
Abstract
Hereditary hemochromatosis is a disorder characterized by enhanced intestinal absorption of dietary iron. Here, we report a heterozygous genotype at two mutation sites in hemojuvelin (HJV) present in two brothers with middle-age-onset hemochromatosis in a Chinese family. To date, only homozygous or compound heterozygous states of HJV gene have been reported as associated with iron overload. However, the patients here were heterozygous for two mutations in one HJV allele in cis: a premature termination mutation (962G>A and 963C>A; C321X) and a mutation in the signal peptide (18G>C; Q6H). Previously unrecognized environmental or other genetic factors may have interacted with the heterozygous genotype in these patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary hemochromatosis (HH) is a disorder characterized by enhanced intestinal absorption of dietary iron. Without therapeutic intervention, iron overload leads to multiple organ damage, such as liver cirrhosis, cardiomyopathy, diabetes, arthritis, hypogonadism and skin pigmentation [1].
Type 1 HH is the most common form of inherited iron overload and is characterized by an autosomal recessive pattern of inheritance and associated with mutation of the HFE gene. HFE is a late-onset, low-penetrance disorder that affects predominantly men. Juvenile hemochromatosis (JH), also classified as type 2 hemochromatosis, is a rare autosomal recessive disorder of iron overload that leads to severe clinical complications typically before age 30. JH is subdivided into types 2A and 2B, which are caused by mutations in the hemojuvelin (HJV) and HAMP genes, respectively. Type 3 HH is also a rare disease caused by mutations in TFR2, and patients had iron overload similar to HFE-HH phenotype. Type 4 hemochromatosis, also called ferroportin disease, is related to mutations in SLC40A1, and it differs from other genetic iron overload disorders in that it is inherited in an autosomal dominant pattern [1, 2].
Juvenile hemochromatosis is reported as a severe hereditary form of iron overload that affects young patients of both sexes [2, 3]. The majority of cases published so far are in populations of European origin [4, 5]. Here, we describe 2 middle-aged patients with hemochromatosis in a Chinese family due to two HJV heterozygous mutations.
Materials and methods
Patients
The proband and his family are from a small village of Jiangsu Province, China. Institutional review board approval for this study was obtained from all participating institutions.
DNA collection
Informed consent for molecular studies was obtained from all persons described here. Venous blood samples of the proband and his family members were collected and genomic DNA was isolated. In addition to these family members, 200 normal unrelated individuals of the same ethnic background (100 male and 100 female) were recruited as controls.
Mutation analysis
To amplify coding sequences and exon–intron boundaries of the HFE, HAMP, TfR2, SLC40A1and HJV genes, primers for polymerase chain reaction (PCR) were synthesized as previously described [3, 6–9]. Amplification and direct sequencing to detect mutations of these genes were performed.
Computed tomography (CT) scanning
Liver biopsy is associated with certain unavoidable risks. Non-invasive CT was performed with a general electric CT (GE-CT) to estimate the density of the liver. The range of normal liver attenuation was from 52 to 68 Hounsfield units (HU).
Western blots
Serum samples were collected and immediately frozen and stored at −80 °C. Western blot analysis using anti-HJV antibody (ab104492, abcam, USA) was performed as previously reported [10]. Briefly, serum (100 μg) was suspended in western sample buffer (100 mM Hepes, pH 6.8, 10 % β-mercaptoethanol. 20 % SDS), boiled, subjected to electrophoresis on 10 % SDS-PAGE, and transferred to PVDF membrane. After blocking with TBS containing 5 % nonfat milk and 0.1 % Tween 20 for 1 h, the membrane was incubated with the primary antibody at 4 °C overnight. After washing with TBS containing 0.1 % Tween 20 three times, each for 5 min, the membrane was then incubated with horseradish peroxidase (HRP)-labeled secondary antibody for 1 h at room temperature. The membrane was then developed using the enhanced chemiluminescent (ECL) detection systems.
Results
Medical history of proband
At the age of 31, he went to the local hospital because of anepithymia and progressive fatigue, and was initially diagnosed as liver cirrhosis and diabetes. He was treated with insulin and liver protection therapy since then. When he was 35, he went to Nanjing City of China and sought better medical care for progressive dyspnea and already developed cardiomyopathy and heart failure as complication of iron overload. He was also noted to have deeply pigmented skin. His blood counts were as follows: WBC 6.3 × 109/L, Hb 135 g/L, RBC 4 × 1012/L, BPC 78 × 109/L. Serum chemistry revealed high blood glucose concentration (18 mmol/L) and high liver transaminase [AST 97 U/L (normal 5–40 U/L); ALT 73 U/L (normal 5–40 U/L)]. Troponin I (TnI) was also high: 0.37 ng/mL (normal <0.2 ng/mL), indicating the damaged heart muscle.
