fibrotic pathology. The major consequence of cGVHD is immune dysregulation and failure to achieve functional tolerance. Existing murine models of cGVHD simulate pathological manifestations involved in cGVHD, including sclerotic cGVHD models, auto-antibody producing models, or less common immune complex deposition models [39, 40]. However, these models fail to accurately represent the full spectrum of human cGVHD symptoms [39]. Several groups have developed promising models representing multi-organ cGVHD involvement and have demonstrated cGVHD pathophysiology involving B-cells [13, 41–45]. Dr. Blazar’s research group has shown that fibrosis developed in the lung and liver in association with CD4+ T-cells and B220 B-cell infiltration and alloantibody deposition [13], and that increased follicular helper T-cells and germinal center B-cells were required for cGVHD development [42]. Dr. Zeng’s research group has shown that donor B-cells augmented clonal expansion of pathogenic CD4+ T-Cells inducing autoimmune-like cGVHD [44], and that thymic damages and cGVHD occurred through both CD4+ and CD8+ T-cells [41]. Dr. MacDonald’s research group has suggested that donor-derived macrophages mediated cGVHD [43]. These experimental models indicate that the constellation of diverse clinical manifestations of cGVHD results from donor T-cell and B-cell allo-immune interactions, as well as fibrosis via macrophages and fibroblasts. In particular, the role of B-cell pathogenicity in cGVHD has recently garnered increased attention from both transplant clinicians and researchers, due to the availability of novel pharmacologic agents targeting B-cells. The role of B-cells in cGVHD pathogenicity will be further described in the section below.
Biomarkers of cGVHD
Along with the advances in experimental cGVHD animal models providing cGVHD pathogenesis insights, human biomarkers of cGVHD are now also contributing to our understanding [8, 46]. Chronic GVHD biomarkers, which are categorized as providing diagnostic, predictive, or prognostic insights, include cellular phenotypes, mediators like cytokines, and antibodies. In terms of cellular biomarkers, regulatory B-cell deficiencies and altered regulatory T-cell homeostasis are observed in cGVHD patients [47, 48]. As an example of a mediator biomarker, elevation of chemokine ligand 9 (CXCL9) correlates with cGVHD diagnosis [49], and high-molecular-weight adiponectin is reported in association with the severity of cGVHD [50]. Antibody (Ab) biomarkers, as a distinctive indicator of allo-immunity, include the development of anti-HY (minor histocompatibility antigens encoded on Y-chromosome) Abs [51, 52]. Other auto-/allo-Abs may be pathogenic like anti-platelet-derived growth factor receptor (PDGFR)-Abs [53, 54]. Gene polymorphisms can be another category of biomarker [55]. Continued biomarker investigations will reveal a broad network of pathophysiology in the development or deterioration of cGVHD, although independent validation studies for each are essential to confirm the role of these biomarkers in cGVHD development.
B-cell immunity and cGVHD
B-cells are multifunctional, providing (1) antigen presentation via both class I and II HLA and T-cell costimulation, (2) cytokine and chemokine production, and (3) diverse antibody production. Antigens are internalized, subsequently processed, and directly presented by B-cells after binding to the B-cell receptor (BCR) [11]. Furthermore, antibody binding can concentrate and transport antigens to lymph nodes and spleen for further antigen presentation. B-cells are activated via BCR binding and produce many kinds of cytokines and chemokines, including IL-4, IL-6, and IL-10, and regulate CD4+ helper T-cells directly and indirectly [11]. Antibody production protects from foreign antigens and pathogens, including bacteria, viruses, and toxoids. The formation of antibody–antigen immune complexes leads to activating complement and effector cells.
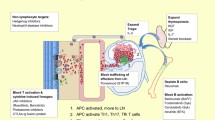
B-cell reconstitution after allo-HCT is generally thought to be slow, and IgG levels do not return to normal for 3–12 months [56, 57]. In fact, hypogammaglobulinemia has been observed with a median nadir of 56 days post-HCT [58], suggesting that novel Ab production and B-cell reconstitution may require critical post-HCT events such as withdrawal of immune suppressive drugs or reconstituted lymph nodes to organize coordinated B and T-cell immunity [59]. A growing body of evidence based on both human clinical observations and animal models has indicated pathogenic potential of B-cells and antibody in cGVHD development [60]. Elements of B-cell reconstitution, such as elevated BAFF during post-HCT lymphopenia, aberrant B-cell reconstitution, and allogeneic B-cell development, have been presumed to associate with cGVHD development (Fig. 1) [8, 11]. Aberrant B-cell homeostasis after allo-HCT has recently been reviewed by Drs. Sarantopoulos and Ritz [60].

a The development of allogeneic and auto-reactive B-cell and antibody responses. b Immune phenotype of B-cell maturation. Allo-HCT allogeneic hematopoietic cell transplantation, BAFF B-cell activating factor, BCR B-cell receptor, GC germinal center, cGVHD chronic graft-versus-host disease. Through allogeneic HCT, donor allogeneic B-cells are adoptively transferred into recipients. Until these donor B-cells are reconstituted in recipients, precursor and naïve B-cells are reduced. This lymphopenic state post-HCT results in elevated BAFF production. Persistent increased BAFF levels may lead to dysregulated B-cell homeostasis post-HCT resulting in the expansion/survival of potentially allogeneic or auto-reactive transitional B-cells. These allogeneic and auto-reactive transitional B-cells encounter allo-/auto-antigens (i.e., H-Y antigens) through circulation of body and lymph nodes, and corresponding BCR signaling enhances maturation and proportionally increases allogeneic and auto-reactive pre-GC B-cells. Eventually, these allogeneic and auto-reactive B-cells survive and escape deletion in lymphoid germinal center and differentiate into plasmablast or plasma cells, producing allogeneic/auto-reactive antibodies. These allogeneic and auto-reactive antibodies could cause inflammation, leading to cGVHD development
One of the first suggestions that B-cells contribute to cGVHD pathogenesis is the observation that allogeneic HY-Ab associate with cGVHD development. Following sex-mismatched allogeneic transplantation that is when male patients have female donors (F→M HCT), allogeneic antibodies develop 3–12 months post-HCT targeting minor histocompatibility antigens encoded on Y-chromosome, including DDX3Y, UTY, ZFY, EIF1AY, RPS4Y, and SMCY, and as a collective group, we call these allogeneic antibodies HY-Ab [52]. Sex-mismatch and H-Y antigens provide a validated and easy to understand human biological model of allo-immune responses in HCT [52]. Dr. Miklos and his colleagues pioneered the identification of HY-Abs and H-Y specific B-cells in association with cGVHD [51, 52, 61, 62]. The concept of H-Y immunity is that naïve lymphocytes from female donors recognize H-Y antigens of male recipients as allo-antigens and respond to attack tissues of male recipients, eventually leading in cGVHD development [52]. Our recent investigation of HY-Abs and cGVHD using a novel microarray system has revealed that half of F→M HCT patients have HY-Abs 3 months post-HCT, and the cumulative number of HY-Abs 3 months post-HCT predicts subsequent cGVHD development and non-relapse mortality [61]. Furthermore, B-cells specific to DBY-2, an immune-dominant peptide of DBY, are also detected in a median of 0.7 % (0.3–1.0 %) of CD19+ B-cells 6–12 months after F→M HCT patients and associated with cGVHD development [62]. Interestingly, these IgM+ B-cells antigen specific to DBY-2 were CD19+IgD+CD27–CD38+ transitional B-cells. The role played by the DBY-2-specific B-cells in cGVHD may drive the expansion of coordinated allo-reactive T-cells that induce cGVHD or through expansion and maturation by virtue of sex-mismatch between the host and recipient. In animal models, Dr. Blazer’s research group has shown that allo-antibody deposition is observed in cGVHD-involved organs, and transplantation from mice deficient in IgG production results in reduced cGVHD incidence. Together these results suggest a conserved pathogenic role of allo-Abs in cGVHD development [13].
A second B-cell role in cGVHD involves autoimmunity and dysregulated immunity. The production of auto-Abs against PDGFR is one of the established auto-Abs associated with the fibrotic change seen in cGVHD, especially extensive cGVHD [53]. These stimulatory PDGFR-Abs are reported to induce tyrosine phosphorylation, reactive oxygen species (ROS), and type 1 collagen gene expression through the Ha-Ras-ERK1/2-ROS signaling pathway [53]. Other various autoantibodies are frequently observed in cGVHD patients, but the pathophysiologic roles of these other autoantibodies remain to be elucidated [54]. In animal models, Dr. Zeng’s research group has shown that high levels of autoantibodies develop in cGVHD mice with sclerodermatous skin damage and glomerulonephritis [63]. Their results show that donor B-cells play the role of efficient APCs, which augment auto-reactive donor CD4+ T-cell expansion, thereby inducing cGVHD with autoimmune manifestations [44].
Third, cytokines affecting B-cell dysregulation are also involved in the network of biological development of cGVHD. In human studies, B-cell activating factor (BAFF) has been reported to be associated with active cGVHD [64]. The persistent high level of BAFF indicates dysregulated B-cell homeostasis and enhances the survival of allogeneic and auto-reactive transitional B-cells (Fig. 1) [14, 64, 65]. Furthermore, increased BCR signaling via contact with allo-/auto-antigens enhances the survival and maturation of these allo-/auto-reactive B-cells that can escape from deletion in lymphoid germinal center. Thus, the failure to achieve functional tolerance with limited B-cell diversity would lead to cGVHD development. B-cells in cGVHD patients are activated with elevated BCR proximal signaling pathways: Bruton’s tyrosine kinase (BTK) and spleen tyrosine kinase (Syk) [66, 67]. These molecular pathways of BAFF and BCR are candidates for newly developing targeted treatments (Fig. 2) [60].

B-cell signaling pathways and novel-targeted drugs. BAFF-R B-cell activating factor receptor, BCR B-cell receptor, Syk spleen tyrosine kinase, BTK Bruton’s tyrosine kinase, ITK IL2-inducible T-cell kinase, BLNK B-cell linker proteins, NF-κB nuclear factor-kappa B, ERK extracellular signal-regulated kinase, TKIs tyrosine kinase inhibitors, TFH T follicular helper cells. Current B-cell-targeted therapies may be categorized as (1) B-cell-targeted antibody mediated depletion, (2) BAFF receptor pathway modulators, and (3) BCR response inhibitors. First, pathogenic B-cells expressing CD20 can be deleted by total B-cell depletion, including rituximab and ofatumumab. Second, BAFF receptor pathways contribute to increased survival of allogeneic/auto-reactive B-cells. BAFF receptor pathways involve signaling to activate ERK, AKT, and NF-κB. ERK can be activated via Syk. Inhibitions of Syk by fostamatinib and NF-κB by bortezomib may modulate allogeneic/auto-reactive B-cell survival. Third, antigen-binding BCR response enhances maturation, differentiation, and survival of allogeneic/auto-reactive B-cells through SyK, BTK, and BLNK signaling, resulting in the activation of NF-κB and ERK. Syk inhibition can be induced by fostamatinib as mentioned above and BTK by ibrutinib and TKIs. Additionally, TFH can provide help for the development, germinal center formation, and maturation of allogeneic/auto-reactive B-cells through IL-21. Increased frequency of TFH correlates with increased GC B-cells and cGVHD development in animal models. Ibrutinib can inhibit ITK in TFH, an important protein for the development of Th2 and Th17. Fostamatinib can also inhibit SyK in TFH. Therefore, ibrutinib and fostamatinib may also regulate the development of allogeneic/auto-reactive B-cells in lymphoid germinal center through regulation of TFH
Targeting B-cells provides therapeutic benefit for cGVHD patients
Given the potential pathogenic role of B-cells in cGVHD development, B-cell-targeted therapies promise to provide cGVHD treatment benefit via (1) total B-cell depletion, (2) BAFF receptor inhibition, and (3) BCR signaling inhibition (Fig. 2). Many investigations have explored the clinical significance of these strategies in cGVHD treatment through pharmacologic agents including rituximab, tyrosine kinase inhibitors (TKI), Bruton’s tyrosine kinase (BTK) inhibitor, and bortezomib (Fig. 2).