At the age of 36, he was admitted to our hospital. The blood was collected for the routine analysis again. The results were as follows: WBC 6.2 × 109/L, Hb 153 g/L, BPC48 × 109/L. Serum chemistry confirmed again the hyperglycemia and elevated liver transaminases. His serum ferritin concentration was >2000 ng/mL, with a transferrin saturation of 60 %. HH was recognized by us finally.
When he was in our hospital, he once had a sudden ventricular tachycardia in hospital and was saved. Therapy was initiated with phlebotomy of 100 mL blood each day. Because it was recognized too late, despite treatment with phlebotomy, 1 month later, he left hospital and died of sudden cardiac arrhythmia at home. He had been married for 10 years and was sterile with no child. Cardiac symptoms dominate the course of the untreated JH, and heart failure and/or major arrhythmias are reported as the most common causes of premature death of this disease.
PCR and mutation detection
We screened the proband by directly sequencing all exons and flanking splice junctions of the HFE, HAMP, TfR2, SLC40A1 and HJV genes; only two mutations in HJV were identified. The sequence analysis of the mutations is provided in Fig. 1. The proband was heterozygous for 2 mutations in HJV: a premature termination mutation (962G>A and 963C>A; C321X) and a mutation in the signal peptide (18G>C; Q6H) (Fig. 1). Then, we screened the other family members and control for mutations in HJV.

HJV sequencing results. a Proband’s HJV mutation in position nt 18. 18G>C results in Q6H; b Proband’s HJV mutations in position nt 962 and nt 963, 962G>A, 963C>A result in C321X
Family investigation
The pedigree of the proband was shown in Fig. 2a, the parents of proband (II7) were consanguineous. We found that heterozygous mutations Q6H and C321X were also present in I2, II1 and II3. So, II1, II3 and II7 all inherited the abnormal HJV allele from their mother I2, but II5 and II9 inherited another normal HJV allele from their mother I2. The abnormal maternal allele contains a missense change (Q6H) and a nonsense mutation (C321X) in cis (Fig. 2b); these two mutations in HJV were absent in 200 control chromosomes.

The result of genetic analysis in a Chinese family with two male JH patients (II3 and II7). a Four individuals (I2, II-1, II-3, II-7) are heterozygous for the HJV mutation, the others are wild type. b The abnormal HJV allele in this family contains a missense change (Q6H) and a nonsense mutation (C321X) in cis
Physical examination
In this family, totally four individuals carried the abnormal HJV allele. Physical examination results showed that the two female carriers, the mother I2 and the sister II1, both were healthy as the other wild type individuals, while the two male carriers, II3 and II7, were iron overload. Clinical and laboratory findings for the two male patients and the two female carriers are summarized in Table 1. II3, the elder brother of the proband, 47 years old, also had an obvious high-level serum iron. Skin pigmentation had already presented and he developed diabetes at age 45. There was still no evidence of cardiac complications in him. His blood routine examination was as follows: Hb 125 g/L, WBC 4.8 × 109/L, BPC 107 × 109/L. AST 107 U/L (normal 5–40 U/L), ALT 118 U/L (normal 5–40 U/L). Two years ago, his examination results were similar: Hb 143 g/L, WBC 5.3 × 109/L, RBC 4.5 × 1012/L, BPC 155 × 109/L. AST 70 U/L, ALT 84 U/L and Testosterone 0.17 μg/L (normal 1.75–7.81 μg/L).
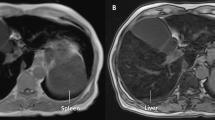
CT was also performed to estimate the density of the liver in 2 patients with hemochromatosis. CT revealed both livers in the patients to be high density area (Fig. 3). The liver attenuation value on CT was 95 HU in the II7 and 91 HU in the II3. Now, we have suggested II3, the elder brother to go to local hospital to get therapeutic phlebotomy to avoid further iron overload.

Computed tomography images of the patients’ livers. CT scanning showed that whole livers of the patients clearly appeared as high-density area because of tissue iron deposition. Hepatic vessels were identified as low-density area, as observed in normal subjects
Western blot analysis of serum hemojuvelin from healthy volunteer and from the four abnormal HJV allele carriers
Hemojuvelin was highly expressed in skeletal muscle and in the liver. It can be expressed both as a membrane form and as a soluble form which have opposite effects on iron regulator hepcidin expression (Fig. 4a, b). Membrane HJV (m-HJV) acts as a co-receptor for bone morphogenetic proteins (BMPs) to enhance hepcidin expression through the smad pathway. Soluble HJV (s-HJV) form competitively down-regulates hepcidin by binding bone morphogenic proteins. Two proteases, furin and matriptase-2 (matriptase-2 was encoded by the TMPRSS6 gene), cleave full-length HJV (426 aa) into s-HJV [11–13].