Rituximab
Rituximab is a monoclonal Ab against CD20 that depletes CD20+ B-cells mainly via antibody-dependent cell cytotoxicity (ADCC). Initially clinical observations and later phase II clinical trials assessed the efficacy of rituximab on cGVHD [68–73] and have well been described [11, 15–17]. Selected clinical trials/observations are summarized in Table 1.
Prevention
Some hypothesized that rituximab infusion early following allo-HCT would deplete allogeneic B-cells adoptively transferred from donors before they are activated by recipient allo-antigens (Fig. 1). However, the prophylactic benefit of rituximab for cGVHD prevention still remains a matter of debate [16]. Previous studies of rituximab-containing conditioning or aGVHD prophylaxis suggested that cGVHD incidences range between 20 and 60 % [16, 71, 74–76]. The variability of this broad range may be due to the heterogeneity of patient characteristics including conditioning intensity, donor sources, and the timing of rituximab administration.
To the best of our knowledge, there have been two prospective studies to assess post-HCT rituximab administrations 2 months or later for the purpose of preventing cGVHD. Researchers at Stanford University investigated the efficacy of weekly rituximab infusion days 56, 63, 70, and 77 post-HCT and reported a promisingly low cGVHD incidence of 20 % with no H-Y antibody development occurring in 10 F→M HCT recipients [71]. However, these patients had received total lymphoid irradiation with anti-thymoglobulin (TLI-ATG) conditioning which results in a relatively low cGVHD incidence of 30 % [37]. Researchers from the Dana-Farber Cancer Institute have shown cGVHD benefit when infusing 375 mg/m2 rituximab 3, 6, 9, and 12 months post-HCT yield in 48 % cGVHD incidence compared with their historical control cGVHD incidence of 60 % (P = 0.1). A reduction of corticosteroid-requiring cGVHD was also observed (31 vs. 49 %, P = 0.015) at 2 years [74]. These limited studies of post-HCT rituximab prophylaxis show potential to reduce cGVHD incidence or severity in accordance with the eradication of allogeneic B-cells (Fig. 1). However, large randomized control studies are necessary to make a conclusion regarding the prophylactic efficacy of rituximab on cGVHD.
Initial treatment
A pilot study of rituximab as an initial therapy with corticosteroids has been conducted and was presented at the 2013 BMT Tandem Meeting [73]. Briefly, the clinically meaningful responses (CR or PR with <0.25 mg/kg/day of prednisone) were observed in 43 % of the 35 patients at 6 months and 1-year post-HCT. Rituximab had a significant steroid-sparing effect. Clinical response was associated with high counts of naïve B-cells and low level of BAFF prior to rituximab treatment. Of the 35 patients, 15 received F→M HCT, and 9 (56 %) had HY-Ab at cGVHD diagnosis. This HY-Ab became undetectable in the responder group, while 3 of the 4 non-responders remained HY-seropositive. DBY2-specific B-cells were also detected in 8 of the 15 F→M HCT patients and became undetectable following rituximab therapy. The HY-Ab and DBY2-specific B-cells recurred about 1.5 years post-rituximab [73]. This study suggests that adding rituximab to corticosteroid for initial cGVHD treatment may be effective 1-year post-rituximab, but the response duration may be limited due to the persistence and recurrence of allogeneic B-cells. Ofatumumab, another CD20 targeting drug, is also under investigation as an initial treatment for cGVHD [77].
Treatment for steroid-refractory cGVHD
There have been many studies to address the efficacy of rituximab treatment for steroid-refractory cGVHD [16, 78, 79]. A systemic review and meta-analysis of rituximab treatment has shown that overall response rate was 0.66, and organ-specific responses were 0.60 in skin, 0.36 in mucosa, 0.29 in liver, 0.31 in gastrointestinal tract, and 0.30 in lung [80]. Recently, a prospective, multi-center, randomized, two-arm phase II crossover trial has been conducted to assess the efficacy of imatinib (200 mg daily) and rituximab (375 mg/m2 intravenously weekly 4× doses, repeatable after 3 months) for cutaneous sclerotic cGVHD (NCT01309997), which were presented at the 2015 BMT tandem meeting. A stringent objective improvement in physical findings was seen in <35 % at 6 months after initial treatment [81].
In total, rituximab appears to have the potential to prevent and treat cGVHD, although randomized comparison studies are needed. Additionally, our H-Y studies in initial treatment trials suggested that future investigation of rituximab to eradicate activated allogenic B-cells should consider other strategies: intermittent rituximab administration post-HCT or incorporation with novel drugs targeting BCR and BAFF signaling (Fig. 2).
B-cell receptor signaling inhibition
BCR stimulation causes maturation and proliferation of allogeneic/auto-reactive B-cells (Fig. 1). Thus, BCR downstream signaling may be effective targets for cGVHD treatment. BTK and Syk are two key signaling components of BCR downstream pathways that are the targets of FDA approved drugs. Inhibitors of BTK and Syk regulate B-cell proliferation and survival and exert anti-tumor effect on B-cell malignancies [82–86]. Ibrutinib is an established BTK inhibitor and has been reported to ameliorate cGVHD in a murine model [87]. The efficacy of ibrutinib in human cGVHD is now under investigation. The serendipitous observation of a CLL patient resolving his mucocutaneous cGVHD when treated for CLL relapse on the initial phase I ibrutinib CLL treatment trial fostered interest [88]. Currently, a phase Ib/II trial is ongoing for steroid-dependent/refractory cGVHD using the well-tolerated phase II dose of 420 mg daily (NCT02195869).
Tyrosine kinase Inhibitor (TKI) drugs including imatinib, dasatinib, and nilotinib were developed for the treatment of chronic myeloid leukemia (CML) or Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL). Relevant to B-cell biology, these TKI block c-Abl kinase activity, leading to inhibition of BCR signaling and reduced B-cell proliferation [38]. In fact, hypogammaglobulinemia develops in CML patients who receive TKI therapy for an extended period of time [38, 89]. A recent investigation has suggested that these TKIs also partially inhibit BTK phosphorylation and impair B-cell immune responses [90]. In addition, TKIs also have a suppressive effect on PDGFR pathways, which are stimulated through auto-PDGFR-Abs in fibrotic cGVHD patients [53]. In summary, TKIs are thought to have an effect on cGVHD through B-cell suppressive effects as well as an inhibition of these PDGFR pathways. Of the available TKIs, imatinib is the most commonly investigated drug for cGVHD treatment (Table 2).
Prevention
A reduced incidence of cGVHD has been reported in several prospective studies of prophylactic TKI administration to reduce relapse of Philadelphia chromosome (Ph)-positive leukemia (Table 2). However, a prospective direct comparison of prophylactic impact on cGVHD development between patients treated with or without imatinib is lacking. There has been only one retrospective analysis designed to compare cGVHD incidence between patients with and without prophylactic imatinib maintenance in 96 Ph+ leukemia patients [24]. The 3 years cumulative cGVHD incidence was significantly reduced in the imatinib treated group (40 %) compared with the no-imatinib group (70 %), and multivariate analyses demonstrated that imatinib post-HCT was significantly associated with the reduced development and severity of cGVHD [24]. Prospective randomized placebo-controlled clinical trials for cGVHD prevention or treatment should be considered.
Initial treatment
To the best of our knowledge, no study has been published to report the efficacy of TKI as an initial therapy for cGVHD.
Salvage treatment for steroid-refractory/dependent cGVHD
The efficacy of imatinib on steroid-refractory sclerotic cGVHD differs by reports, and the overall response rate ranges between 30 and 80 %. The variability in responses may be due to trial differences in dose, duration, and day of imatinib treatment initiation post-HCT (Table 2).
A phase I study demonstrated that imatinib treatment for steroid-refractory/dependent cGVHD was tolerated at a dose of 400 mg/day in 9 of 15 patients and was administrated for 6 months and beyond in 10 patients. The overall response rate was 40 % (6 of 15) [91]. The investigators suggested that 200 mg/day may be an adequate dose for cGVHD treatment, as the dose is limited by both side effects and consent withdrawal. In this study, anti-PDGFR antibodies were also assessed and detected in 7 of 11 evaluable subjects but correlated with clinical activity in only 4 patients.
A phase II study assessed 40 steroid-refractory cGVHD patients with cGVHD involvement of skin in 32 (80 %), lung in 33 (83 %), mouth in 23 (58 %), and gut in 9 (23 %) [92]. Imatinib administration was initiated with a dose of 100 mg/day and escalated as high as the patients tolerated. Most patients received imatinib at 200 mg/day, and the median duration of imatinib administration was 16 months (range 1–45 months). Of the evaluable 32 patients at 6 months, the overall response rate was about 50 %, and the best response rates were observed in the gut and lungs, with 50 and 35 % of affected patients. The researchers also investigated anti-PDGF-R antibody activity in 11 patients (responder in 7 and non-responder in 4) and evaluated the relative decrease in PDGFR agonistic activity in terms of ROS stimulation (ROS index) compared to baseline. ROS index was decreased in the responder group, but it did not change substantially in the non-responder group [92].
The use of imatinib as a salvage treatment appears feasible and effective, achieving an overall response in half of the patients. Larger randomized controlled trials are required to confirm the results of previous studies. Regarding dasatinib and nilotinib, only case series are available [93], and several phase I/II trials are ongoing to assess their role for salvage treatment of cGVHD (NCT01155817 and NCT01810718).
Bortezomib
Treatment with bortezomib, an established proteasome inhibitor agent, causes B-cells and plasma cells to cell cycle arrest and subsequent apoptosis. Bortezomib has well been investigated in B-cell malignancies, especially in multiple myeloma and mantle cell lymphoma [94]. In addition to the cytotoxic effects against B-cell tumors, bortezomib is also known to have immune-modulatory functions through inhibitions of nuclear factor-kappa B (NF-κB) pathways, leading to a decrease of pro-inflammatory cytokines and allo-reactive T-cells [94]. Several investigations in human and animal studies have been published to assess the efficacy of bortezomib on cGVHD (Table 3).