Analysis of HJV expression in the family. a Structure of the HJV protein, showing the positions of structural domains and motifs. SP signal peptide, RGD RGD motif, vWF partial von Willebrand factor type D domain, TM transmembrane domain. The dotted line indicates the proteolytic cleavage site. Positions of a missense mutation Q6H and a nonsense mutation C321X are marked with the red arrow. The furin cleavage motif is also shown. b Model for release of sHJV. Several isoforms of the hemojuvelin protein are generated by multiple proteolytic cleavages. Membrane hemojuvelin can be represented by both single and two chain species that can remain bound through the disulfide bond. Soluble HJV (40–42 kD) and its shorter fragment of 33 kD are potentially through the action of a furin protease. Other soluble form of HJV can be released by the action of the protease matriptase 2 (TMPRSS6). c Western blot analysis of the serum from healthy volunteer (N) and from four carriers (II7, II3, I2, II1). 100 μg serum was subjected to electrophoresis on 10 % SDS-PAGE, followed by western blot using the antibody against HJV. Black arrows indicate the full-length sHJV (42 kDa) and the processed forms found in serum. The stars represent N-linked glycosylation sites, and the thin lines, disulfide bonds; scissors shape, cleavage sites. C321X was very close to the furin cleavage site
Western analysis of serum HJV from healthy volunteer and from the four abnormal HJV allele carriers is shown in Fig. 4c. The healthy volunteer and the four abnormal HJV allele carriers have the similar migration pattern and the similar expression level. Because C321X truncated site was very close to the furin cleavage site, it was undistinguishable by comparing the sizes of the normal soluble HJV and the C321X truncated form in the serum western blot. Our result was consistent with previous report [10, 14]. The full-length normal HJV (426 aa) and the C321X truncated form (320 aa) can be separated in the western blot with the whole cell lysate of the liver or muscle sample [12, 13], but these samples are not acquired from the family without their permission.
Discussion
Hemojuvelin mutations are the major cause of JH, which is a severe hereditary form of iron overload that manifests itself rather earlier in life (2–3rd decade) [1]. Only homozygous or compound heterozygous states are associated with iron overload, while heterozygous subjects with HJV mutations are symptomless and have normal iron parameters [4, 15, 16].
In the Chinese population, hemochromatosis has been rarely encountered. Reports are even fewer with only several cases [17, 18]. At present, there is difficulty with recognizing HH in some countryside of China; furthermore, the high prevalence of iron deficiency in Chinese populations might have prevented or delayed their clinical expression. It was also reported 3 middle-aged Japanese patients (clinical signs were present around age 50) with hemochromatosis due to HJV mutations [19]. In this investigated Chinese family, the proband II7, especially his elder brother II3, in comparison to the other patients with JH described previously, appears to have later presentation of hemochromatosis [20–22]. In one previously reported case, the woman carried the same mutated HJV allele as occurred in our investigated family was also a Chinese [23]. She was 46 and had normal findings on physical examination [23]. Her genotype of HJV and phenotype are the same as I2 and II1 in our investigated family. Her 19 years old daughter, the hemochromatosis patient, was compound heterozygote in HJV. Until now, no man carrying the same mutated HJV allele was reported. Progressive iron overload associated with HJV mutation is usually observed in the early stages of JH regardless of sex. But why the men and women display different phenotype with this HJV genotype in this Chinese family? Environmental and/or other genetic factors might also contribute to the clinical disease progression in HH. So that is why even all carrying the same mutation, still a phenotypic variation in terms of both ages at diagnosis and organ damage is observed [4].
It was reported that R326X, a similar mutant to C321X truncated form, lacks the GPI anchor motif, is not present on the cell surface, but it is still efficiently secreted, and its soluble form has the same MW of the cellular form [14]. So only one normal allele produces the full-length HJV and binds the membrane while the soluble HJV levels are the same as the wild type in the abnormal allele carriers. It was also reported that soluble HJV injection in mice causes iron overload [24]. We hypothesize that s-HJV and m-HJV must be in balance to modulate the bone morphogenetic protein signaling in vivo regulating systemic iron balance. Therefore, the reduction of m-HJV or relative high level of s-HJV can cause the iron overload. Clearly, much remains to be learned about the phenotypes of various ethnic groups in different environments who have abnormal HJV but no mutations of the HFE gene.
References
Franchini M. Hereditary iron overload: update on pathophysiology, diagnosis, and treatment. Am J Hematol. 2006;81:202–9.
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dubé MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q–linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82.
Lee PL, Beutler E, Rao SV, Barton JC. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood. 2004;103:4669–71.
Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H, et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood. 2004;103:4317–21.
Gehrke SG, Pietrangelo A, Kascák M, Braner A, Eisold M, Kulaksiz H, et al. HJV gene mutations in European patients with juvenile hemochromatosis. Clin Genet. 2005;67:425–8.