Prevention
The efficacy of bortezomib-based GVHD prophylaxis has recently been assessed in the phase I/II study among 45 RIC patients who underwent HCT form HLA-mismatched unrelated donors [95]. The dose schedule was 1.3 mg/m2 intravenous administration (iv) on days 1, 4, and 7 post-HCT. The 1-year cGVHD incidence was 29 % in this cohort, comparable with that in HLA-matched RIC patients [95]. The study also suggested that bortezomib enhanced immune reconstitution of CD8 T-cells and natural killer cells [95].
Initial treatment
A phase II study of bortezomib with corticosteroid as an initial treatment for cGVHD patients was recently conducted [96]. Twenty-two patients were enrolled, and bortezomib was administered at 1.3 mg/m2 intravenously on days 1, 8, 15, and 22 of each 35-day cycle for 3 cycles (15 weeks), with a dose at 0.5–1 mg/kg/day of prednisone. The overall response rate was 80 %, including 10 % of CR and 70 % of PR. In detail, organ-specific response was observed in 73 % for skin, 53 % for liver, 75 % for gastrointestinal tract, and 33 % for joint or muscle involvements [96]. Only one patient experienced grade 3 sensory neuropathy.
Salvage treatment for steroid-refractory/dependent cGVHD
A pilot study investigating the use of bortezomib as salvage therapy for steroid-refractory/dependent cGVHD was recently conducted based on favorable results from mice experiments [97]. Ten patients were enrolled in the dose-escalating study, which started from 0.2 mg/m2 subcutaneously once a week, and then increased every 2 weeks by 0.2 mg/m2 until the patients responded or grade III to IV dose-limited toxicity occurred. Six of the 10 patients could receive >60 % of the scheduled doses, and five patients responded and achieved at least PR. The remaining five patients experienced adverse events: a progressive bronchiolitis obliterans and multiple respiratory infections, neuropathy, viral mouth infection, relapse of underlying disease, or worsening of thrombocytopenia [97].
In total, bortezomib may be effective and tolerable to prevent and treat cGVHD but the use of bortezomib may be limited by toxicities. However, direct comparison with other strategies is necessary to make any further conclusion regarding its use, and clinical trials combining bortezomib with other B-cell agents remain to be explored.
Summary and future perspectives
A growing body of evidence supports a pathogenic B-cell immunity role in cGVHD development. Allogeneic and auto-reactive B-cells develop through elevated BAFF and subsequent increased BCR response to allo-/auto-antigens in recipients. Rituximab clinical trials suggested that B-cell depletion could eradicate allogeneic B-cells and decrease cGVHD severity, but these allogenic B-cells seem to overcome Rituximab therapy of new onset cGVHD and often recur with cGVHD progression. Studies testing extended retuximab scheduling or combination with drugs targeting BCR pathways seem worthy to investigate. Furthermore, while B-cell targeting or modulating therapy would be effective to prevent and treat cGVHD, the optimal dose and timing has yet to be established. For the purpose of optimization, it is necessary to understand when the allo-immune response occurs and further elucidate B-cell pathogenesis in cGVHD. Therefore, monitoring allo-immune response like H-Y-Abs could shed light on how a novel B-cell drug affects allo-humoral reaction. We will assess the dynamics of H-Y-Abs and H-Y-specific B-cells in ongoing TKI, BTK, or rituximab clinical trials for cGVHD. These studies will provide important pharmacodynamic insights of B-cell targeting drugs in cGVHD treatment for optimal timing or doses of the drugs, which may be applied to all types of allo-HCT.
References
Powles R. 50 years of allogeneic bone-marrow transplantation. Lancet Oncol. 2010;11(4):305–6.
Gratwohl A, Baldomero H, Aljurf M, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA. 2010;303(16):1617–24.
Vigorito AC, Campregher PV, Storer BE, et al. Evaluation of NIH consensus criteria for classification of late acute and chronic GVHD. Blood. 2009;114(3):702–8.
Thepot S, Zhou J, Perrot A, et al. The graft-versus-leukemia effect is mainly restricted to NIH-defined chronic graft-versus-host disease after reduced intensity conditioning before allogeneic stem cell transplantation. Leukemia. 2010;24(11):1852–8.
Cho BS, Min CK, Eom KS, et al. Feasibility of NIH consensus criteria for chronic graft-versus-host disease. Leukemia. 2009;23(1):78–84.
Kanda J, Nakasone H, Atsuta Y, et al. Risk factors and organ involvement of chronic GVHD in Japan. Bone Marrow Transpl. 2014;49(2):228–35.
Arai S, Arora M, Wang T, et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the center for international blood and marrow transplant research. Biol Blood Marrow Transpl. 2015;21(2):266–74.
Socié G, Ritz J. Current issues in chronic graft-versus-host disease. Blood. 2014;124(3):374–84.
Pidala J, Anasetti C, Jim H. Quality of life after allogeneic hematopoietic cell transplantation. Blood. 2009;114(1):7–19.
Pidala J, Vogelsang G, Martin P, et al. Overlap subtype of chronic graft-versus-host disease is associated with an adverse prognosis, functional impairment, and inferior patient-reported outcomes: a chronic graft-versus-host disease consortium study. Haematologica. 2012;97(3):451–8.
Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, von Bergwelt-Baildon MS. The role of B cells in the pathogenesis of graft-versus-host disease. Blood. 2009;114(24):4919–27.
Ratanatharathorn V, Pavletic S, Uberti JP. Clinical applications of rituximab in allogeneic stem cell transplantation: anti-tumor and immunomodulatory effects. Cancer Treat Rev. 2009;35(8):653–61.
Srinivasan M, Flynn R, Price A, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood. 2012;119(6):1570–80.
Sarantopoulos S, Blazar BR, Cutler C, Ritz J. B cells in chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2015;21(1):16–23.
Alousi AM, Uberti J, Ratanatharathorn V. The role of B cell depleting therapy in graft versus host disease after allogeneic hematopoietic cell transplant. Leuk Lymphoma. 2010;51(3):376–89.
Kharfan-Dabaja MA, Cutler CS. Rituximab for prevention and treatment of graft-versus-host disease. Int J Hematol. 2011;93(5):578–85.
Arai S, Miklos DB. Rituximab in hematopoietic cell transplantation. Expert Opin Biol Ther. 2010;10(6):971–82.
Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2003;9(4):215–33.
Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 diagnosis and staging working group report. Biol Blood Marrow Transpl. 2015;21(3):389–401.
Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117(11):3214–9.
Inamoto Y, Flowers ME. Treatment of chronic graft-versus-host disease in 2011. Curr Opin Hematol. 2011;18(6):414–20.
Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transpl. 2005;11(12):945–56.
Nakasone H, Onizuka M, Suzuki N, et al. Pre-transplant risk factors for cryptogenic organizing pneumonia/bronchiolitis obliterans organizing pneumonia after hematopoietic cell transplantation. Bone Marrow Transpl. 2013;48(10):1317–23.
Nakasone H, Ito A, Endo H, Kida M, Koji I, Usuki K. Pancreatic atrophy is associated with gastrointestinal chronic GVHD following allogeneic PBSC transplantation. Bone Marrow Transpl. 2010;45(3):590–2.
Brook OR, Mullan CP, Mendiratta-Lala M, et al. Pancreatic atrophy in patients with chronic graft-versus-host disease. Abdom Imaging. 2014;39(2):342–7.
Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA. 2009;302(3):306–14.
Nakasone H, Kanda J, Yano S, et al. A case-control study of bronchiolitis obliterans syndrome following allogeneic hematopoietic stem cell transplantation. Transpl Int. 2013;26(6):631–9.
Chien JW, Duncan S, Williams KM, Pavletic SZ. Bronchiolitis obliterans syndrome after allogeneic hematopoietic stem cell transplantation-an increasingly recognized manifestation of chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2010;16(1 Suppl):S106–14.
Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550–61.
Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285(5426):412–5.
Teshima T, Ordemann R, Reddy P, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8(6):575–81.
Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90(8):3204–13.
Hill GR, Teshima T, Rebel VI, et al. The p55 TNF-alpha receptor plays a critical role in T cell alloreactivity. J Immunol. 2000;164(2):656–63.
Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95(9):2754–9.
Nakasone H, Fukuda T, Kanda J, et al. Impact of conditioning intensity and TBI on acute GVHD after hematopoietic cell transplantation. Bone Marrow Transpl. 2014. doi:10.1038/bmt.2014.293.
Kanda J, Lopez RD, Rizzieri DA. Alemtuzumab for the prevention and treatment of graft-versus-host disease. Int J Hematol. 2011;93(5):586–93.
Kohrt HE, Turnbull BB, Heydari K, et al. TLI and ATG conditioning with low risk of graft-versus-host disease retains antitumor reactions after allogeneic hematopoietic cell transplantation from related and unrelated donors. Blood. 2009;114(5):1099–109.
Olivieri J, Coluzzi S, Attolico I, Olivieri A. Tirosin kinase inhibitors in chronic graft versus host disease: from bench to bedside. Sci World J. 2011;11:1908–31.
Chu YW, Gress RE. Murine models of chronic graft-versus-host disease: insights and unresolved issues. Biol Blood Marrow Transpl. 2008;14(4):365–78.
Blazar B, White ES, Couriel D. Understanding chronic GVHD from different angles. Biol Blood Marrow Transpl. 2012;18(1 Suppl):S184–8.
Wu T, Young JS, Johnston H, et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J Immunol. 2013;191(1):488–99.
Flynn R, Du J, Veenstra RG, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood. 2014;123(25):3988–98.
Alexander KA, Flynn R, Lineburg KE, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest. 2014;124(10):4266–80.
Young JS, Wu T, Chen Y, et al. Donor B cells in transplants augment clonal expansion and survival of pathogenic CD4+ T cells that mediate autoimmune-like chronic graft-versus-host disease. J Immunol. 2012;189(1):222–33.
Zhao D, Young JS, Chen YH, et al. Alloimmune response results in expansion of autoreactive donor CD4+ T cells in transplants that can mediate chronic graft-versus-host disease. J Immunol. 2011;186(2):856–68.
Schultz KR, Miklos DB, Fowler D, et al. Toward biomarkers for chronic graft-versus-host disease: National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: III. Biomarker Working Group Report. Biol Blood Marrow Transpl. 2006;12(2):126–37.
Khoder A, Sarvaria A, Alsuliman A, et al. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood. 2014;124(13):2034–45.
Matsuoka K, Kim HT, McDonough S, et al. Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J Clin Invest. 2010;120(5):1479–93.
Kitko CL, Levine JE, Storer BE, et al. Plasma CXCL9 elevations correlate with chronic GVHD diagnosis. Blood. 2014;123(5):786–93.
Nakasone H, Binh PN, Yamazaki R, et al. Association between serum high-molecular-weight adiponectin level and the severity of chronic graft-versus-host disease in allogeneic stem cell transplantation recipients. Blood. 2011;117(12):3469–72.
Miklos DB, Kim HT, Miller KH, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005;105(7):2973–8.