Barton JC, Sawada-Hirai R, Rothenberg BE, Acton RT. Two novel missense mutations of the HFE gene (I105T and G93R) and identification of the S65C mutation in Alabama hemochromatosis probands. Blood Cells Mol Dis. 1999;25:147–55.
Lee PL, Halloran C, West C, Beutler E. Mutation analysis of the transferrin receptor-2 gene in patients with iron overload. Blood Cells Mol Dis. 2001;27:285–9.
Majore S, Binni F, Pennese A, De Santis A, Crisi A, Grammatico P. HAMP gene mutation c.208T>C (p. C70R) identified in an Italian patient with severe hereditary hemochromatosis. Hum Mutat. 2004;23:400.
Koyama C, Wakusawa S, Hayashi H, Ueno T, Suzuki R, Yano M, et al. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med. 2005;44:990–3.
Brasse-Lagnel C, Poli M, Lesueur C, Grandchamp B, Lavoinne A, Beaumont C, et al. Immunoassay for human serum hemojuvelin. Haematologica. 2010;95:2031–7.
Kuninger D, Kuns-Hashimoto R, Nili M, Rotwein P. Pro-protein convertases control the maturation and processing of the iron-regulatory protein, RGMc/hemojuvelin. BMC Biochem. 2008;9:9.
Maxson JE, Chen J, Enns CA, Zhang AS. Matriptase-2- and proprotein convertase-cleaved forms of hemojuvelin have different roles in the down-regulation of hepcidin expression. J Biol Chem. 2010;285:39021–8.
Lin L, Nemeth E, Goodnough JB, Thapa DR, Gabayan V, Ganz T. Soluble hemojuvelin is released by proprotein convertase-mediated cleavage at a conserved polybasic RNRR site. Blood Cells Mol Dis. 2008;40:122–31.
Silvestri L, Pagani A, Fazi C, Gerardi G, Levi S, Arosio P, et al. Defective targeting of hemojuvelin to plasma membrane is a common pathogenetic mechanism in juvenile hemochromatosis. Blood. 2007;109:4503–10.
De Gobbi M, Roetto A, Piperno A, Mariani R, Alberti F, Papanikolaou G, et al. Natural history of juvenile haemochromatosis. Br J Haematol. 2002;117:973–9.
Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008;111:5195–204.
Lin A, Yan WH, Xu HH, Zhu M, Zhou MY. Analysis of the HFE gene (C282Y, H63D and S65C) mutations in a general Chinese Han population. Tissue Antigens. 2007;70:252–5.
Kng C, Ng FH, Ng WF, Wong BC, Grosso LE, Brunt EM, et al. A Chinese patient with non-HFE-linked iron overload. J Clin Gastroenterol. 2001;33:69–71.
Koyama C, Hayashi H, Wakusawa S, Ueno T, Yano M, Katano Y, et al. Three patients with middle-age-onset hemochromatosis caused by novel mutations in the hemojuvelin gene. J Hepatol. 2005;43:740–2.
Aguilar-Martinez P, Lok CY, Cunat S, Cadet E, Robson K, Rochette J. Juvenile hemochromatosis caused by a novel combination of hemojuvelin G320V/R176C mutations in a 5-year old girl. Haematologica. 2007;92:421–2.
Janosi A, Andrikovics H, Vas K, Bors A, Hubay M, Sápi Z, et al. Homozygosity for a novel nonsense mutation (G66X) of the HJV gene causes severe juvenile hemochromatosis with fatal cardiomyopathy. Blood. 2005;105:432.
Daraio F, Ryan E, Gleeson F, Roetto A, Crowe J, Camaschella C. Juvenile hemochromatosis due to G320V/Q116X compound heterozygosity of hemojuvelin in an Irish patient. Blood Cells Mol Dis. 2005;35:174–6.
Huang FW, Rubio-Aliaga I, Kushner JP, Andrews NC, Fleming MD. Identification of a novel mutation (C321X) in HJV. Blood. 2004;104:2176–7.
Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–9.
Acknowledgments
This work was supported by grant from the Chinese National Nature Science Foundation (31070706) and by the Fund from Nanjing Health Bureau (ykk10082). We thank Dr. Sheng Zhao for his valuable comments on the manuscript.
Conflict of interest
None of the authors has conflict of interest with regard to the manuscript submitted.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Li, J. Xue, B. Chen, Q. Wang, M. Shi authors contributed equally to this work.
About this article
Cite this article
Li, S., Xue, J., Chen, B. et al. Two middle-age-onset hemochromatosis patients with heterozygous mutations in the hemojuvelin gene in a Chinese family. Int J Hematol 99, 487–492 (2014). https://doi.org/10.1007/s12185-014-1547-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-014-1547-5