Popli R, Sahaf B, Nakasone H, Lee JY, Miklos DB. Clinical impact of H-Y alloimmunity. Immunol Res. 2014;58(2–3):249–58.
Svegliati S, Olivieri A, Campelli N, et al. Stimulatory autoantibodies to PDGF receptor in patients with extensive chronic graft-versus-host disease. Blood. 2007;110(1):237–41.
Kuzmina Z, Gounden V, Curtis L, et al. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. Am J Hematol. 2015;90(2):114–9.
Boukouaci W, Busson M, Peffault de Latour R, et al. MICA-129 genotype, soluble MICA, and anti-MICA antibodies as biomarkers of chronic graft-versus-host disease. Blood. 2009;114(25):5216–24.
Elfenbein GJ, Anderson PN, Humphrey RL, et al. Immune system reconstitution following allogeneic bone marrow transplantation in man: a multiparameter analysis. Transpl Proc. 1976;8(4):641–6.
Noel DR, Witherspoon RP, Storb R, et al. Does graft-versus-host disease influence the tempo of immunologic recovery after allogeneic human marrow transplantation? An observation on 56 long-term survivors. Blood. 1978;51(6):1087–105.
Frangoul H, Min E, Wang W, et al. Incidence and risk factors for hypogammaglobulinemia in pediatric patients following allo-SCT. Bone Marrow Transpl. 2013;48(11):1456–9.
Müller AM, Linderman JA, Florek M, Miklos D, Shizuru JA. Allogeneic T cells impair engraftment and hematopoiesis after stem cell transplantation. Proc Natl Acad Sci USA. 2010;107(33):14721–6.
Sarantopoulos S, Ritz J. Aberrant B cell homeostasis in chronic GVHD. Blood. 2015;125(11):1703–7.
Nakasone H, Tian L, Sahaf B, et al. Allogeneic HY antibodies 3 months following female to male sex-mismatched HCT predicts chronic GVHD and non-relapse mortality. Blood. 2015 [Epub ahead of print].
Sahaf B, Yang Y, Arai S, Herzenberg LA, Miklos DB. H-Y antigen-binding B cells develop in male recipients of female hematopoietic cells and associate with chronic graft vs. host disease. Proc Natl Acad Sci USA. 2013;110(8):3005–10.
Zhang C, Todorov I, Zhang Z, et al. Donor CD4+ T and B cells in transplants induce chronic graft-versus-host disease with autoimmune manifestations. Blood. 2006;107(7):2993–3001.
Sarantopoulos S, Stevenson KE, Kim HT, et al. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin Cancer Res. 2007;13(20):6107–14.
Sarantopoulos S, Stevenson KE, Kim HT, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009;113(16):3865–74.
Allen JL, Tata PV, Fore MS, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood. 2014;123(13):2108–15.
Allen JL, Fore MS, Wooten J, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood. 2012;120(12):2529–36.
Cutler C, Miklos D, Kim HT, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood. 2006;108(2):756–62.
Ratanatharathorn V, Logan B, Wang D, et al. Prior rituximab correlates with less acute graft-versus-host disease and better survival in B-cell lymphoma patients who received allogeneic peripheral blood stem cell transplantation. Br J Haematol. 2009;145(6):816–24.
van Dorp S, Pietersma F, Wölfl M, et al. Rituximab treatment before reduced-intensity conditioning transplantation associates with a decreased incidence of extensive chronic GVHD. Biol Blood Marrow Transpl. 2009;15(6):671–8.
Arai S, Sahaf B, Narasimhan B, et al. Prophylactic rituximab after allogeneic transplantation decreases B-cell alloimmunity with low chronic GVHD incidence. Blood. 2012;119(25):6145–54.
Cutler C, Kim HT, Bindra B, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase 2 trial. Blood. 2013;122(8):1510–7.
Sahaf B, Arai S, Otani J, Schoenrock K, Logan A, Miklos DB. Rituximab provides steroid-sparing therapy in new-onset chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2013;19(2):S140 (Abstract 155).
Cutler C, Kim HT, Bindra B, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase II trial. Blood. 2013;122(8):1510-7.
Sauter CS, Barker JN, Lechner L, et al. A phase II study of a nonmyeloablative allogeneic stem cell transplant with peritransplant rituximab in patients with B cell lymphoid malignancies: favorably durable event-free survival in chemosensitive patients. Biol Blood Marrow Transpl. 2014;20(3):354–60.
Khouri IF, McLaughlin P, Saliba RM, et al. Eight-year experience with allogeneic stem cell transplantation for relapsed follicular lymphoma after nonmyeloablative conditioning with fludarabine, cyclophosphamide, and rituximab. Blood. 2008;111(12):5530–6.
Pidala J, Kim J, Yue B, et al. Ofatumumab in combination with glucocorticoids for primary therapy of chronic graft vs. host disease. Biol Blood Marrow Transpl. 2015;21(2):S343–S344 (Abstract 500).
Zaja F, Bacigalupo A, Patriarca F, et al. Treatment of refractory chronic GVHD with rituximab: a GITMO study. Bone Marrow Transpl. 2007;40(3):273–7.
Kim SJ, Lee JW, Jung CW, et al. Weekly rituximab followed by monthly rituximab treatment for steroid-refractory chronic graft-versus-host disease: results from a prospective, multicenter, phase II study. Haematologica. 2010;95(11):1935–42.
Kharfan-Dabaja MA, Mhaskar AR, Djulbegovic B, Cutler C, Mohty M, Kumar A. Efficacy of rituximab in the setting of steroid-refractory chronic graft-versus-host disease: a systematic review and meta-analysis. Biol Blood Marrow Transpl. 2009;15(9):1005–13.
Arai S, Pidala J, Pusic I, et al. A randomized phase II study of imatinib and rituximab for cutaneous sclerosis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2015;21(2):S324 (Abstract 469).
Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–7.
Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94.
Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1 selective pressure in T-lymphocytes. Blood. 2013;122(15):2539–49.
Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219–32.
Dubovsky JA, Flynn R, Du J, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;124(11):4867–76.
Ryan C, Logan A, Rezvani A, et al. Ibrutinib treatment of relapsed CLL following allogeneic transplantation: sustained disease response and promising donor immune modulation. Biol Blood Marrow Transpl. 2015;21(S2):S307–8.
Santachiara R, Maffei R, Martinelli S, et al. Development of hypogammaglobulinemia in patients treated with imatinib for chronic myeloid leukemia or gastrointestinal stromal tumor. Haematologica. 2008;93(8):1252–5.
de Lavallade H, Khoder A, Hart M, et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood. 2013;122(2):227–38.
Chen GL, Arai S, Flowers ME, et al. A phase 1 study of imatinib for corticosteroid-dependent/refractory chronic graft-versus-host disease: response does not correlate with anti-PDGFRA antibodies. Blood. 2011;118(15):4070–8.
Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood. 2013;122(25):4111–8.
Sánchez-Ortega I, Servitje O, Arnan M, et al. Dasatinib as salvage therapy for steroid refractory and imatinib resistant or intolerant sclerotic chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2012;18(2):318–23.
Koreth J, Alyea EP, Murphy WJ, Welniak LA. Proteasome inhibition and allogeneic hematopoietic stem cell transplantation: a review. Biol Blood Marrow Transpl. 2009;15(12):1502–12.
Koreth J, Stevenson KE, Kim HT, et al. Bortezomib-based graft-versus-host disease prophylaxis in HLA-mismatched unrelated donor transplantation. J Clin Oncol. 2012;30(26):3202–8.
Herrera AF, Kim HT, Bindra B, et al. A phase II study of bortezomib plus prednisone for initial therapy of chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2014;20(11):1737–43.
Pai CC, Chen M, Mirsoian A, et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood. 2014;124(10):1677–88.
Khouri IF, Saliba RM, Giralt SA, et al. Nonablative allogeneic hematopoietic transplantation as adoptive immunotherapy for indolent lymphoma: low incidence of toxicity, acute graft-versus-host disease, and treatment-related mortality. Blood. 2001;98(13):3595–9.
Sahaf B, Arai S, Otani J, Schoenrock K, Logan A, Miklos DB. Rituximab provides steroid-sparing therapy in new-onset chronic graft-versus-host disease. Biol Blood Marrow Transpl. 2013;19(2):S140 (Abstract 155).
Olavarria E, Siddique S, Griffiths MJ, et al. Posttransplantation imatinib as a strategy to postpone the requirement for immunotherapy in patients undergoing reduced-intensity allografts for chronic myeloid leukemia. Blood. 2007;110(13):4614–7.
Nakasone H, Kanda Y, Takasaki H, et al. Prophylactic impact of imatinib administration after allogeneic stem cell transplantation on the incidence and severity of chronic graft versus host disease in patients with Philadelphia chromosome-positive leukemia. Leukemia. 2010;24(6):1236–9.
Caocci G, Vacca A, Ledda A, et al. Prophylactic and preemptive therapy with dasatinib after hematopoietic stem cell transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia. Biol Blood Marrow Transpl. 2012;18(4):652–4.
Zhao Y, Luo Y, Shi J, Cai Z, Huang H. Second-generation tyrosine kinase inhibitors combined with stem cell transplantation in patients with imatinib-refractory chronic myeloid leukemia. Am J Med Sci. 2014;347(6):439–45.
Shimoni A, Volchek Y, Koren-Michowitz M, et al. Phase 1/2 study of nilotinib prophylaxis after allogeneic stem cell transplantation in patients with advanced chronic myeloid leukemia or Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2015;121(6):863–71.
Magro L, Mohty M, Catteau B, et al. Imatinib mesylate as salvage therapy for refractory sclerotic chronic graft-versus-host disease. Blood. 2009;114(3):719–22.
Olivieri A, Locatelli F, Zecca M, et al. Imatinib for refractory chronic graft-versus-host disease with fibrotic features. Blood. 2009;114(3):709–18.
de Masson A, Bouaziz JD, Peffault de Latour R, et al. Limited efficacy and tolerance of imatinib mesylate in steroid-refractory sclerodermatous chronic GVHD. Blood. 2012;120(25):5089–90.
Mateos-Mazon J, Pérez-Simón JA, Lopez O, Hernández E, Etxebarria J, San Miguel JF. Use of bortezomib in the management of chronic graft-versus-host disease among multiple myeloma patients relapsing after allogeneic transplantation. Haematologica. 2007;92(9):1295–6.
Acknowledgments
This work was supported by National Heart Lung and Blood Institute Grant R21 HL084318, National Cancer Institute Grant P01 CA049605, The Leukemia and Lymphoma Society TRA 6204-06, and Stanford University Cancer Institute Support Grant 1P030CA124435-01.
Conflict of interest
Dr. Miklos has performed sponsored research funded by Novartis, Roche, and Pharmacyclics Inc.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Nakasone, H., Sahaf, B. & Miklos, D.B. Therapeutic benefits targeting B-cells in chronic graft-versus-host disease. Int J Hematol 101, 438–451 (2015). https://doi.org/10.1007/s12185-015-1782-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1782-4